Reviews:
Cell Stress, Vol. 3, No. 3, pp. 70 - 85; doi: 10.15698/cst2019.03.178
Role of protein phosphatases PP1, PP2A, PP4 and Cdc14 in the DNA damage response

1 Cell Cycle and Genome Stability Group. Institute of Functional Biology and Genomics (IBFG). Spanish National Research Council (CSIC), University of Salamanca (USAL), C/ Zacarías González 2, Salamanca 37007, SPAIN.
Keywords: phosphatases, DNA damage response, PP1, PP2A, PP4, Cdc14.
Abbreviations:
CLL – chronic lymphocytic leukemia,
DDR – DNA damage response,
DSB – double strand break,
DUSP – dual-specificity phosphatase,
FEAR – fourteen early anaphase release,
HR – homologous recombination,
HU – hydroxyurea,
MEF – mouse embryonic fibroblasts,
MMS – methyl methanesulfonate
NHEJ – non-homologous endjoining,
NHL – non-Hodgkin lymphoma,
PPP – phosphoprotein phosphatase,
SAC – spindle assemble checkpoint,
SPB – spindle pole body, ss – single stranded.
Received originally: 15/11/2018 Received in revised form: 18/01/2019
Accepted: 28/01/2019
Published: 21/02/2019
Correspondence:
Andrés Clemente Blanco, Cell Cycle and Genome Stability Group. Institute of Functional Biology and Genomics (IBFG). Spanish National Research Council (CSIC), University of Salamanca (USAL), C/ Zacarías González 2, Salamanca 37007, SPAIN andresclemente@usal.es
Conflict of interest statement: The authors declare no competing financial interests. Corre-spondence should be addressed to A.C-B.
Please cite this article as: Facundo Ramos, María Teresa Villoria, Esmeralda Alonso-Rodríguez and Andrés Clemente-Blanco (2019). Role of protein phosphatases PP1, PP2A, PP4 and Cdc14 in the DNA damage response. Cell Stress 3(3): 70-85. doi: 10.15698/cst2019.03.178
Abstract
Maintenance of genome integrity is fundamental for cellular physiology. Our hereditary information encoded in the DNA is intrinsically susceptible to suffer variations, mostly due to the constant presence of endogenous and environmental genotoxic stresses. Genomic insults must be repaired to avoid loss or inappropriate transmission of the genetic information, a situation that could lead to the appearance of developmental anomalies and tumorigenesis. To safeguard our genome, cells have evolved a series of mechanisms collectively known as the DNA damage response (DDR). This surveillance system regulates multiple features of the cellular response, including the detection of the lesion, a transient cell cycle arrest and the restoration of the broken DNA molecule. While the role of multiple kinases in the DDR has been well documented over the last years, the intricate roles of protein dephosphorylation have only recently begun to be addressed. In this review, we have compiled recent information about the function of protein phosphatases PP1, PP2A, PP4 and Cdc14 in the DDR, focusing mainly on their capacity to regulate the DNA damage checkpoint and the repair mechanism encompassed in the restoration of a DNA lesion.
INTRODUCTION
Cells are constantly suffering endogenous and exogenous stresses that affect the integrity of the genetic material. It has been estimated that every cell of our body is exposed to about 105 lesions per day. In response to such levels of DNA damage, cells have evolved sophisticated mechanisms that safeguard the stability of our genome. These mechanisms, collectively enclosed under the name of DNA damage response (DDR), are constantly surveying the genome to detect DNA errors and fix them [1][2]. When these mechanisms fail or the rate of DNA damage exceeds the capacity of the cell to deal with it, the increased accumulation of genetic alterations can overwhelm the cell resulting in the appearance of a malignant transformation. As a matter of fact, multiple congenital human disorders have been directly linked to a failure in executing this surveillance pathway, mirroring the importance of the DDR in the maintenance of genome integrity for health and development in humans [3][4][5][6].
–
During the last years, there have been rapid progresses in the characterization of the mechanisms governed by the DNA damage response. Upon generation of a DNA lesion, the main function of the DDR is to couple cell cycle with DNA repair. This is attained by triggering two inter-connected pathways: 1) the DNA damage checkpoint, a molecular mechanism that restrains cell cycle progression to avoid the segregation of the duplicated chromosomes until the broken DNA has been restored, and 2) the activation of specific repair factors responsible for the correct execution of the different phases encompassed in the repair process. The correct activation and coordination of both routes by the DDR ensures a proficient and timely restoration of the DNA molecule. Today we know that the transmission of the signal along these pathways is mainly driven by phosphorylation events by specific kinases that phosphorylate DDR components predominantly at serine and threonine residues [7][8][9]. However, less is known about the role of protein dephosphorylation by protein phosphatases and their implication in the restoration of a DNA lesion. Still, it is reasonable to think that the fine-tuning of the response relies on the activity of phosphatases in order to prevent illegitimate activation of the DDR in the absence of damage as well as to allow a rapid cessation of the signal once the DNA lesion has been fixed. Because of this perspective, most of the studies involving protein phosphatases have focused on their role in counterbalancing DDR-kinases to stimulate cell cycle re-entry upon repair. However, in the last years several studies have revealed that these enzymes are also able to directly modulate the DDR at the repair level, a discovery that has changed the perception of protein dephosphorylation in the response to DNA damage.
–
To date, four different phosphatases have been mainly involved in the DDR: the Ser/Thr protein phosphatase-1 (PP1), the protein phosphatase 2A (PP2A), the protein phosphatase 4 (PP4) and the Cdk-antagonizing phosphatase CDC14. These phosphatases can be classified into two groups on the basis of their sequence, structure and biological activity. PP1, PP4 and PP2A are comprised in the classic Ser/Thr phosphoprotein phosphatases (PPPs) family while Cdc14 forms part of the dual-specificity phosphatase (DUSP) family. One peculiarity of these phosphatases is their ability to counteract a great number of kinases. It has been estimated in human cells that there are about 500 protein kinases, while only 150 phosphatases have been described up to date [10][11]. Due to this difference between the number of kinases and phosphatases, it has always been considered that phosphatases are promiscuous enzymes. Today we know that these proteins are indeed selective and tightly regulated enzymes. The discrimination in target recognition by protein phosphatases is attained by their ability to form specific complexes between a catalytic subunit and multiple regulatory elements that confer the specificity to the holoenzyme [12][13][14]. Each of these multimeric holoenzymes works as a distinct signalling entity by modulating the activity of the catalytic subunit and creating their own substrate specificity. Actually, taking into account the great number of phosphatase complexe rearrangements identified in vivo recent studies postulated that protein phosphatases exhibit similar complexity and specificity as protein kinases.
–
It is important to note that regulation of protein phosphorylation/dephosphorylation during the DDR is critical to maintain genome integrity and prevent the development of diseases such as cancer. Phosphatases are involved in the control of DDR activation after a DNA lesion is generated, as well as to its inactivation when the DNA adduct has been repaired. It is generally accepted that this control might be hijacked by cancer cells to elude the activation of checkpoint pathways during tumorigenesis, allowing tumor cells to grow uncontrolled. Supporting this notion, several types of cancer show an altered regulation of the DDR, a fact that may explain the accumulation of high levels of DNA damage at later stages of the disease. In addition, most oncogenes encode for protein kinases and phosphatases, reflecting the importance of protein phosphorylation in cancer development and progression. Interestingly, protein phosphatases can also operate as tumor suppressors through positive regulation of the DDR [13][15]. In this regard, these enzymes have been implicated not only in the control of the DNA damage checkpoint, but also in the regulation of the repair mechanisms operating in the response. Thus, even though it is quite premature to consider protein phosphatases as specific targets to tackle cancer progression, it is nevertheless an attractive field to work on.
–
In this review, we summarize recent advances in the fundamental principles behind the main DDR-phosphatases PP1, PP4, PP2A and CDC14 in the repair of a DNA lesion and their physiological significance in the regulation of the DNA damage response (Figure 1). We also discuss the potential role of these phosphatases in cancer progression and treatment.
–
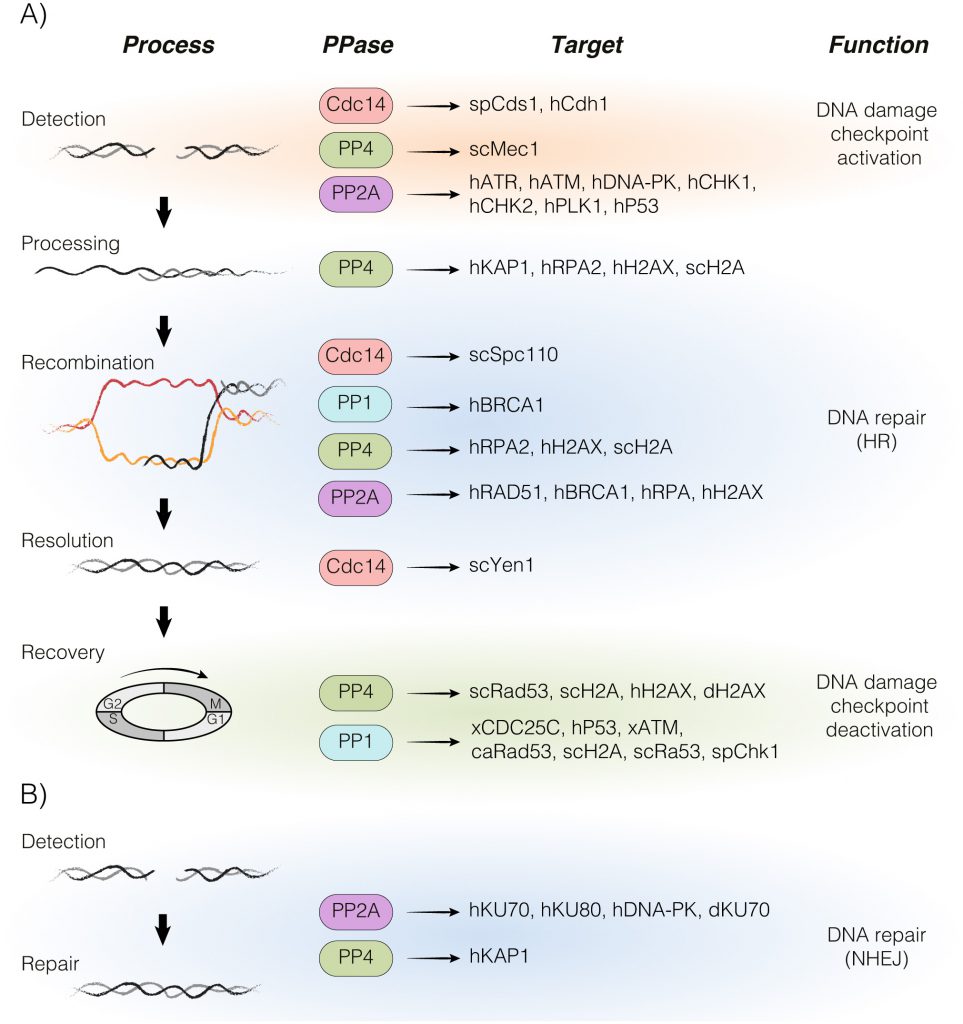 | FIGURE 1: A global overview of the protein dephosphorylation landscape in the DDR. The figure summarizes the participation of PP1, PP2A, PP4 and Cdc14 in each step of the DNA damage response. All phosphatase´s targets identified in different model organisms are also depicted (sc, Saccharomyces cerevisae; sp, Schizosaccharomyces pombe; ca, Candida albicans; d, Drosophila melanogaster; x, Xenopus; h, Human). The involvement of each protein phosphatase in the homologous recombination (A) and non-homologous end joining (B) pathways is portrayed. HR – homologous recombination, NHEJ – non-homologous end joining. |
PP1: FROM CELL CYCLE REGULATOR TO A KEYSTONE MEMBER OF THE DNA DAMAGE RESPONSE
Among all Ser/Thr phosphatases, protein phosphatase-1 (PP1) is perhaps the most broadly and best studied. PP1 is responsible for the majority of the dephosphorylation events taking place in the cell. It has been estimated that one third of all phosphoproteins are dephosphorylated by this phosphatase [16]. As other phosphatase complexes, PP1 is formed by a catalytic subunit and multiple regulatory elements that provide specificity for multiple targets. The central region of the catalytic subunit is practically identical in amino acid sequence between different species, while specific species-dependent variations are observed in the N- and C-terminal domains. Interestingly, the conserved central domain of PP1´s catalytic subunit is shared with PP2A, a feature that could explain the redundancy in protein functions observed between both complexes. As mentioned above, a hallmark of the PP1 enzyme is that its catalytic subunit always works in complex with regulatory elements. Most PP1 regulatory elements interact with the catalytic subunit through a conserved binding region known as the RVxF motif [17]. Up to date, there have been identified more than 200 PP1´s interacting proteins, a feature that reflects the vast number of cellular functions attributed to this holoenzyme including glucose metabolism, transcription, cytoskeleton organization, cell cycle and meiosis [18].
–
One of the first observations that underlined a possible role for PP1 in controlling checkpoints activity was the discovery that its overexpression bypassed the spindle assemble checkpoint (SAC) arrest triggered during the response to spindle-unattached chromosomes [19]. Sister chromatids without tension generate a signal that is transmitted by the Ipl1 and Mps1 kinases to stabilize the separase inhibitor Pds1, thus restraining nuclear segregation [20]. At present, we know that SAC inactivation by PP1 is carried out by its ability to dephosphorylate multiple Ipl1´s substrates [21]. A similar role was also found for PP1 in the control of meiosis. Red1 phosphorylation by Mek1 activates the pachytene checkpoint in response to defects in meiotic recombination and/or chromosome synapsis. However, when meiotic recombination has been accomplished, Red1 dephosphorylation by PP1 relieves the checkpoint arrest to stimulate cell cycle progression [22].
–
In the last years, several lines of investigation have involved PP1 in the DNA damage checkpoint by directly modulating the steady state phosphorylation of DDR factors. One of the first evidences involving PP1 in the DNA damage response came from a screening in Schizosaccharomyces pombe to isolate genes that, when overexpressed, resulted in premature mitotic entry in the presence of genotoxic stress. In this screening, Dis2 (main subunit of the PP1 complex in the fission yeast) was identified as the only requirement to endorse cell cycle re-entry upon DNA repair by dephosphorylating the DNA damage checkpoint effector Chk1 [23]. Interestingly, PP1 was not required for cell cycle resumption in response to replication stress, suggesting that the role of the phosphatase in the control of the DDR in the fission yeast was restricted to enhance cell recovery from G2/M arrested cells responding to physical DNA lesions [23]. In Saccharomyces cerevisiae, PP1 is required for the dephosphorylation of histone γ-H2A and Rad53, a prerequisite for cell cycle resumption from replication stress, extending its role in DDR silencing to the budding yeast [24]. Excitingly, the role of PP1 in checkpoint deactivation must be regulated by its interaction with regulatory elements since Sds22, a well-known regulatory subunit of PP1 in the fungal pathogen Candida albicans, mediates Rad53 dephosphorylation in response to alkylating agents [25].
–
It has been well documented that the DNA damage checkpoint silencing function of PP1 is evolutionary conserved in higher eukaryotes. PP1 is responsible for controlling the threshold of DNA damage checkpoint activation by opposing ATM activity in Xenopus [26]. Importantly, PP1-dependent regulation of ATM is mediated by its regulatory subunit Repo-Man, a factor that stimulates the binding of the holoenzyme to chromatin [26]. The role of PP1 in controlling the levels of checkpoint activation has also been extended to human cells, since it has been demonstrated that inhibition of the damage-dependent p53 signalling pathway depends on PP1 activity [27][28]. Importantly, p53 attenuation is attained by its direct dephosphorylation at Ser15 and Ser37 [29] in a process mediated by different PP1 regulatory factors such as PNUTS [30], p53BP2 [31] and GADD34 [28]. Another well-known substrate of PP1 during the DNA damage checkpoint in higher eukaryotes is the mitotic phosphatase CDC25. CDC25 stimulates mitotic entry by removing inhibitory phosphorylation on the CDK1 subunit Cyclin B [32]. In addition, human CDC25C is phosphorylated by CHK1 and CHK2 in response to DNA damage to prevent its transport into the nucleus, thus restraining mitotic entry [33]. Taking into account that PP1 dephosphorylates CDC25C during the G2/M transition in Xenopus, it is tempting to speculate that cell cycle re-entry might be enhanced by the positive effect that PP1 exerts over CDC25C. However, as CDC25C has been shown to be dispensable for DNA damage checkpoint activation in human and mouse cells [34][35], the molecular significance of PP1-dependent CDC25C dephosphorylation remains controversial.
–
Even though the principal function of PP1 in the DDR seems to be cell cycle re-entry upon repair, it has recently been postulated that PP1 could have also a role in the direct modulation of the repair machinery. In this line, human PP1 binds and dephosphorylates hCds1/Chk2-phosphorylated BRCA1, a function that is essential for its role in promoting recombinational DNA repair following γ-radiation exposure [36][37]. Moreover, depletion of PP1 affects NHEJ (Non-homologous end joining) in both Xenopus and humans [38], establishing a direct role of the phosphatase in the physical restoration of double-strand breaks (DSBs). Importantly, the regulation of PP1 activity by its regulatory subunits along the DDR is also important for the correct execution of the repair process, as the expression of a covalent fusion of PP1 with the regulatory element NIPP1 results in the generation of RNA-DNA hybrids (R-loops), enhanced chromatin compaction, slow replication fork progression and a reduced capacity to deal with DNA lesions [39].
PP2A: AN ESSENTIAL FACTOR FOR THE REGULATION OF DNA DAMAGE CHECKPOINT AND DNA REPAIR
PP2A is a Ser/Thr protein phosphatase belonging to the PPP family of phosphatases conserved in higher eukaryotes. It operates as a heterotrimeric complex consisting of a catalytic subunit (PPP2Cα and PPP2Cβ), a scaffolding subunit (Aα and Aβ), and multiple regulatory subunits (B). The large number of regulatory elements allows the formation of numerous functionally distinct PP2A complexes, explaining the vast number of cellular functions attributed to PP2A [40][41][42]. It is believed that these regulatory subunits dictate the subcellular localization of the holoenzyme and confer substrate specificity. In the eukaryotic model S. cerevisiae, PP2AC is encoded by two identical genes, PPH21 and PPH22, each of them contributing to approximately half of the PP2A activity in the cell [43][44]. TPD3 codifies the subunit A [45] and the regulatory subunits are encoded only by two known distinct genes, CDC55 and RTS1 [46][47].
–
PP2A is one of the most well-studied phosphatases and has been implicated in the regulation of many cellular processes including cell cycle progression [48][49], DNA replication, gene transcription/translation [40], cell differentiation [50] and DNA damage response [51]. Of all these functions, probably the best characterized is the regulation of the G2/M transition. PP2A involvement in cell cycle regulation was originally suggested by several findings showing that its inactivation promoted premature mitotic entry in fission yeast [52]. This observation was also reproduced in budding yeast experiments demonstrating that elimination of the PP2A regulatory subunit Cdc55 resulted in a similar premature mitotic entry due to the loss of function of the holoenzyme [53]. In Xenopus, PP2A regulates the G2/M transition by modulating the phosphorylation levels of the mitotic phosphatase Cdc25 [54], while in S. cerevisiae it seems that its main effector is the kinase Swe1 [55]. A similar molecular mechanism has also been postulated in S. pombe [52] and humans [56]. Importantly, Cdc25 and Swe1 are not the only targets of the phosphatase during the G2/M transition. It has also been reported in S. cerevisiae that PP2A acts as a negative regulator of sister chromatids separation by counteracting polo-kinase phosphorylation of Scc1 to inhibit cohesin cleavage [57]. A similar mechanism has been described in centromeric cohesion during mitosis in human cells [58].
–
The awareness of a role of PP2A in the DDR came from the observation that elimination of its function by treating mice with okadaic acid in a two-stage carcinogenesis experiment led to an increase in tumorigenesis [59][60]. Today we know that PP2A impacts on the damage response by regulating the activity of the primary (ATM, ATR and DNA-PK) and secondary (CHK1 and CHK2) kinases involved in the signalling cascade. It has been reported that PP2A-deficient cells display an increased level of ATM auto-phosphorylation/activation accompanied by an upregulation of the ATM downstream kinase CHK2 [61][62]. This results in the activation of a G1/S arrest and the down-regulation of RAD51 and BRCA1, which mediate DNA repair by homologous recombination (HR) [62]. Consistent with a general role of PP2A in the DDR, several studies have reported that PP2A dephosphorylates not only ATM and CHK2 but also the ATR kinase and its downstream target CHK1. Surprisingly, this function of PP2A is directly involved in the maintenance of a low checkpoint activation in an unperturbed cell cycle while allowing a rapid release from this regulation immediately after DNA damage induction [63][64][65]. A similar hypothesis has been postulated in endothelial cells in response to oxidative stress. In this regard, PP2A induces a rapid dephosphorylation of the protein nucleophosmin, which is translocated from the cytoplasm into the nucleus, thus preventing the formation of γ-H2AX foci [66]. Furthermore, the role of PP2A in controlling the DNA damage checkpoint is reinforced by its ability to dephosphorylate p53. PP2A binds and dephosphorylates p53 at Ser37 after DNA damage, thereby controlling its transcriptional activity [67]. Moreover, dephosphorylation of p53 at Thr55 upon DNA damage stabilizes the protein to enhance a proficient DNA damage checkpoint activation [68]. It is important to remark that PP2A-dependent DDR inhibition is also operating to integrate metabolic signals within the response. In cells experiencing replication stress, PP2A/PP2A-like acts in a network with Irc21 (cytochrome b5 reductase implicated in the production of ceramide) and TORC1 to attenuate the ATR cascade [69]. Once the DNA damage checkpoint has been activated, PP2A also cooperates in its maintenance by dephosphorylating and inhibiting Polo-like kinase 1 (Plk1), a positive regulator of the G2/M transition, thus stimulating a G2 arrest [70][71][72].
–
Excitingly, PP2A is not only required for DNA damage checkpoint regulation, but also for DNA repair. It is already known that in response to DNA damage induced by hydroxyurea (HU), the single-stranded (ss)DNA-binding protein RPA32 is phosphorylated at Thr21 and Ser33 in an ATM/ATR-dependent manner. It is believed that RPA32 phosphorylation suppresses DNA replication and enhances the recruitment of other checkpoint/repair proteins to DNA lesions. Interestingly, PP2A-mediated RPA32 dephosphorylation is required for the completion of the DNA repair process [73]. This result is reinforced by the observation that PP2A is involved in regulation of the steady-state phosphorylation of H2AX. Indeed, it has been demonstrated that dephosphorylation of γ-H2AX during DNA repair removes it from nuclear foci near the break sites and elicits DNA repair. PP2A-dependent γ-H2AX removal from chromatin is directly linked to its role in enhancing DNA repair as PP2A-deficient cells are affected in the restoration of a DNA lesion and are hypersensitive to DNA-damaging agents [74][75]. Modulation of RPA and H2AX phosphorylation levels directly connects this phosphatase with the accomplishment of the HR pathway. However, PP2A has also been involved in stimulating NHEJ since it dephosphorylates DNA-PKcs, Ku70 and Ku80, in vitro [76]. Similar data were also obtained in Drosophila melanogaster, where failure of the B55-mediated dephosphorylation generates abnormal and/or untimely phosphorylation of Ku70, which interferes with DNA repair and causes the appearance of chromosome aberrations [77].
PP4: A PHOSPHATASE COMPLEX WITH MULTIPLE ROLES IN THE DNA DAMAGE RESPONSE
Over two decades since its discovery in the nineties, protein phosphatase 4 (Ppp4/PP4/PPX) has earned a recognised place as a ubiquitous Ser/Thr phosphatase that regulates many cellular functions independently of other protein phosphatases of the PPP family. Mammalian Ppp4 was predicted from several cDNAs and identified for the first time in a library screening with a PP1 cDNA looking for distinct forms of this enzyme in different tissues. The new protein phosphatase, termed protein phosphatase X or protein phosphatase 4 in the human genome nomenclature, was 41% identical to PP1 and 65% identical to PP2A [78][79]. Despite its ≈65% amino acid identity to PPP2Cα and PPP2Cβ isoforms and the high similarity with D. melanogaster PP4, it was early proposed that mammalian Ppp4 developed distinct cellular roles from PP2A. However, the main subunit of the PP4 complex in S. cerevisiae (Pph3) is not essential and was isolated for the first time as a new PP2A-related protein. Whereas double inactivation of the catalytic subunits Pph21 and Pph22 showed a weak defect on spores growth, triple disruption of Pph3 with Pph21 and Pph22 completely prevented growth, indicating that Pph3 provides some PP2A-complementing activity that contributes to the viability of PP2A-deficient cells [44]. According to the overlapping functions with the PP2A orthologues, Pph3 shares a great similarity in amino acid sequence with both Pph21 and Pph22 (82.18% and 82.51%, respectively), implying that this protein could be part of the PP2A holoenzyme. Both mammalian Ppp4 and its counterpart Pph3 contain all the conserved motifs indicative of the PPP family of protein Ser/Thr phosphatases, and they are also closely related to human Ppp6, D. melanogaster PPV-6A and S. cerevisiae Sit4 [80].
–
Like most Ser/Thr phosphatases, the substrate specificity of Ppp4 depends on its interaction with its regulatory subunits. In mammals, Ppp4, the catalytic subunit of PP4, is accompanied by two structurally distinct regulatory subunits, termed R1 (105 kDa) and R2 (55 kDa), which do not interact with PP2A. In contrast, the regulatory subunit α4 (39 kDa) dimerises with protein phosphatase catalytic subunits of Ppp4, PP2A and Ppp6 [80]. In S. cerevisiae, the open reading frame (ORF) YBL046w, named Psy4, was identified as a putative mammalian R2 orthologue, and Tap42p (two A phosphatase associated protein) as an α4 orthologue [80]. Psy2 (for platinum sensitivity 2) has also been identified as a Pph3 regulatory subunit in S. cerevisiae. This protein has two orthologues in humans, named R3A and R3B, which exist in complex with R2-Ppp4 and share an overall sequence similarity with Psy2 [80][81].
–
Regarding its molecular functions, PP4 has been implicated in numerous cellular processes. Among them, it is implicated in organelle assembly through its role in centrosome maturation in D. melanogaster and Caenorhabditis elegans [82][83], and for spliceosomal assembly via its interaction with the survival of motor neurons (SMNs) complex in mammals [84]. Ppp4 is also involved in several cellular signalling routes including the NF-κB pathway, a key factor in immune and inflammatory responses and tumorigenesis. Ppp4 is thought to dephosphorylate and activate NF-κB in response to cisplatin. This activation is associated with the development of drug resistance against this anticancer treatment. These data suggest that the phosphatase could be the reason behind the increased resistance of some tumor cells to cisplatin [85]. Besides, PP4 is also involved in regulating cell growth both in yeast and mammals in response to nutrients by controlling the TOR pathway. When cells grow on a poor source of nitrogen, Pph3 dissociates from Tap42 and binds to Psy4 to dephosphorylate Gln3 and Ure2. Dephosphorylated Gln3 can move to the nucleus and activate the transcription of genes needed for growth in poor nitrogen sources [86]. In addition, it has been described that Ppp4-R1 complexes decrease the activity of the histone deacetylase HDAC3 by dephosphorylating Ser424, implicating Ppp4 in the regulation of histone acetylation and chromatin remodelling [87]. Finally, budding yeast PP4 controls centromere pairing in meiosis by counteracting Mec1-dependent phosphorylation of Zip1 during the meiotic prophase [88].
–
The first implication of the PP4 complex in the DDR came from a genome-wide screening to identify proteins required for the repair of DNA lesions generated by UV light. PP4 regulatory subunit Psy2 was identified as a binding partner of Wss1p and Tof1p, which are involved in the response to replication stress. The analysis of genetic interactions between these proteins concluded that Wss1, Tof1 and Psy2 (probably together with the Pph3-Psy4 complex) are implicated in the stabilization of stalled or collapsed replication forks [89]. Over the years, it has become evident that the main role of PP4 in the DDR is to promote cell recovery once the DNA lesion has been repaired. It is well known that DNA damage checkpoint activation requires Rad53 phosphorylation by the DDR-specific kinases Mec1 and Tel1 (yeast homologues of mammalian ATR/ATM), as well as its own auto-phosphorylation. Deactivation of Rad53, a prerequisite to resume cell cycle after DNA repair, is attained through its dephosphorylation by different phosphatases, among them PP4. Pph3 forms a complex with its regulatory subunit Psy2 to dephosphorylate activated Rad53 during recovery from methyl methanesulfonate (MMS)-mediated DNA damage both in vitro and in vivo [90]. Together with Rad53 deactivation, dephosphorylation of γ-H2AX by PP4 is also necessary for an efficient recovery from the DNA damage checkpoint in Drosophila, S. cerevisiae and humans [77][91][92]. Besides, depletion of human Ppp4 outcomes in a prolonged checkpoint arrest, in part due to the persistence of MDC1 (mediator of DNA damage checkpoint 1) bound to γ-H2AX at the sites of DNA lesions [92]. These data have been reinforced in budding yeast experiments, demonstrating that the delay in checkpoint recovery manifested in pph3Δ cells were alleviated by the expression of a non-phosphorylatable hta1-S129A version [91]. Taking into account that there is an additive increase in MMS sensitivity when PPH3 is deleted in an hta1-S129A mutant, it seems that Pph3 function in γ-H2AX dephosphorylation is mechanistically independent of its role in Rad53 deactivation. Confirming this hypothesis, hyper-phosphorylation of γ-H2AX in psy4Δ cells does not affect Rad53 dephosphorylation [90]. Surprisingly, PP4 is not only required for DDR deactivation but also for its activation. Pph3 and Psy2 were identified as Mec1-Ddc2 (ATR-ATRIP homologs in humans) regulators in two independent screenings using a mec1-100 mutant, which is compromised in Rad53 phosphorylation specifically in S-phase arrested cells. Phosphoproteomic analysis revealed that PP4 has the ability to dephosphorylate Ser1991 from Mec1. Interestingly, this function is attained by the physical interaction between Ddc2-Mec1 and the Pph3-Psy2 phosphatase complex at sites of replication fork collapse and DSBs, thus facilitating a coordinated action between the kinase and the phosphatase over many targets in response to replication stress caused by HU treatment [81].
–
Recently, it has been demonstrated that beyond its functions in checkpoint regulation, PP4 is also involved in DNA repair. Ppp4-R2 complex mediates NHEJ repair of I-SceI-induced DSBs at least partially through regulation of KAP1 (KRAB-associated protein 1) phosphorylation. KAP1 phosphorylation upon DNA damage enhances its binding to chromatin and relaxes it, thereby facilitating DNA repair. Immunoprecipitation assays have revealed that Ppp4-R2 physically associates with KAP1 to dephosphorylate it, since depletion of PPP4 or PP4R2 leads to an increase of KAP1 phosphorylation at Ser824 upon camptothecin (CPT) or etoposide treatment [93]. Interestingly, PP4 is also required for recombinational DNA repair by HR. This process requires DNA end resection to allow homology search. The ssDNA generated during resection is immediately protected by the binding of several RPA subunits, including RPA2, which is another target of the human Ppp4-R2 complex in response to replication stress or damage. Depletion of PP4C or PP4R2 results in an increased RPA2 phosphorylation levels, a situation that impedes HR-mediated DSB repair by affecting the loading of the essential factor RAD51 [94]. As for RPA2 dephosphorylation, PP4´s role in dephosphorylating H2AX is also essential to stimulate DNA repair. Human phosphatase complex Ppp4-R2-R3B eliminates ATR-mediated γ-H2AX formed with or without exogenous DNA damage generated during DNA replication to enhance a proficient repair of the DNA molecule [92][95]. This function of PP4 must be evolutionary conserved since yeast Pph3 also regulates basal γ-H2A levels even in the absence of exogenous damage. Importantly, considering that Pph3 does not localize at an engineered DSB, PP4 function in γ-H2A dephosphorylation probably occurs once γ-H2AX has been removed from chromatin [91].
CDC14: A NEW PLAYER IN THE RESPONSE TO DNA DAMAGE
The cell division cycle 14 (Cdc14) is one of the most studied families within the DUSPs. These proteins are characterized by their ability to dephosphorylate both phosphotyrosine and phosphoserine/phosphothreonine residues in their substrates. The Cdc14 phosphatases family is highly conserved, and orthologs have been described in several organisms. One special feature of this family is its predisposition to dephosphorylate targets of the cyclin-dependent kinase (Cdk). In particular, the Ser/Thr phosphatase Cdc14 was firstly identified in S. cerevisiae as an essential cell cycle protein required for Cdk inactivation and mitotic exit [96]. In the budding yeast, the function of this protein is highly regulated by cell cycle-dependent changes in its localization. During interphase, Cdc14 is retained at the nucleolus by interacting with Net1 (also called Cfi1), a subunit of the RENT (Regulator of the nucleolar silencing and telophase exit) complex. As cells enter in early anaphase, Cdc14 spreads first to the nucleus and then to the cytoplasm by the activation of two regulatory networks known as FEAR (Fourteen early anaphase release) and MEN (Mitotic exit network), respectively [97]. In contrast to the high dependence of Cdc14 for exiting mitosis in the budding yeast, the S. pombe homologue Cdc14-like phosphatase 1 (Clp1; also known as Flp1) is not essential for accomplishing mitosis [98][99]. This different requirement of Cdc14/Clp1 between budding and fission yeasts has always been attributed to the distinctive roles of both phosphatases in the execution of the mitotic exit program. However, the fact that a severe depletion of Cdc14 in S. cerevisiae by using an auxin-inducible degron does not affect mitosis exit has challenged this point of view, and has reunited both organisms under the common idea of multiple phosphatases cooperating in the removal of Cdk phosphoresidues throughout mitosis [100]. Regarding Flp1 localization during the cell cycle, it localizes at the nucleolus and the spindle pole body (SPB) during G1- and S-phases. However, there are some differences between the budding and the fission yeast in terms of release and activation of the phosphatase. Flp1 release from the nucleolus takes place in metaphase and does not depend on the FEAR pathway [101]. Nevertheless, once released it relocates first to the mitotic spindle and kinetochores and later at the contractile ring, similarly to that in the budding yeast [102]. In vertebrates, three homologous of Cdc14 yeast have been characterized (CDC14A, CDC14B and CDC14C). During interphase, Cdc14A is localized at centrosomes while Cdc14B is mainly nucleolar. As in their yeast counterparts, Cdc14B is also liberated from the nucleolus in anaphase to relocate to the sister chromatids [103]. Little is known about the cellular localization and spatial regulation of Cdc14C. However, taking into account the great similarity in protein sequence with Cdc14B, it is thought that both phosphatases might be genetically redundant.
–
Regarding Cdc14´s molecular functions, multiple studies on different orthologues from yeast to humans have revealed a large number of roles in different cellular processes such as cytokinesis [104][105], chromosome segregation [106][107], transcription [106][108][109], centrosome duplication [107], ciliogenesis [110] and in resolving linked DNA intermediates [111-113]. Interestingly, it has been demonstrated that Cdc14A and Cdc14B can rescue the lack of Flp1 in fission yeast [114]. In addition, Cdc14A is able to complement the essential function of S. cerevisiae Cdc14 in cells lacking the activity of the phosphatase [115]. These evidences suggest that some functional properties of these phosphatases are conserved along different species. In this section we will not discuss the role of Cdc14 in mitotic exit and its regulation during the cell cycle since excellent reviews focused on these topics have been published [107][116][117][118][119][120].
–
As mention before, one special feature of the Cdc14 phosphatase family is its predisposition to dephosphorylate targets that have previously been phosphorylated by the Cdk. Since the Cdk controls multiple aspects of the DDR pathway, it is tempting to speculate that the main role of Cdc14 in these processes might be related to its capacity to revert the phosphorylation events imposed by this kinase. In this line, efforts have been focused on understanding the role of this phosphatase family by counteracting Cdk substrates of the DDR. One of the first evidences regarding the role of Cdc14 in DNA damage came from the fission yeast, where Flp1 is translocated from the nucleolus to the nucleus after DNA replication stress induced by HU [121]. Nucleolar release of Flp1 is regulated by the checkpoint kinase Cds1, which is recruited to stalled forks during replication stress. Surprisingly, Flp1 seems to regulate the complete activation of Cds1 through a positive feedback loop that allows a proficient execution of the DNA damage checkpoint in response to replication stress [121]. Corrobo-rating the phosphatase release/activation in response to genotoxic stress, mammalian Cdc14B is also translocated from the nucleolus to the nucleoplasm. Cdc14B liberation promotes the degradation of Plk1 by the ubiquitin ligase APC/C-Cdh1, resulting in the stabilization of the DNA damage activator Claspin and the cell cycle inhibitor Wee1, allowing the proficient G2 checkpoint activation [122]. Together with previous observation demonstrating that Cdc14 is required for Cds1 activation in the fission yeast, it seems that the function of the phosphatase is exclusively related to the activation of the DNA damage checkpoint both in yeast and mammalian cells. Unexpectedly, the publication of two independent studies created controversy about the function of the Cdc14 phosphatases family in the DDR. Using both avian and human somatic cell lines it was demonstrated that neither Cdc14A nor Cdc14B are required for DNA damage checkpoint activation. Indeed, Cdc14A/B knockout cells arrested efficiently in G2-phase without affecting the activation of Chk1 and Chk2 in response to irradiation. However, these cells showed defects in repairing endogenous and exogenous DNA damage, accumulated γ-H2AX foci (as marker for DSBs) and developed hypersensitivity to irradiation [123]. Supporting these results, Cdc14b-deficient mouse embryonic fibroblasts (MEFs) exposed to DNA damage also accumulated endogenous DNA damage and triggered senescence. However, no defects in DNA damage checkpoint activation were observed, indicating that the function of the phosphatase is only restricted to promote efficient DNA repair [124]. The precise mechanism by which Cdc14b is operating in the repair of a DNA lesion is unclear but it seems that Cdc14a is also required for the same process, suggesting that both phosphatases could share at least some substrates and be redundant in terms of function. Supporting this idea, it has been demonstrated that Cdc14B knockout MEFs have defects in repairing DSBs induced by ionizing radiation (IR) but only at late passages when Cdc14A levels are low [125].
–
One of best-characterized models for the study of the DDR is S. cerevisiae. However, the limited evidences regarding a function of Cdc14 in DNA damage response in this organism makes it difficult to understand whether the phosphatase is involved in DNA checkpoint regulation, DNA repair, or both. In this line, it has recently been shown that Cdc14 is transiently released under different sources of DNA damage in the budding yeast [126]. Mass spectrometry data of cells exposed to a DNA lesion revealed multiple targets of the phosphatase in the DDR, suggesting that Cdc14 might have multiple functions in the damage response. Interestingly, it has been found that Cdc14 activity is required for DSB recruitment to the proximities of the SPBs, a vital feature that stimulates DNA repair by HR [126]. Cdc14 stimulates DSB-SPB tethering by dephosphorylating the intranuclear receptor for the γ-tubulin complex Spc110 during the response to DNA damage. Accordingly, both lack of Cdc14 activity and alteration of the steady-state phosphorylation of Spc110 disrupt DSB-SPB interaction and impair DNA repair by HR [126]. These pieces of evidences put forward a new line of Cdc14-dependent DDR regulation by acting over the spatial distribution of its components along the damage response. In this regard, it has been proposed that this layer of regulation is likely to be advantageous for cells to coordinate cell cycle progression with DNA repair, as well as to recruit factors that are required for certain steps during the damage response. This is in line with previous observations demonstrating that unrepairable or slowly repaired DSBs are recruited to the nuclear periphery by interacting with the nuclear pore component Nup84 [127] or the nuclear envelope protein Mps3 [128][129] in yeast. These mechanisms are believed to collaborate in promoting legitimate DNA repair by HR. It is unclear if this regulation exists in mammals, but the fact that DSB ends are positionally stable in mammalian cells could explain why these cells have a predilection for the NHEJ pathway. Nevertheless, it would be interesting to demonstrate if this new layer of Cdc14-dependent regulation of the DDR is also extended to high eukaryotes.
–
In addition to the role of Cdc14 in DNA damage checkpoint and DNA repair, it has been proposed that the phosphatase is also required for the resolution of intermediates that are generated during the response to DNA lesions. Interestingly, Cdc14 activity is responsible for the accumulation of the active form of the budding yeast Holliday junction resolvase Yen1 [111][112][113]. The precise activation of this protein is crucial for the coordination of the DNA repair with chromosome segregation in order to maintain genome stability. These evidences suggest that Cdc14 function in the DDR may be not only restricted to control the damage checkpoint and repair processes, but also the precise resolution of recombination intermediates generated during the response to DNA damage. If Yen1 activation by Cdc14 is an active process that accompanies the repair of a DNA lesion during its damage-dependent release or a passive mechanism operating during the FEAR/MEN activation is an interesting question for the future.
–
Despite all the information regarding the role of Cdc14 in the DDR, the molecular mechanism by which this phosphatase exerts its function and its regulation during the damage response is still unclear and more effort is required to determine its precise role in the damage response. Remarkably, taking into account the functional redundancy of Cdc14 between different model organisms, it is reasonable to think that the combination of all information can be the answer for the interrogation concerning its role in the DDR.
INVOLVEMENT OF DDR PHOSPHATASES IN CANCER DEVELOPMENT
During the last years, the repercussion of protein phosphatases in the regulation of the DNA damage response has become clearer. Numerous studies have set the basis for the role of protein dephosphorylation in the regulation of DNA repair at each step of the repair pathway. These observations, together with the fact that protein phosphatases have been implicated in multiple cellular processes, strongly suggest that these enzymes are essential for the maintenance of genome integrity. Supporting this hypothesis, alteration in the expression pattern of several phosphatases and mutations in their sequence have been described in numerous carcinoma cells, highlighting the importance of these enzymes in genome stability. In this regard, protein phosphatases are believed to act either as tumor suppressors or oncogenes [13][15]. Therefore, they are considered as good candidates for cancer therapy. Accordingly, some experiments have pointed out the use of phosphatases activating drugs to antagonize cancer development and progression. In addition, reactivation of some phosphatases kills cancer cells while spanning normal cells.
–
One of the first phosphatases directly related to cancer development was PP1. As mention above, PP1 has been involved in the regulation of different cellular pathways, including cell cycle progression and apoptosis, by modulating their main components AKT, APAF-1, AURK, BCL-2, BRCA1, Caspases, CDC25, pRb and p53 [130]. Any disruption in the normal execution of these pathways can lead to the development of cancer so efforts have been focused on understanding the role of PP1 and its regulatory subunits in cell homeostasis. One clear example of its role in tumorigenesis is the fact that PP1 interacts with the breast cancer susceptibility gene BRCA1 [36][131][132]. It is worth mentioning that any alteration in the expression of genes that regulate or interact with BRCA1 can negatively affect to the function of this protein and promote the development of breast cancer. In this line, it has been demonstrated that sporadic breast tumors have variable levels of PP1. Strikingly, some PP1 isoforms are less expressed in breast tumors cells when compared to the normal tissue [132], reflecting the essential role of this phosphatase in cancer development. Supporting these observations, it has recently been postulated that SDS22, a regulatory subunit of PP1, inhibits the growth of breast cancer cells by inducing apoptosis, demonstrating the molecular mechanism by which PP1 negatively regulates the AKT pathway [133]. It is important to note that, due to the role of PP1 in numerous cellular processes, it is difficult to consider this phosphatase as a target for therapy, since the use of drugs that affect the activity of the main catalytic subunit produces a wide range of undesirable toxic side effects. Therefore, the current therapeutic methods are focused on targeting PP1-interacting regulators to alter specific functions of the phosphatase in the DDR. This novel approach was inspired by the realization that most PP1 inhibitors bind to the catalytic subunit through interaction motifs, such as the RVxF docking site. These discoveries have postulated the possibility to modulate a concrete function of PP1 by targeting the RVxF sequence of a particular regulatory element in order to provide a greater selectivity. As a matter of fact, disruption of PPP1C interaction with the regulators GADD34 and HDAC1/6 by treating with the small molecules compounds Salubrinal and trichostaitin A, respectively, has successfully been used in several therapeutic approaches [134].
–
Together with PP1, PP2A regulates most of the phosphoproteome of the cell. This explains the large number of functions attributed, including modulation of apoptosis, DDR, immune checkpoint signalling and cell proliferation and survival. Interestingly, all these mechanisms are affected during tumorigenesis, mirroring the importance of PP2A in controlling cell homeostasis. Historically, the potential role of PP2A in cancer was postulated when it was reported that okadaic acid, a potent inhibitor of the holoenzyme, had a drastic effect in tumor progression through B-catenin transcription and AKT activation [135]. This evidence was supported by early genetic models of malignant transformation that described the role of PP2A as a tumor suppressor. In addition, several mechanisms that inhibit this phosphatase in cancer have been described [136][137]. Based on these observations, anticancer therapies involving PP2A have been focused on the activation of the phosphatase in order to prevent cancer development, progression and resistance to other treatments [136][138][139]. In this regard, preclinical studies have shown that PP2A reactivation by using FTY720 effectively prevents cancer development, progression and resistance to other treatments [136][138][139]. Conversely, inhibition of PP2A to complement chemotherapy and radiation-induced cancer cell death is also an area of active investigation. Direct impairment of PP2A blocks critical defense pathways, rendering cells susceptible to accumulate high levels of DNA damage which ultimately induce apoptosis. On this subject, it has been demonstrated that PP2A´s role in cell homeostasis and DNA damage repair has different vulnerabilities in cancer cells, where the inhibition of PP2A coupled with additional DNA damaging strategies may be therapeutically beneficial [139][140][141].
–
Alteration of levels or function of PP4 and Cdc14 is also related with malignant transformation and tumorigenesis. PP4 participates in the regulation of microtubule organization, apoptosis, tumor necrosis, immune system signaling and DDR [81][95][142][143][144][145], pathways involved in cancer development. One of the first evidences that associates the lack of PP4 with tumorigenesis came from S. cerevisiae, where pph3 and psy2 mutants showed a high sensitivity to cisplatin [146][147], a platinum-derived drug used in the treatment of solid tumors. Moreover, PP4 was found to be over-expressed in human breast and lung tumors [148], and high levels of this phosphatase are related with aggressive colorectal carcinoma [149]. There are not clear evidences about the involvement of Cdc14 in cancer development. However, due to the important function of the phosphatase in cell cycle and DNA repair, it is reasonable to think that this phosphatase might also be involved in tumor development and progression. Supporting this notion, it has been shown that inactivation of Cdc14 in S. cerevisiae leads to chromosome rearrangements and genome instability [150]. Surprisingly, human CDC14A activity is required to control cell adhesion and migration, since the lack of this phosphatase may induce tumor proliferation and metastasis [151]. Indeed, several types of cancer present a down-regulation of CDC14A expression, corroborating the role of this phosphatase in tumorigenesis. Interestingly, overexpression of Cdc14b in mouse cells induces cell transformation due to changes in morphology by disrupting F-actin organization [152]. These data suggest that Cdc14 can act both as an oncogene or a tumor suppressor depending on the context and the cell line. Importantly, it has been postulated that Cdc14b-deficient mouse cells are prone to develop a premature-aging phenotype probably due to the role of the phosphatase in DNA repair [124].
DDR PHOSPHATASES AND THERAPEUTIC APPROACHES
There are a large number of strategies to modulate phosphatase activity, most of them relying on the use of small molecules that bind to different subunits of the complex to endorse conformational changes that lead to the activation/inhibition of the holoenzyme. To date, several compounds have been identified as inhibitors of the PPP phosphatases. Most of them were originally extracted from natural products generated by organisms with a great biological variability. Probably, the most known inhibitor of protein phosphatases is the okadaic acid, a toxin complex polyether fatty acid produced by dinoflagellates. Its discovery meant a huge breakthrough in the study of Ser/Thr phosphatases function. Multiple studies demonstrated that okadaic acid is a potent inhibitor of PP1, PP2A and PP4 [153]. Similarly, calyculin A, identified from a marine sponge extract, is a potent inhibitor of these phosphatases [154]. Interestingly, inhibition of PP1/PP2A by using okadaic acid or calyculin A has been reported to reduce tumor resistance to radiation or chemotherapy [155], confirming the veracity of these phosphatase inhibitors at the therapeutic level. A variety of cyanophyte strains produce cyclic peptide inhibitors, known as microcystins, which are responsible for the hepatotoxicity of certain species of cyanobacteria. These toxic compounds potently inhibit the activity of PP1, PP2A [156][157] and PP4 [153]. On the other hand, tautomycin is the only naturally occurring toxin that presents slightly higher affinity for PP1 than PP2A [158][159]. There are also some antitumor agents able to inhibit phosphatases of the PPP family, like cantharidin or fostriecin. Cantharidin, which is naturally produced by insects, owns inhibitory activity over PP1, PP2A and PP4 [153][160]. Interestingly, cantharidin has been successfully applied in lung and bladder cancer models, making this new drug a promising alternative for the treatment of these diseases [161]. Regarding fostriecin, identified in Streptomyces as tautomycin, is a strong inhibitor of PP2A and PP4, and a weak inhibitor of PP1 [153][162][163]. Both of them lead to a G2/M-phase arrest, but their use in oncology is limited due to toxicity or stability problems. Even though phosphatase inhibitors could be idyllic for the cancer treatment, the restricted selectivity of these compounds and the different existing isoforms of some protein phosphatases in the cell, turn the development of specific PPase inhibitors into a very hard task.
–
While phosphatase inhibition has been the most common therapeutic treatment employed, during the last decade there has been a drastic emergence of a large number of phosphatase activators used as new strategies in cancer therapy. Recently, there have been identified several small molecules that bind to the scaffolding subunit of PP2A to induce the holoenzyme activity, including perphenazine, a tricyclic neuroleptic, SMAP, and a re-engineered version of tricyclic sulfonamide [164][165][166]. Interestingly, the activation of PP2A can also be tackled by controlling the regulatory subunits of the holoenzyme. The oncoprotein SET is a potent inhibitor of the PP2A holoenzyme, whose levels are dramatically increased in primary chronic lymphocytic leukemia (CLL) and non-Hodgkin lymphoma (NHL) cells. It was reported that COG449, a peptide that binds antagonistically to SET, increased cellular PP2A activity, decreased Mcl-1 expression, and displayed selective cytotoxicity for CLL and NHL cells in vitro [167]. These results demonstrate that reactivation of PP2A by modulating its regulatory elements can be considered as a novel treatment in B-cell malignancies. Finally, FTY720, a synthetic sphingosine immunosuppressant that has extensively been used for the treatment of multiple sclerosis, has also been proven to be effective in multiple pre-clinical models of cancer, including leukemia, colon cancer, non-small cell lung cancer, breast cancer, hepatocellular carcinoma and prostate cancer [168]. Importantly, the anti-tumor effect of FTY720 has also been attributed to its ability to disrupt the interaction between SET and PP2A, resulting in an increased PP2A activity and cancer cell death [169]. The same mechanism of action has been proven for ApoE (Apolipoprotein E) and TGI1002. However, the use of these small molecules needs to be further investigated to provide better insights into their mechanism of action and possible side effects.
–
We have just started to have an idea of the role of protein phosphatases in DNA repair, and much work is required to understand the physiological significance of these enzymes during the repair of a DNA lesion. In this regard, the understanding of the specific functions of each phosphatase in the DDR pathway is vital for the development of new approaches in the treatment of different types of cancer characterized by the alteration of protein phosphatases activity.
REFERENCES
- Ciccia A, Elledge SJ (2010). The DNA damage response: making it safe to play with knives. Mol Cell 40(2): 179-204. doi: 10.1016/j.molcel.2010.09.019
- Panier S, Durocher D (2013). Push back to respond better: regulatory inhibition of the DNA double-strand break response. Nat Rev Mol Cell Biol 14(10): 661-672. doi: 10.1038/nrm3659
- Jackson SP, Bartek J (2009). The DNA-damage response in human biology and disease. Nature 461(7267): 1071-1078. doi: 10.1038/nature08467
- O’Connor MJ (2015). Targeting the DNA Damage Response in Cancer. Mol Cell 60(4): 547-560. doi: 10.1016/j.molcel.2015.10.040
- O’Driscoll M (2012). Diseases associated with defective responses to DNA damage. Cold Spring Harb Perspect Biol 4(12): a012773- a012773. doi: 10.1101/cshperspect.a012773
- Shimizu I, Yoshida Y, Suda M, Minamino T (2014). DNA damage response and metabolic disease. Cell Metab 20(6): 967-977. doi: 10.1016/j.cmet.2014.10.008
- Bensimon A, Aebersold R, Shiloh Y (2011). Beyond ATM: the protein kinase landscape of the DNA damage response. FEBS Lett 585(11): 1625-1639. doi: 10.1016/j.febslet.2011.05.013
- Finn K, Lowndes NF, Grenon M (2012). Eukaryotic DNA damage checkpoint activation in response to double-strand breaks. Cell Mol Life Sci 69(9): 1447-1473. doi: 10.1007/s00018-011-0875-3
- Trovesi C, Manfrini N, Falcettoni M, Longhese MP (2013). Regulation of the DNA damage response by cyclin-dependent kinases. J Mol Biol 425(23): 4756-4766. doi: 10.1016/j.jmb.2013.04.013
- Alonso A, Sasin J, Bottini N, Friedberg I, Friedberg I, Osterman A, Godzik A, Hunter T, Dixon J, Mustelin T (2004). Protein tyrosine phosphatases in the human genome. Cell 117(6): 699-711. doi: 10.1016/j.cell.2004.05.018
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S (2002). The protein kinase complement of the human genome. Science 298(5600): 1912-1934. doi: 10.1126/science.1075762
- Freeman AK, Monteiro AN (2010). Phosphatases in the cellular response to DNA damage. Cell Commun Signal 8: 27. doi: 10.1186/1478-811X-8-27
- Peng A, Maller JL (2010). Serine/threonine phosphatases in the DNA damage response and cancer. Oncogene 29(45): 5977-5988. doi: 10.1038/onc.2010.371
- Shimada M, Nakanishi M (2013). Response to DNA damage: why do we need to focus on protein phosphatases? Front Oncol 3: 8. doi: 10.3389/fonc.2013.00008
- Stebbing J, Lit LC, Zhang H, Darrington RS, Melaiu O, Rudraraju B, Giamas G (2014). The regulatory roles of phosphatases in cancer. Oncogene 33(8): 939-953. doi: 10.1038/onc.2013.80
- Heroes E, Lesage B, Gornemann J, Beullens M, Van Meervelt L, Bollen M (2013). The PP1 binding code: a molecular-lego strategy that governs specificity. FEBS J 280(2): 584-595. doi: 10.1111/j.1742-4658.2012.08547.x
- Egloff MP, Johnson DF, Moorhead G, Cohen PT, Cohen P, Barford D (1997). Structural basis for the recognition of regulatory subunits by the catalytic subunit of protein phosphatase 1. EMBO J 16(8): 1876-1887. doi: 10.1093/emboj/16.8.1876
- Cannon JF (2010). Function of protein phosphatase-1, Glc7, in Saccharomyces cerevisiae. Adv Appl Microbiol 73: 27-59. doi: 10.1016/S0065-2164(10)73002-1
- Francisco L, Wang W, Chan CS (1994). Type 1 protein phosphatase acts in opposition to IpL1 protein kinase in regulating yeast chromosome segregation. Mol Cell Biol 14(7): 4731-4740. doi: 10.1128/mcb.14.7.4731
- Kang J, Yu H (2009). Kinase signaling in the spindle checkpoint. J Biol Chem 284(23): 15359-15363. doi: 10.1074/jbc.R900005200
- Pinsky BA, Kotwaliwale CV, Tatsutani SY, Breed CA, Biggins S (2006). Glc7/protein phosphatase 1 regulatory subunits can oppose the Ipl1/aurora protein kinase by redistributing Glc7. Mol Cell Biol 26(7): 2648-2660. doi: 10.1128/MCB.26.7.2648-2660.2006
- Bailis JM, Roeder GS (2000). Pachytene exit controlled by reversal of Mek1-dependent phosphorylation. Cell 101(2): 211-221. doi: 10.1016/S0092-8674(00)80831-4
- den Elzen NR, O’Connell MJ (2004). Recovery from DNA damage checkpoint arrest by PP1-mediated inhibition of Chk1. EMBO J 23(4): 908-918. doi: 10.1038/sj.emboj.7600105
- Bazzi M, Mantiero D, Trovesi C, Lucchini G, Longhese MP (2010). Dephosphorylation of gamma H2A by Glc7/protein phosphatase 1 promotes recovery from inhibition of DNA replication. Mol Cell Biol 30(1): 131-145. doi: 10.1128/MCB.01000-09
- Yao G, Wan J, Mu C, Liu Q, Wang Y, Sang J (2016). Sds22 participates in Glc7 mediated Rad53 dephosphorylation in MMS- induced DNA damage in Candida albicans. Fungal Genet Biol 93: 50- 61. doi: 10.1016/j.fgb.2016.06.003
- Peng A, Lewellyn AL, Schiemann WP, Maller JL (2010). Repo-man controls a protein phosphatase 1-dependent threshold for DNA damage checkpoint activation. Curr Biol 20(5): 387-396. doi: 10.1016/j.cub.2010.01.020
- Lu Z, Wan G, Guo H, Zhang X, Lu X (2013). Protein phosphatase 1 inhibits p53 signaling by dephosphorylating and stabilizing Mdmx. Cell Signal 25(4): 796-804. doi: 10.1016/j.cellsig.2012.12.014
- Haneda M, Kojima E, Nishikimi A, Hasegawa T, Nakashima I, Isobe K (2004). Protein phosphatase 1, but not protein phosphatase 2A, dephosphorylates DNA-damaging stress-induced phospho-serine 15 of p53. FEBS Lett 567(2-3): 171-174. doi: 10.1016/j.febslet.2004.04.066
- Li DW, Liu JP, Schmid PC, Schlosser R, Feng H, Liu WB, Yan Q, Gong L, Sun SM, Deng M, Liu Y (2006). Protein serine/threonine phosphatase-1 dephosphorylates p53 at Ser-15 and Ser-37 to modulate its transcriptional and apoptotic activities. Oncogene 25(21): 3006-3022. doi: 10.1038/sj.onc.1209334
- Lee SJ, Lim CJ, Min JK, Lee JK, Kim YM, Lee JY, Won MH, Kwon YG (2007). Protein phosphatase 1 nuclear targeting subunit is a hypoxia inducible gene: its role in post-translational modification of p53 and MDM2. Cell Death Differ 14(6): 1106-1116. doi: 10.1038/sj.cdd.4402111
- Helps NR, Barker HM, Elledge SJ, Cohen PT (1995). Protein phosphatase 1 interacts with p53BP2, a protein which binds to the tumour suppressor p53. FEBS Lett 377(3): 295-300. doi: 10.1016/0014-5793(95)01347-4
- Millar JB, Blevitt J, Gerace L, Sadhu K, Featherstone C, Russell P (1991). p55CDC25 is a nuclear protein required for the initiation of mitosis in human cells. Proc Natl Acad Sci U S A 88(23): 10500-10504. doi: 10.1073/pnas.88.23.10500
- Dalal SN, Schweitzer CM, Gan J, DeCaprio JA (1999). Cytoplasmic localization of human cdc25C during interphase requires an intact 14- 3-3 binding site. Mol Cell Biol 19(6): 4465-4479. doi: 10.1128/mcb.19.6.4465
- van Vugt MA, Bras A, Medema RH (2004). Polo-like kinase-1 controls recovery from a G2 DNA damage-induced arrest in mammalian cells. Mol Cell 15(5): 799-811. doi: 10.1016/j.molcel.2004.07.015
- Chen MS, Hurov J, White LS, Woodford-Thomas T, Piwnica-Worms H (2001). Absence of apparent phenotype in mice lacking Cdc25C protein phosphatase. Mol Cell Biol 21(12): 3853-3861. doi: 10.1128/MCB.21.12.3853-3861.2001
- Hsu LC (2007). Identification and functional characterization of a PP1-binding site in BRCA1. Biochem Biophys Res Commun 360(2): 507-512. doi: 10.1016/j.bbrc.2007.06.090
- Chen BY, Huang CH, Lin YH, Huang CC, Deng CX, Hsu LC (2014). The K898E germline variant in the PP1-binding motif of BRCA1 causes defects in DNA Repair. Sci Rep 4: 5812. doi: 10.1038/srep05812
- Zhu S, Fisher LA, Bessho T, Peng A (2017). Protein phosphatase 1 and phosphatase 1 nuclear targeting subunit-dependent regulation of DNA-dependent protein kinase and non-homologous end joining. Nucleic Acids Res 45(18): 10583-10594. doi: 10.1093/nar/gkx686
- Winkler C, Rouget R, Wu D, Beullens M, Van Eynde A, Bollen M (2018). Overexpression of PP1-NIPP1 limits the capacity of cells to repair DNA double-strand breaks. J Cell Sci 131(13): jcs214932. doi: 10.1242/jcs.214932
- Janssens V, Goris J (2001). Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J 353(Pt 3): 417-439. doi: 10.1042/0264-6021:3530417
- Janssens V, Longin S, Goris J (2008). PP2A holoenzyme assembly: in cauda venenum (the sting is in the tail). Trends Biochem Sci 33(3): 113-121. doi: 10.1016/j.tibs.2007.12.004
- Westermarck J, Hahn WC (2008). Multiple pathways regulated by the tumor suppressor PP2A in transformation. Trends Mol Med 14(4): 152-160. doi: 10.1016/j.molmed.2008.02.001
- Sneddon AA, Cohen PT, Stark MJ (1990). Saccharomyces cerevisiae protein phosphatase 2A performs an essential cellular function and is encoded by two genes. EMBO J 9(13): 4339-4346. doi: 10.1002/j.1460-2075.1990.tb07883.x
- Ronne H, Carlberg M, Hu GZ, Nehlin JO (1991). Protein phosphatase 2A in Saccharomyces cerevisiae: effects on cell growth and bud morphogenesis. Mol Cell Biol 11(10): 4876-4884. doi: 10.1128/mcb.11.10.4876
- van Zyl W, Huang W, Sneddon AA, Stark M, Camier S, Werner M, Marck C, Sentenac A, Broach JR (1992). Inactivation of the protein phosphatase 2A regulatory subunit A results in morphological and transcriptional defects in Saccharomyces cerevisiae. Mol Cell Biol 12(11): 4946-4959. doi: 10.1128/mcb.12.11.4946
- Shu Y, Yang H, Hallberg E, Hallberg R (1997). Molecular genetic analysis of Rts1p, a B’ regulatory subunit of Saccharomyces cerevisiae protein phosphatase 2A. Mol Cell Biol 17(6): 3242-3253. doi: 10.1128/mcb.17.6.3242
- Wang Y, Burke DJ (1997). Cdc55p, the B-type regulatory subunit of protein phosphatase 2A, has multiple functions in mitosis and is required for the kinetochore/spindle checkpoint in Saccharomyces cerevisiae. Mol Cell Biol 17(2): 620-626. doi: 10.1128/mcb.17.2.620
- Jeong AL, Yang Y (2013). PP2A function toward mitotic kinases and substrates during the cell cycle. BMB Rep 46(6): 289-294. doi: 10.5483/bmbrep.2013.46.6.041
- Hunt T (2013). On the regulation of protein phosphatase 2A and its role in controlling entry into and exit from mitosis. Adv Biol Regul 53(2): 173-178. doi: 10.1016/j.jbior.2013.04.001
- Janssens V, Goris J, Van Hoof C (2005). PP2A: the expected tumor suppressor. Curr Opin Genet Dev 15(1): 34-41. doi: 10.1016/j.gde.2004.12.004
- Lambrecht C, Haesen D, Sents W, Ivanova E, Janssens V (2013). Structure, regulation, and pharmacological modulation of PP2A phosphatases. Methods Mol Biol 1053: 283-305. doi: 10.1007/978-1-62703-562-0_17
- Kinoshita N, Ohkura H, Yanagida M (1990). Distinct, essential roles of type 1 and 2A protein phosphatases in the control of the fission yeast cell division cycle. Cell 63(2): 405-415. doi: 10.1016/0092-8674(90)90173-c
- Wang Y, Ng TY (2006). Phosphatase 2A negatively regulates mitotic exit in Saccharomyces cerevisiae. Mol Biol Cell 17(1): 80-89. doi: 10.1091/mbc.E04-12-1109
- Kumagai A, Dunphy WG (1992). Regulation of the cdc25 protein during the cell cycle in Xenopus extracts. Cell 70(1): 139-151. doi: 10.1016/0092-8674(92)90540-s
- Harvey SL, Enciso G, Dephoure N, Gygi SP, Gunawardena J, Kellogg DR (2011). A phosphatase threshold sets the level of Cdk1 activity in early mitosis in budding yeast. Mol Biol Cell 22(19): 3595-3608. doi: 10.1091/mbc.E11-04-0340
- Slupe AM, Merrill RA, Strack S (2011). Determinants for Substrate Specificity of Protein Phosphatase 2A. Enzyme Res 2011: 398751. doi: 10.4061/2011/398751
- Yaakov G, Thorn K, Morgan DO (2012). Separase biosensor reveals that cohesin cleavage timing depends on phosphatase PP2A(Cdc55) regulation. Dev Cell 23(1): 124-136. doi: 10.1016/j.devcel.2012.06.007
- Liu H, Rankin S, Yu H (2013). Phosphorylation-enabled binding of SGO1-PP2A to cohesin protects sororin and centromeric cohesion during mitosis. Nat Cell Biol 15(1): 40-49. doi: 10.1038/ncb2637
- Bialojan C, Takai A (1988). Inhibitory effect of a marine-sponge toxin, okadaic acid, on protein phosphatases. Specificity and kinetics. Biochem J 256(1): 283-290. doi: 10.1042/bj2560283
- Suganuma M, Fujiki H, Suguri H, Yoshizawa S, Hirota M, Nakayasu M, Ojika M, Wakamatsu K, Yamada K, Sugimura T (1988). Okadaic acid: an additional non-phorbol-12-tetradecanoate-13-acetate-type tumor promoter. Proc Natl Acad Sci U S A 85(6): 1768-1771. doi: 10.1073/pnas.85.6.1768
- Goodarzi AA, Jonnalagadda JC, Douglas P, Young D, Ye R, Moorhead GB, Lees-Miller SP, Khanna KK (2004). Autophosphorylation of ataxia-telangiectasia mutated is regulated by protein phosphatase 2A. EMBO J 23(22): 4451-4461. doi: 10.1038/sj.emboj.7600455
- Kalev P, Simicek M, Vazquez I, Munck S, Chen L, Soin T, Danda N, Chen W, Sablina A (2012). Loss of PPP2R2A inhibits homologous recombination DNA repair and predicts tumor sensitivity to PARP inhibition. Cancer Res 72(24): 6414-6424. doi: 10.1158/0008-5472.CAN-12-1667
- Leung-Pineda V, Ryan CE, Piwnica-Worms H (2006). Phosphorylation of Chk1 by ATR is antagonized by a Chk1-regulated protein phosphatase 2A circuit. Mol Cell Biol 26(20): 7529-7538. doi: 10.1128/MCB.00447-06
- Yan Y, Cao PT, Greer PM, Nagengast ES, Kolb RH, Mumby MC, Cowan KH (2010). Protein phosphatase 2A has an essential role in the activation of gamma-irradiation-induced G2/M checkpoint response. Oncogene 29(30): 4317-4329. doi: 10.1038/onc.2010.187
- Freeman AK, Dapic V, Monteiro AN (2010). Negative regulation of CHK2 activity by protein phosphatase 2A is modulated by DNA damage. Cell Cycle 9(4): 736-747. doi: 10.4161/cc.9.4.10613
- Guillonneau M, Paris F, Dutoit S, Estephan H, Beneteau E, Huot J, Corre I (2016). Oxidative stress disassembles the p38/NPM/PP2A complex, which leads to modulation of nucleophosmin-mediated signaling to DNA damage response. FASEB J 30(8): 2899-2914. doi: 10.1096/fj.201500194R
- Dohoney KM, Guillerm C, Whiteford C, Elbi C, Lambert PF, Hager GL, Brady JN (2004). Phosphorylation of p53 at serine 37 is important for transcriptional activity and regulation in response to DNA damage. Oncogene 23(1): 49-57. doi: 10.1038/sj.onc.1207005
- Li HH, Cai X, Shouse GP, Piluso LG, Liu X (2007). A specific PP2A regulatory subunit, B56gamma, mediates DNA damage-induced dephosphorylation of p53 at Thr55. EMBO J 26(2): 402-411. doi: 10.1038/sj.emboj.7601519
- Ferrari E, Bruhn C, Peretti M, Cassani C, Carotenuto WV, Elgendy M, Shubassi G, Lucca C, Bermejo R, Varasi M, Minucci S, Longhese MP, Foiani M (2017). PP2A Controls Genome Integrity by Integrating Nutrient-Sensing and Metabolic Pathways with the DNA Damage Response. Mol Cell 67(2): 266-281 e264. doi: 10.1016/j.molcel.2017.05.027
- Jang YJ, Ji JH, Choi YC, Ryu CJ, Ko SY (2007). Regulation of Polo-like kinase 1 by DNA damage in mitosis. Inhibition of mitotic PLK-1 by protein phosphatase 2A. J Biol Chem 282(4): 2473-2482. doi: 10.1074/jbc.M605480200
- Lee HJ, Hwang HI, Jang YJ (2010). Mitotic DNA damage response: Polo-like kinase-1 is dephosphorylated through ATM-Chk1 pathway. Cell Cycle 9(12): 2389-2398. doi: 10.4161/cc.9.12.11904
- Wang L, Guo Q, Fisher LA, Liu D, Peng A (2015). Regulation of polo- like kinase 1 by DNA damage and PP2A/B55alpha. Cell Cycle 14(1): 157-166. doi: 10.4161/15384101.2014.986392
- Feng J, Wakeman T, Yong S, Wu X, Kornbluth S, Wang XF (2009). Protein phosphatase 2A-dependent dephosphorylation of replication protein A is required for the repair of DNA breaks induced by replication stress. Mol Cell Biol 29(21): 5696-5709. doi: 10.1128/MCB.00191-09
- Chowdhury D, Keogh MC, Ishii H, Peterson CL, Buratowski S, Lieberman J (2005). gamma-H2AX dephosphorylation by protein phosphatase 2A facilitates DNA double-strand break repair. Mol Cell 20(5): 801-809. doi: 10.1016/j.molcel.2005.10.003
- Li X, Nan A, Xiao Y, Chen Y, Lai Y (2015). PP2A-B56 complex is involved in dephosphorylation of gamma-H2AX in the repair process of CPT-induced DNA double-strand breaks. Toxicology 331: 57-65. doi: 10.1016/j.tox.2015.03.007
- Douglas P, Moorhead GB, Ye R, Lees-Miller SP (2001). Protein phosphatases regulate DNA-dependent protein kinase activity. J Biol Chem 276(22): 18992-18998. doi: 10.1074/jbc.M011703200
- Merigliano C, Marzio A, Renda F, Somma MP, Gatti M, Verni F (2017). A Role for the Twins Protein Phosphatase (PP2A-B55) in the Maintenance of Drosophila Genome Integrity. Genetics 205(3): 1151- 1167. doi: 10.1534/genetics.116.192781
- da Cruz e Silva OB, da Cruz e Silva EF, Cohen PT (1988). Identification of a novel protein phosphatase catalytic subunit by cDNA cloning. FEBS Lett 242(1): 106-110. doi: 10.1016/0014-5793(88)80995-5
- Brewis ND, Cohen PT (1992). Protein phosphatase X has been highly conserved during mammalian evolution. Biochim Biophys Acta 1171(2): 231-233. doi: 10.1016/0167-4781(92)90129-n
- Cohen PT, Philp A, Vazquez-Martin C (2005). Protein phosphatase 4–from obscurity to vital functions. FEBS Lett 579(15): 3278-3286. doi: 10.1016/j.febslet.2005.04.070
- Hustedt N, Seeber A, Sack R, Tsai-Pflugfelder M, Bhullar B, Vlaming H, van Leeuwen F, Guenole A, van Attikum H, Srivas R, Ideker T, Shimada K, Gasser SM (2015). Yeast PP4 interacts with ATR homolog Ddc2-Mec1 and regulates checkpoint signaling. Mol Cell 57(2): 273- 289. doi: 10.1016/j.molcel.2014.11.016
- Helps NR, Brewis ND, Lineruth K, Davis T, Kaiser K, Cohen PT (1998). Protein phosphatase 4 is an essential enzyme required for organisation of microtubules at centrosomes in Drosophila embryos. J Cell Sci 111 ( Pt 10)(1331-1340. PMID: 9570751
- Sumiyoshi E, Sugimoto A, Yamamoto M (2002). Protein phosphatase 4 is required for centrosome maturation in mitosis and sperm meiosis in C. elegans. J Cell Sci 115(Pt 7): 1403-1410. PMID: 11896188
- Carnegie GK, Sleeman JE, Morrice N, Hastie CJ, Peggie MW, Philp A, Lamond AI, Cohen PT (2003). Protein phosphatase 4 interacts with the Survival of Motor Neurons complex and enhances the temporal localisation of snRNPs. J Cell Sci 116(Pt 10): 1905-1913. doi: 10.1242/jcs.00409
- Yeh PY, Yeh KH, Chuang SE, Song YC, Cheng AL (2004). Suppression of MEK/ERK signaling pathway enhances cisplatin-induced NF-kappaB activation by protein phosphatase 4-mediated NF-kappaB p65 Thr dephosphorylation. J Biol Chem 279(25): 26143-26148. doi: 10.1074/jbc.M402362200
- Bertram PG, Choi JH, Carvalho J, Ai W, Zeng C, Chan TF, Zheng XF (2000). Tripartite regulation of Gln3p by TOR, Ure2p, and phosphatases. J Biol Chem 275(46): 35727-35733. doi: 10.1074/jbc.M004235200
- Zhang X, Ozawa Y, Lee H, Wen YD, Tan TH, Wadzinski BE, Seto E (2005). Histone deacetylase 3 (HDAC3) activity is regulated by interaction with protein serine/threonine phosphatase 4. Genes Dev 19(7): 827-839. doi: 10.1101/gad.1286005
- Falk JE, Chan AC, Hoffmann E, Hochwagen A (2010). A Mec1- and PP4-dependent checkpoint couples centromere pairing to meiotic recombination. Dev Cell 19(4): 599-611. doi: 10.1016/j.devcel.2010.09.006
- O’Neill BM, Hanway D, Winzeler EA, Romesberg FE (2004). Coordinated functions of WSS1, PSY2 and TOF1 in the DNA damage response. Nucleic Acids Res 32(22): 6519-6530. doi: 10.1093/nar/gkh994
- O’Neill BM, Szyjka SJ, Lis ET, Bailey AO, Yates JR, 3rd, Aparicio OM, Romesberg FE (2007). Pph3-Psy2 is a phosphatase complex required for Rad53 dephosphorylation and replication fork restart during recovery from DNA damage. Proc Natl Acad Sci U S A 104(22): 9290- 9295. doi: 10.1073/pnas.0703252104
- Keogh MC, Kim JA, Downey M, Fillingham J, Chowdhury D, Harrison JC, Onishi M, Datta N, Galicia S, Emili A, Lieberman J, Shen X, Buratowski S, Haber JE, Durocher D, Greenblatt JF, Krogan NJ (2006). A phosphatase complex that dephosphorylates gammaH2AX regulates DNA damage checkpoint recovery. Nature 439(7075): 497-501. doi: 10.1038/nature04384
- Nakada S, Chen GI, Gingras AC, Durocher D (2008). PP4 is a gamma H2AX phosphatase required for recovery from the DNA damage checkpoint. EMBO Rep 9(10): 1019-1026. doi: 10.1038/embor.2008.162
- Liu J, Xu L, Zhong J, Liao J, Li J, Xu X (2012). Protein phosphatase PP4 is involved in NHEJ-mediated repair of DNA double-strand breaks. Cell Cycle 11(14): 2643-2649. doi: 10.4161/cc.20957
- Lee DH, Pan Y, Kanner S, Sung P, Borowiec JA, Chowdhury D (2010). A PP4 phosphatase complex dephosphorylates RPA2 to facilitate DNA repair via homologous recombination. Nat Struct Mol Biol 17(3): 365-372. doi: 10.1038/nsmb.1769
- Chowdhury D, Xu X, Zhong X, Ahmed F, Zhong J, Liao J, Dykxhoorn DM, Weinstock DM, Pfeifer GP, Lieberman J (2008). A PP4- phosphatase complex dephosphorylates gamma-H2AX generated during DNA replication. Mol Cell 31(1): 33-46. doi: 10.1016/j.molcel.2008.05.016
- Visintin R, Craig K, Hwang ES, Prinz S, Tyers M, Amon A (1998). The phosphatase Cdc14 triggers mitotic exit by reversal of Cdk-dependent phosphorylation. Mol Cell 2(6): 709-718. doi: 10.1016/s1097-2765(00)80286-5
- Stegmeier F, Visintin R, Amon A (2002). Separase, polo kinase, the kinetochore protein Slk19, and Spo12 function in a network that controls Cdc14 localization during early anaphase. Cell 108(2): 207-220. doi: 10.1016/s0092-8674(02)00618-9
- Cueille N, Salimova E, Esteban V, Blanco M, Moreno S, Bueno A, Simanis V (2001). Flp1, a fission yeast orthologue of the s. cerevisiae CDC14 gene, is not required for cyclin degradation or rum1p stabilisation at the end of mitosis. J Cell Sci 114(Pt 14): 2649-2664. PMID: 11683392
- Trautmann S, Wolfe BA, Jorgensen P, Tyers M, Gould KL, McCollum D (2001). Fission yeast Clp1p phosphatase regulates G2/M transition and coordination of cytokinesis with cell cycle progression. Curr Biol 11(12): 931-940. doi: 10.1016/s0960-9822(01)00268-8
- Powers BL, Hall MC (2017). Re-examining the role of Cdc14 phosphatase in reversal of Cdk phosphorylation during mitotic exit. J Cell Sci 130(16): 2673-2681. doi: 10.1242/jcs.201012
- Chen CT, Peli-Gulli MP, Simanis V, McCollum D (2006). S. pombe FEAR protein orthologs are not required for release of Clp1/Flp1 phosphatase from the nucleolus during mitosis. J Cell Sci 119(Pt 21): 4462-4466. doi: 10.1242/jcs.03220
- Clifford DM, Wolfe BA, Roberts-Galbraith RH, McDonald WH, Yates JR, 3rd, Gould KL (2008). The Clp1/Cdc14 phosphatase contributes to the robustness of cytokinesis by association with anillin-related Mid1. J Cell Biol 181(1): 79-88. doi: 10.1083/jcb.200709060
- Berdougo E, Nachury MV, Jackson PK, Jallepalli PV (2008). The nucleolar phosphatase Cdc14B is dispensable for chromosome segregation and mitotic exit in human cells. Cell Cycle 7(9): 1184- 1190. doi: 10.4161/cc.7.9.5792
- Meitinger F, Palani S, Pereira G (2012). The power of MEN in cytokinesis. Cell Cycle 11(2): 219-228. doi: 10.4161/cc.11.2.18857
- Meitinger F, Petrova B, Lombardi IM, Bertazzi DT, Hub B, Zentgraf H, Pereira G (2010). Targeted localization of Inn1, Cyk3 and Chs2 by the mitotic-exit network regulates cytokinesis in budding yeast. J Cell Sci 123(Pt 11): 1851-1861. doi: 10.1242/jcs.063891
- Clemente-Blanco A, Mayán-Santos M, Schneider D, Machín F, Jarmuz A, Tschochner H, Aragón L (2009). Cdc14 inhibits transcription by RNA polymerase I during anaphase. Nature 458(7235): 219-222. doi: 10.1038/nature07652
- Mocciaro A, Schiebel E (2010). Cdc14: a highly conserved family of phosphatases with non-conserved functions? J Cell Sci 123(Pt 17): 2867-2876. doi: 10.1242/jcs.074815
- Clemente-Blanco A, Sen N, Mayan-Santos M, Sacristan MP, Graham B, Jarmuz A, Giess A, Webb E, Game L, Eick D, Bueno A, Merkenschlager M, Aragon L (2011). Cdc14 phosphatase promotes segregation of telomeres through repression of RNA polymerase II transcription. Nat Cell Biol 13(12): 1450-1456. doi: 10.1038/ncb2365
- Guillamot M, Manchado E, Chiesa M, Gomez-Lopez G, Pisano DG, Sacristan MP, Malumbres M (2011). Cdc14b regulates mammalian RNA polymerase II and represses cell cycle transcription. Sci Rep 1: 189. doi: 10.1038/srep00189
- Clement A, Solnica-Krezel L, Gould KL (2011). The Cdc14B phosphatase contributes to ciliogenesis in zebrafish. Development 138(2): 291-302. doi: 10.1242/dev.055038
- Garcia-Luis J, Clemente-Blanco A, Aragon L, Machin F (2014). Cdc14 targets the Holliday junction resolvase Yen1 to the nucleus in early anaphase. Cell Cycle 13(9): 1392-1399. doi: 10.4161/cc.28370
- Eissler CL, Mazon G, Powers BL, Savinov SN, Symington LS, Hall MC (2014). The Cdk/cDc14 module controls activation of the Yen1 holliday junction resolvase to promote genome stability. Mol Cell 54(1): 80-93. doi: 10.1016/j.molcel.2014.02.012
- Blanco MG, Matos J, West SC (2014). Dual control of Yen1 nuclease activity and cellular localization by Cdk and Cdc14 prevents genome instability. Mol Cell 54(1): 94-106. doi: 10.1016/j.molcel.2014.02.011
- Vazquez-Novelle MD, Esteban V, Bueno A, Sacristan MP (2005). Functional homology among human and fission yeast Cdc14 phosphatases. J Biol Chem 280(32): 29144-29150. doi: 10.1074/jbc.M413328200
- Li L, Ernsting BR, Wishart MJ, Lohse DL, Dixon JE (1997). A family of putative tumor suppressors is structurally and functionally conserved in humans and yeast. J Biol Chem 272(47): 29403-29406. doi: 10.1074/jbc.272.47.29403
- De Wulf P, Montani F, Visintin R (2009). Protein phosphatases take the mitotic stage. Curr Opin Cell Biol 21(6): 806-815. doi: 10.1016/j.ceb.2009.08.003
- Queralt E, Uhlmann F (2008). Cdk-counteracting phosphatases unlock mitotic exit. Curr Opin Cell Biol 20(6): 661-668. doi: 10.1016/j.ceb.2008.09.003
- Amon A (2008). A decade of Cdc14–a personal perspective. Delivered on 9 July 2007 at the 32nd FEBS Congress in Vienna, Austria. FEBS J 275(23): 5774-5784. doi: 10.1111/j.1742-4658.2008.06693.x
- Wurzenberger C, Gerlich DW (2011). Phosphatases: providing safe passage through mitotic exit. Nature Rev Mol Cell Biol 12(8): 469-482. doi: 10.1038/nrm3149
- Stegmeier F, Amon A (2004). Closing mitosis: the functions of the Cdc14 phosphatase and its regulation. Annu Rev Genet 38: 203-232. doi: 10.1146/annurev.genet.38.072902.093051
- Diaz-Cuervo H, Bueno A (2008). Cds1 controls the release of Cdc14-like phosphatase Flp1 from the nucleolus to drive full activation of the checkpoint response to replication stress in fission yeast. Mol Biol Cell 19(6): 2488-2499. doi: 10.1091/mbc.E07-08-0737
- Bassermann F, Frescas D, Guardavaccaro D, Busino L, Peschiaroli A, Pagano M (2008). The Cdc14B-Cdh1-Plk1 axis controls the G2 DNA- damage-response checkpoint. Cell 134(2): 256-267. doi: 10.1016/j.cell.2008.05.043
- Mocciaro A, Berdougo E, Zeng K, Black E, Vagnarelli P, Earnshaw W, Gillespie D, Jallepalli P, Schiebel E (2010). Vertebrate cells genetically deficient for Cdc14A or Cdc14B retain DNA damage checkpoint proficiency but are impaired in DNA repair. J Cell Biol 189(4): 631-639. doi: 10.1083/jcb.200910057
- Wei Z, Peddibhotla S, Lin H, Fang X, Li M, Rosen JM, Zhang P (2011). Early-onset aging and defective DNA damage response in Cdc14b-deficient mice. Mol Cell Biol 31(7): 1470-1477. doi: 10.1128/MCB.01330-10
- Lin H, Ha K, Lu G, Fang X, Cheng R, Zuo Q, Zhang P (2015). Cdc14A and Cdc14B Redundantly Regulate DNA Double-Strand Break Repair. Mol Cell Biol 35(21): 3657-3668. doi: 10.1128/MCB.00233-15
- Villoria MT, Ramos F, Duenas E, Faull P, Cutillas PR, Clemente- Blanco A (2017). Stabilization of the metaphase spindle by Cdc14 is required for recombinational DNA repair. EMBO J 36(1): 79-101. doi: 10.15252/embj.201593540
- Nagai S, Dubrana K, Tsai-Pflugfelder M, Davidson MB, Roberts TM, Brown GW, Varela E, Hediger F, Gasser SM, Krogan NJ (2008). Functional targeting of DNA damage to a nuclear pore-associated SUMO-dependent ubiquitin ligase. Science 322(5901): 597-602. doi: 10.1126/science.1162790
- Oza P, Peterson CL (2010). Opening the DNA repair toolbox: localization of DNA double strand breaks to the nuclear periphery. Cell Cycle 9(1): 43-49. doi: 10.4161/cc.9.1.10317
- Horigome C, Oma Y, Konishi T, Schmid R, Marcomini I, Hauer MH, Dion V, Harata M, Gasser SM (2014). SWR1 and INO80 chromatin remodelers contribute to DNA double-strand break perinuclear anchorage site choice. Mol Cell 55(4): 626-639. doi: 10.1016/j.molcel.2014.06.027
- Figueiredo J, da Cruz ESOA, Fardilha M (2014). Protein phosphatase 1 and its complexes in carcinogenesis. Curr Cancer Drug Targets 14(1): 2-29. doi: 10.2174/15680096113136660106
- Liu Y, Virshup DM, White RL, Hsu LC (2002). Regulation of BRCA1 phosphorylation by interaction with protein phosphatase 1alpha. Cancer research 62(22): 6357-6361. PMID: 12438214
- Winter SL, Bosnoyan-Collins L, Pinnaduwage D, Andrulis IL (2007). The interaction of PP1 with BRCA1 and analysis of their expression in breast tumors. BMC Cancer 7: 85. doi: 10.1186/1471-2407-7-85
- Paul D, Bargale AB, Rapole S, Shetty PK, Santra MK (2019). Protein Phosphatase 1 Regulatory Subunit SDS22 Inhibits Breast Cancer Cell Tumorigenesis by Functioning as a Negative Regulator of the AKT Signaling Pathway. Neoplasia 21(1): 30-40. doi: 10.1016/j.neo.2018.10.009
- McConnell JL, Wadzinski BE (2009). Targeting protein serine/threonine phosphatases for drug development. Mol Pharmacol 75(6): 1249-1261. doi: 10.1124/mol.108.053140
- Sablina AA, Hector M, Colpaert N, Hahn WC (2010). Identification of PP2A complexes and pathways involved in cell transformation. Cancer Res 70(24): 10474-10484. doi: 10.1158/0008-5472.CAN-10-2855
- Perrotti D, Neviani P (2013). Protein phosphatase 2A: a target for anticancer therapy. Lancet Oncol 14(6): e229-238. doi: 10.1016/S1470-2045(12)70558-2
- Eichhorn PJ, Creyghton MP, Bernards R (2009). Protein phosphatase 2A regulatory subunits and cancer. Biochim Biophys Acta 1795(1): 1-15. doi: 10.1016/j.bbcan.2008.05.005
- Ruvolo PP (2016). The broken “Off” switch in cancer signaling: PP2A as a regulator of tumorigenesis, drug resistance, and immune surveillance. BBA Clin 6: 87-99. doi: 10.1016/j.bbacli.2016.08.002
- Mazhar S, Taylor SE, Sangodkar J, Narla G (2019). Targeting PP2A in cancer: Combination therapies. Biochim Biophys Acta Mol Cell Res 1866(1): 51-63. doi: 10.1016/j.bbamcr.2018.08.020
- Chung V, Mansfield AS, Braiteh F, Richards D, Durivage H, Ungerleider RS, Johnson F, Kovach JS (2017). Safety, Tolerability, and Preliminary Activity of LB-100, an Inhibitor of Protein Phosphatase 2A, in Patients with Relapsed Solid Tumors: An Open-Label, Dose Escalation, First-in-Human, Phase I Trial. Clin Cancer Res 23(13): 3277- 3284. doi: 10.1158/1078-0432.CCR-16-2299
- Lu J, Kovach JS, Johnson F, Chiang J, Hodes R, Lonser R, Zhuang Z (2009). Inhibition of serine/threonine phosphatase PP2A enhances cancer chemotherapy by blocking DNA damage induced defense mechanisms. Proc Natl Acad Sci U S A 106(28): 11697-11702. doi: 10.1073/pnas.0905930106
- Shui JW, Hu MC, Tan TH (2007). Conditional knockout mice reveal an essential role of protein phosphatase 4 in thymocyte development and pre-T-cell receptor signaling. Mol Cell Biol 27(1): 79-91. doi: 10.1128/MCB.00799-06
- Mihindukulasuriya KA, Zhou G, Qin J, Tan TH (2004). Protein phosphatase 4 interacts with and down-regulates insulin receptor substrate 4 following tumor necrosis factor-alpha stimulation. J Biol Chem 279(45): 46588-46594. doi: 10.1074/jbc.M408067200
- Toyo-oka K, Mori D, Yano Y, Shiota M, Iwao H, Goto H, Inagaki M, Hiraiwa N, Muramatsu M, Wynshaw-Boris A, Yoshiki A, Hirotsune S (2008). Protein phosphatase 4 catalytic subunit regulates Cdk1 activity and microtubule organization via NDEL1 dephosphorylation. J Cell Biol 180(6): 1133-1147. doi: 10.1083/jcb.200705148
- Lee J, Adelmant G, Marto JA, Lee DH (2015). Dephosphorylation of DBC1 by Protein Phosphatase 4 Is Important for p53-Mediated Cellular Functions. Mol Cells 38(8): 697-704. doi: 10.14348/molcells.2015.0066
- Gingras AC, Caballero M, Zarske M, Sanchez A, Hazbun TR, Fields S, Sonenberg N, Hafen E, Raught B, Aebersold R (2005). A novel, evolutionarily conserved protein phosphatase complex involved in cisplatin sensitivity. Mol Cell Proteomics 4(11): 1725-1740. doi: 10.1074/mcp.M500231-MCP200
- Wu HI, Brown JA, Dorie MJ, Lazzeroni L, Brown JM (2004). Genome-wide identification of genes conferring resistance to the anticancer agents cisplatin, oxaliplatin, and mitomycin C. Cancer Res 64(11): 3940-3948. doi: 10.1158/0008-5472.CAN-03-3113
- Wang B, Zhao A, Sun L, Zhong X, Zhong J, Wang H, Cai M, Li J, Xu Y, Liao J, Sang J, Chowdhury D, Pfeifer GP, Yen Y, Xu X (2008). Protein phosphatase PP4 is overexpressed in human breast and lung tumors. Cell Res 18(9): 974-977. doi: 10.1038/cr.2008.274
- Li X, Liang L, Huang L, Ma X, Li D, Cai S (2015). High expression of protein phosphatase 4 is associated with the aggressive malignant behavior of colorectal carcinoma. Mol Cancer 14: 95. doi: 10.1186/s12943-015-0356-7
- Quevedo O, Ramos-Perez C, Petes TD, Machin F (2015). The Transient Inactivation of the Master Cell Cycle Phosphatase Cdc14 Causes Genomic Instability in Diploid Cells of Saccharomyces cerevisiae. Genetics 200(3): 755-769. doi: 10.1534/genetics.115.177626
- Chen NP, Uddin B, Voit R, Schiebel E (2016). Human phosphatase CDC14A is recruited to the cell leading edge to regulate cell migration and adhesion. Proc Natl Acad Sci U S A 113(4): 990-995. doi: 10.1073/pnas.1515605113
- Chiesa M, Guillamot M, Bueno MJ, Malumbres M (2011). The Cdc14B phosphatase displays oncogenic activity mediated by the Ras- Mek signaling pathway. Cell Cycle 10(10): 1607-1617. doi: 10.4161/cc.10.10.15566
- Hastie CJ, Cohen PT (1998). Purification of protein phosphatase 4 catalytic subunit: inhibition by the antitumour drug fostriecin and other tumour suppressors and promoters. FEBS Lett 431(3): 357-361. doi: 10.1016/s0014-5793(98)00775-3
- Matsunaga S, Wakimoto T, Fusetani N (1997). Isolation of Four New Calyculins from the Marine Sponge Discodermia calyx(1). J Org Chem 62(8): 2640-2642. PMID: 11671611
- Hamilton J, Grawenda AM, Bernhard EJ (2009). Phosphatase inhibition and cell survival after DNA damage induced by radiation. Cancer Biol Ther 8(16): 1577-1586. doi: 10.4161/cbt.8.16.8962
- Honkanen RE, Zwiller J, Moore RE, Daily SL, Khatra BS, Dukelow M, Boynton AL (1990). Characterization of microcystin-LR, a potent inhibitor of type 1 and type 2A protein phosphatases. J Biol Chem 265(32): 19401-19404. PMID: 2174036
- Matsushima R, Yoshizawa S, Watanabe MF, Harada K, Furusawa M, Carmichael WW, Fujiki H (1990). In vitro and in vivo effects of protein phosphatase inhibitors, microcystins and nodularin, on mouse skin and fibroblasts. Biochem Biophys Res Commun 171(2): 867-874. doi: 10.1016/0006-291x(90)91226-i
- Choy MS, Swingle M, D’Arcy B, Abney K, Rusin SF, Kettenbach AN, Page R, Honkanen RE, Peti W (2017). PP1:Tautomycetin Complex Reveals a Path toward the Development of PP1-Specific Inhibitors. J Am Chem Soc 139(49): 17703-17706. doi: 10.1021/jacs.7b09368
- MacKintosh C, Klumpp S (1990). Tautomycin from the bacterium Streptomyces verticillatus. Another potent and specific inhibitor of protein phosphatases 1 and 2A. FEBS Lett 277(1-2): 137-140. doi: 10.1016/0014-5793(90)80828-7
- Honkanen RE (1993). Cantharidin, another natural toxin that inhibits the activity of serine/threonine protein phosphatases types 1 and 2A. FEBS Lett 330(3): 283-286. doi: 10.1016/0014-5793(93)80889-3
- Wang G, Dong J, Deng L (2018). Overview of Cantharidin and its Analogues. Curr Med Chem 25(17): 2034-2044. doi: 10.2174/0929867324666170414165253
- Cheng A, Balczon R, Zuo Z, Koons JS, Walsh AH, Honkanen RE (1998). Fostriecin-mediated G2-M-phase growth arrest correlates with abnormal centrosome replication, the formation of aberrant mitotic spindles, and the inhibition of serine/threonine protein phosphatase activity. Cancer Res 58(16): 3611-3619. PMID: 9721869
- Walsh AH, Cheng A, Honkanen RE (1997). Fostriecin, an antitumor antibiotic with inhibitory activity against serine/threonine protein phosphatases types 1 (PP1) and 2A (PP2A), is highly selective for PP2A. FEBS Lett 416(3): 230-234. doi: 10.1016/s0014-5793(97)01210-6
- Gutierrez A, Pan L, Groen RW, Baleydier F, Kentsis A, Marineau J, Grebliunaite R, Kozakewich E, Reed C, Pflumio F, Poglio S, Uzan B, Clemons P, VerPlank L, An F, Burbank J, Norton S, Tolliday N, Steen H, Weng AP, Yuan H, Bradner JE, Mitsiades C, Look AT, Aster JC (2014). Phenothiazines induce PP2A-mediated apoptosis in T cell acute lymphoblastic leukemia. J Clin Invest 124(2): 644-655. doi: 10.1172/JCI65093
- Kastrinsky DB, Sangodkar J, Zaware N, Izadmehr S, Dhawan NS, Narla G, Ohlmeyer M (2015). Reengineered tricyclic anti-cancer agents. Bioorg Med Chem 23(19): 6528-6534. doi: 10.1016/j.bmc.2015.07.007
- Sangodkar J, Perl A, Tohme R, Kiselar J, Kastrinsky DB, Zaware N, Izadmehr S, Mazhar S, Wiredja DD, O’Connor CM, Hoon D, Dhawan NS, Schlatzer D, Yao S, Leonard D, Borczuk AC, Gokulrangan G, Wang L, Svenson E, Farrington CC, Yuan E, Avelar RA, Stachnik A, Smith B, Gidwani V, Giannini HM, McQuaid D, McClinch K, Wang Z, Levine AC, et al. (2017). Activation of tumor suppressor protein PP2A inhibits KRAS-driven tumor growth. J Clin Invest 127(6): 2081-2090. doi: 10.1172/JCI89548
- Christensen DJ, Chen Y, Oddo J, Matta KM, Neil J, Davis ED, Volkheimer AD, Lanasa MC, Friedman DR, Goodman BK, Gockerman JP, Diehl LF, de Castro CM, Moore JO, Vitek MP, Weinberg JB (2011). SET oncoprotein overexpression in B-cell chronic lymphocytic leukemia and non-Hodgkin lymphoma: a predictor of aggressive disease and a new treatment target. Blood 118(15): 4150-4158. doi: 10.1182/blood-2011-04-351072
- O’Connor CM, Perl A, Leonard D, Sangodkar J, Narla G (2018). Therapeutic targeting of PP2A. Int J Biochem Cell Biol 96: 182-193. doi: 10.1016/j.biocel.2017.10.008
- Neviani P, Santhanam R, Oaks JJ, Eiring AM, Notari M, Blaser BW, Liu S, Trotta R, Muthusamy N, Gambacorti-Passerini C, Druker BJ, Cortes J, Marcucci G, Chen CS, Verrills NM, Roy DC, Caligiuri MA, Bloomfield CD, Byrd JC, Perrotti D (2007). FTY720, a new alternative for treating blast crisis chronic myelogenous leukemia and Philadelphia chromosome-positive acute lymphocytic leukemia. J Clin Invest 117(9): 2408-2421. doi: 10.1172/JCI31095
ACKNOWLEDGMENTS
We thank “Ministerio de Economía y Competitividad” for the grants (BFU2013-41216-P and BFU2016-77081-P) conceded to A.C-B. The IBFG is supported in part by an institutional grant from the “Junta de Castilla y León” (CLU-2017-03). M.T.V. was recipient of a predoctoral fellowship from the “Junta de Castilla y León”. F.R. was recip-ient of a predoctoral fellowship from the “Ministerio de Economía y Competitividad”.
COPYRIGHT
© 2019

Role of protein phosphatases PP1, PP2A, PP4 and Cdc14 in the DNA damage response by Ramos et al. is licensed under a Creative Commons Attribution 4.0 International License.


