Reviews:
Cell Stress, Vol. 2, No. 1, pp. 4 - 13; doi: 10.15698/cst2018.01.119
MST kinases in innate immune signaling

1 State Key Laboratory of Cell Biology, CAS Center for Excellence in Molecular Cell Science, Institute of Biochemistry and Cell Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai, China.
2 School of Life Science and Technology, ShanghaiTech University, Shanghai, China.
Keywords: MST kinases, innate immunity, TLR signaling, antiviral signaling.
Abbreviations:
dsRNA – double-stranded RNA
GCK – germinal center kinase,
HCC – hepatocellular carcinoma,
IFN – interferon,
LPS – lipopolysaccharide,
mROS – mitochondrial ROS
Poly(I:C) – polyinosinic-polycytidylic acid,
RLR – RIG-I-like receptor,
ROS – reactive oxygen species,
STRN – striatin,
TLR – Toll-like receptor.
Received originally: 05/11/2017 Received in revised form: 06/12/2017
Accepted: 07/12/2017
Published: 23/12/2017
Correspondence:
Zhaocai Zhou, Institute of Biochemistry and Cell Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 Yue-Yang Road, Shanghai 200031, China. Phone/Fax: 86-21-54921291 zczhou@sibcb.ac.cn
Conflict of interest statement: The authors have no financial conflict of interest.
Please cite this article as: Zhubing Shi and Zhaocai Zhou (2017). MST kinases in innate immune signaling. Cell Stress 2(1): 4-13. doi: 10.15698/cst2018.01.119
Abstract
The mammalian STE20-like (MST) protein kinases are composed of MST1, MST2, MST3, MST4 and YSK1. They play crucial roles in cell growth, migration, polarity and apoptosis. Dysfunction of these kinases often leads to diseases. MST kinases are extensively involved in development and function of immune system. Here, we review recent progresses on the regulatory function of MST kinases in innate immune signaling.
INTRODUCTION
The MST kinases are evolutionarily conserved homologues of yeast Sterile20 (STE20) kinase [1]. Mammalian STE20 kinases can be divided into GCK (germinal center kinase) and PAK (p21-activated kinase) families [2]. The GCK family includes eight subfamilies, named GCKI to GCKVIII. They share a conserved kinase domain at N-terminal region, but possess diverse C-terminal regions that mediate protein-protein interaction. In mammal, the GCKII subfamily consists of MST1 (also named STK4) and MST2 (also named STK3); the GCKIII subfamily consists of MST3 (also named STK24), MST4 (also named STK26 or MASK) and YSK1 (also named STK25 and SOK1). The GCKII and GCKIII subfamilies constitute MST kinases.
–
MST1 and MST2 are major components of the mammalian Hippo signaling pathway, which controls cell number, organ size and tissue homeostasis [3][4][5][6][7][8]. Together with SAV1 and MOB1A/B, MST1/2 can activate downstream kinases LATS1/2, leading to the phosphorylation and inhibition of the transcriptional coactivators YAP and TAZ. Unphosphorylated YAP/TAZ can freely translocate from the cytoplasm to the nucleus and then activate TEAD family transcription factors, causing the expression of pro-proliferative and anti-apoptotic genes. Therefore, MST1/2 are considered as tumor suppressors, and YAP/TAZ as oncoproteins. Upregulation of YAP or/and TAZ has been identified in many types of cancers, such as breast, colorectal, gastric, liver and lung cancers [9][10].
–
MST3, MST4 and YSK1 are implicated in the regulation of cell growth, migration, polarity and apoptosis. MST3 was firstly identified as a protein involved in caspase-mediated apoptosis [11]. Following studies found that MST3 could phosphorylate NDR1/2, which are paralogs of LATS1/2 and also the substrates of MST1/2, to regulate cell cycle progression [12][13][14]. MST3 promotes dendritic filopodia and spine synapse development via phosphorylating TAO1/2 [15], while the MST3b isoform regulates axon regeneration [16]. One of the major physiological functions of MST4 is acting downstream of the LKB1−MO25−STRAD polarization complex, which also works as tumor suppressor, to induce brush border formation in intestinal epithelial cells [17]. YSK1 can function with LKB1 together, regulating the neuronal polarization via influencing the dynamics of the GM130-mediated Golgi apparatus [18][19]. The LKB1−STRAD scaffold and activator MO25 can directly bind GCKIII kinases and significantly enhance their activities [20][21][22]. However, the functional interplay between GCKIII kinases and LKB1 has not been fully addressed.
–
Recently, MST3 and MST4 were reported to promote cancer cell migration together with their adaptor CCM3 [23][24][25][26]. MST kinases, as well as CCM3, are components of supramolecular complexes termed “striatin (STRN)-interacting phosphatase and kinase (STRIPAK)” that contain both MST kinases and phosphatase PP2A with STRN proteins as scaffolds for complex assembly [27]. STRN proteins recruit PP2A to negatively regulate the functions of MST kinases in cell growth and cancer metastasis most likely via dephosphorylating these kinases and therefore inhibition of their kinase activities [23][28].
–
Besides their roles in development and tumorigenesis, the MST kinases have been increasingly implicated in immune response, especially innate immune signaling during the last few years. These kinases modulate antimicrobial and antiviral responses at multi-levels. Here, we firstly introduce the known functions of MST kinases in immune system, and then summarize recent progresses on the regulatory relationship between MST kinases and innate immune signaling.
FUNCTIONS OF MST KINASES IN IMMUNE SYSTEM
MST kinases especially MST1/2 are extensively involved in immune regulation [29]. MST1 is enriched in lymphoid organs including thymus, spleen and lymph nodes [30]. It regulates the proliferation of naïve T cells together with its partner RAPL (also named RASSF5 or NORE1). It is also suggested that MST1 protects naïve T cells from oxidative stress via phosphorylating and activating transcription factors FOXO1/3 [31][32]. Lymphocyte trafficking is a central event during immunological response and controlled by chemokines and adhesion receptors such as integrins [33]. Upon sphingosine-1 phosphate and chemokine stimulation, MST1/2 governs Rho family GTPase Rac1 activation via phosphorylating MOB1A/B to promote thymocyte egress [34][35]. The homing and egress abilities of T lymphocytes to peripheral lymphoid organs were impaired in MST1 or MST1/2 knockout mice [34][35][36]. MST1 can be regulated by Rap1−RAPL signal to promote lymphocyte polarization and adhesion through inducing the translocation of LFA-1 (also named αLβ2 integrin) to the leading edge and immunological synapse [37][38][39]. Recently, the well-known substrates NDR1/2 kinases and newly identified substrate L-plastin of MST1 were considered to mediate its function on thymocyte egress and T cell migration [40][41][42].
–
Treg cells are required for maintaining immune tolerance and homeostasis via inhibiting immune response of other cells [43]. Treg cells can limit the development of autoimmune and chronic inflammatory diseases, and also have negative effects on cancer immunity. MST1/2 can regulate the development and function of Treg cells through phosphorylating and stabilizing FOXO1/3 [44][45]. FOXP3 is a specific marker of Treg cells and essential for their function [46]. The function of FOXP3 is regulated by acetylation [47]. Both lysine acetyltransferases TIP60 and p300 can promote FOXP3 acetylation and stability to mediate its function on transcriptional repression, while lysine deacetylases SIRT1 and HDAC6 have an opposite role. MST1 can enhance FOXP3 acetylation through suppressing the deacetylase activity of SIRT1 and the interaction between SIRT1 and FOXP3 [48]. Recent study found that the MST1/2 downstream effector TAZ suppresses FOXP3 acetylation and promotes its degradation, and therefore attenuates Treg cell differentiation [49]. Th17 cells can protect the host against infection via its pro-inflammatory role. Geng et al. found that TAZ functions as a coactivator of the transcription factor RORγt, but not TEADs, to induce Th17 cell differentiation. However, TEAD1 can sequester TAZ from RORγt and FOXP3 to counterturn its function and promote Treg cell differentiation. Moreover, the MST1/2 kinases are supposed to be involved in these processes. Thus, both the MST1/2 kinases and the TAZ-TEAD transcription factors in the Hippo pathway play key roles in the maintenance of T cell homeostasis.
–
Neutrophils constitute the most part of white blood cells in circulation. Upon pathogen invasion, neutrophils migrate to inflammatory sites and execute the program of degranulation to release granular antimicrobial molecules [50]. MST3 and its partner CCM3 can regulate neutrophil degranulation via modulating UNC13D-driven vesicle exocytosis [51]. Furthermore, MST1-dependent vesicle trafficking is required for neutrophil extravasation [52].
INNATE IMMUNE SIGNALING
Innate immunity is the first line of defense against pathogen invasion. Innate immune system utilizes multiple pathogen pattern receptors to sense pathogen-associated molecular patterns (PAMPs) from pathogens and trigger immune response. Pathogen pattern receptors include membrane-anchored Toll-like receptors (TLRs) and C-type lectin receptors, and cytosolic RIG-I-like receptors (RLRs), NOD-like receptors and AIM2-like receptors [53][54]. TLR- and RLR-mediated signaling has been reported to associate with MST kinases. There are ten TLRs in humans. TLR1, TLR2, TLR4, TLR5, TLR6 and TLR10 are localized on plasma membrane where they sense mainly microbial membrane components, while TLR3, TLR7, TLR8 and TLR 9 are expressed in endosomal compartments where they recognize microbial nucleic acids [55][56]. TLR2 can sense multiple microbial components such as lipoproteins, peptidoglycans and hemagglutinin from bacteria, fungi and viruses. TLR2 usually functions via forming heterodimers with other TLRs such as TLR1 and TLR6. TLR4 recognizes bacterial lipopolysaccharide (LPS). TLR5 is a receptor for bacterial flagellin. Endosomal TLR3 recognizes viral double-stranded RNA (dsRNA) and its synthetic analogue polyinosinic–polycytidylic acid (poly(I:C)), while TLR7 and TLR8 recognize viral single-stranded RNA. TLR9 specifically senses unmethylated CpG motifs in bacterial and viral DNA.
–
Upon activated by their ligands, TLRs can recruit adaptors MyD88 or/and TRIF that bind different downstream signal molecules [55] (Figure 1A). MyD88 recruits IRAK4, IRAK1 and IRAK2 forming the Myddosome, which bind and activate E3 ligase TRAF6. TRAF6 in turn activates the IKK complex including NEMO, IKKα and IKKβ, which phosphorylates IκB and promotes its degradation, causing the activation of transcription factor NF-κB and the induction of pro-inflammatory cytokines. TRIF can recruit E3 ligase TRAF3, resulting in the activation of transcription factor IRF3 and the induction of type I interferons (IFNs). The TLR2−TLR1/TLR6 heterodimers, TLR5, TLR7, TLR8 and TLR9 recruit MyD88 to activate NF-κB, while TLR3 recruits TRIF to activate IRF3. TLR7, TLR8 and TLR9 also induce MyD88-mediated TRAF3 activation that causes IRF7-mediated transcription. The activation of TLR4 induces Myddosome formation and NF-κB activation. In the help of CD14, TLR4 can translocate to endosome where it recruits TRIF and activates both IRF3 and NF-κB.
–
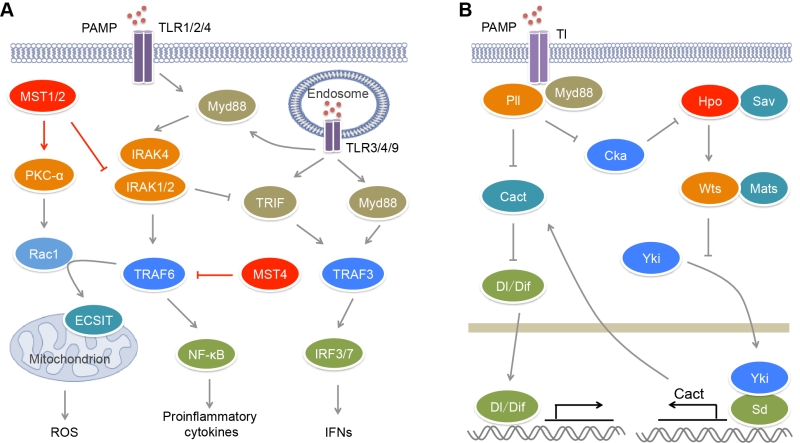 | FIGURE 1: MST kinases in the regulation of TLR signaling. (A) In mammals, upon microbial infection, TLRs sense different kinds of PAMPs and promote the production of pro-inflammatory cytokines and IFNs through activating E3 ligases TRAF3/6 and transcription factors NF-κB and IRF3/7. TRAF6 also promotes mROS production in a Rac1- and ECSIT-dependent manner. MST1/2 could stimulate mROS production through PKC-α−Rac1−TRAF6−ECSIT pathway to clear pathogens. On the contrary, MST1 suppresses the upstream kinase IRAK1 of TRAF6 to limit the production of pro-inflammatory cytokines. MST4 also attenuates TLR-mediated inflammatory responses through directly phosphorylating and inhibiting TRAF6. The inhibitory roles of MST1 and MST4 in proinflammatory response protect body against inflammatory damage and chronic inflammation-driven HCC. (B) In Drosophila, upon microbial infection, Tl receptor promotes the activation of Hpo kinase, which in turn restricts the transcription of Dl/Dif inhibitor Cact through Hippo signaling, and thus promotes Tl-mediated antimicrobial response. |
–
RLRs are composed of three members, RIG-I, MDA5 and LGP2. RIG-I and MDA5 recognize viral dsRNA [57] (Figure 2). RIG-I prefers short 5’ppp and 5’pp dsRNA, while MDA5 recognizes long dsRNA. During viral infection, activated RIG-I and MDA5 bind to a mitochondrion-located adaptor MAVS and induce its oligomerization [58]. The resultant MAVS filament further recruits multiple TRAF proteins such as TRAF2, TRAF3 and TRAF6 to activate TBK1, IKKε, IKKα and IKKβ, leading to the activation of IRF3 and NF-κB and the production of type I and type III IFNs. Cytosolic DNA sensors include cGAS, IFI16, DAI and DDX41 [59]. Viral DNA binds and activates cGAS, which utilizes ATP and GTP to generate cyclic di-GMP/AMP [60] (Figure 2). Cyclic di-GMP/AMP induces endoplasmic reticulum-resident STING dimerization to activate TBK1−IRF3 signaling, causing the production of antiviral type I IFNs.
–
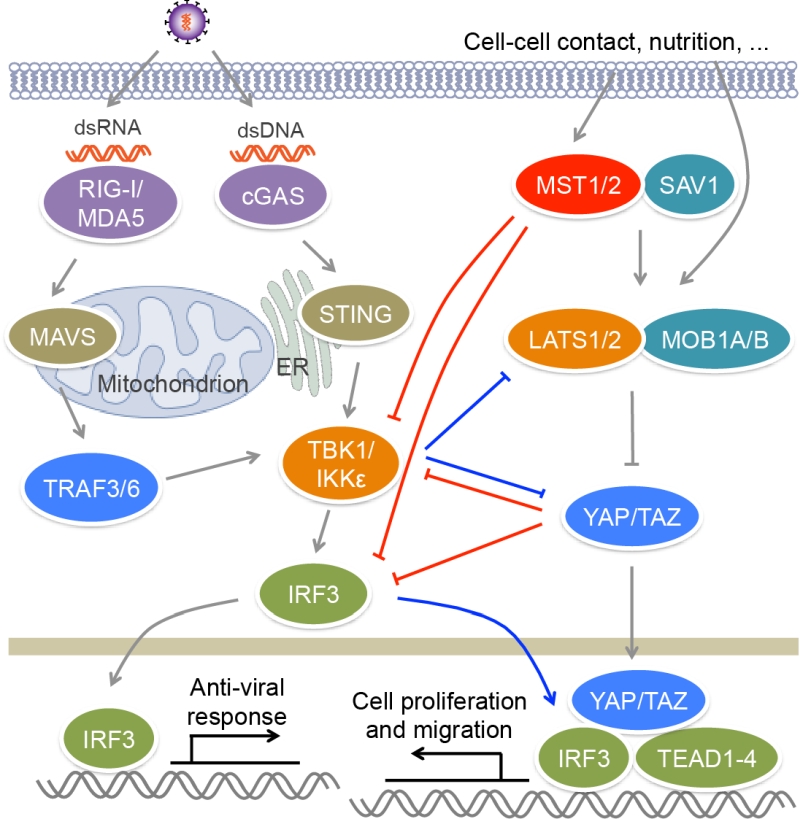 | FIGURE 2: MST kinases and Hippo signaling regulate antiviral signaling. Upon viral infection, RIG-I/MDA5 and cGAS recognize viral dsRNA and dsDNA respectively, and activate TBK1/IKKε via adaptors MAVS and TRIF, as well as E3 lig-ases TRAF3/6, leading to the activation of IRF3 and the production of IFNs. MST1 can phosphorylate and suppress the activation of IRF3, as well as TBK1, to impair antiviral response. The downstream effectors YAP/TAZ of the Hippo pathway also negatively regulate antiviral signaling through inactivating TBK1 and IRF3. Viral infection induces the phosphorylation and degradation of YAP by IKKε, relieving its inhibitory role in antiviral immunity. IKKε can also induce the ubiquitination and degradation of LATS1/2 in glioblastoma multiforme cell lines, causing YAP activation. In addition, IRF3 can promote YAP−TEAD-driven gastric caner growth. |
MST KINASES AND TLR SIGNALING
Recently, we and others have reported that MST kinases play crucial roles on the modulation of TLR signaling and inflammation [61][62][63] (Figure 1A). The expression of MST4 responds dynamically to LPS stimulation and/or bacterial infection in immune cells and organs of mice [61]. Overexpression of MST4 suppresses LPS- or bacteria-induced production of pro-inflammatory cytokines IL-6 and TNF-α in human THP-1 cells and mouse peritoneal exudate macrophages (PEMs), while knockdown of MST4 has an opposite role. MST4 activator MO25 can further enhance the inhibitory effect of MST4 on TLR signaling. Upon LPS stimulation, MST4 expression is increased, and it directly binds and phosphorylates the TRAF_C domain of TRAF6 at Thr463 and Thr486, which impairs the oligomerization of TRAF6. Due to the important role of oligomerization on the E3 ligase activity of TRAF6 [64][65], TRAF6 phosphorylation by MST4 impairs its autoubiquitination and the formation of signaling complex, causing the suppression of TLR-mediated inflammatory responses. Clinical data showed that MST4 expression is markedly downregulated and inversely correlates with IL6 expression in the peripheral blood samples of patients with sepsis [61]. In a mouse model developing septic shock, MST4 protects mice against exacerbated inflammation in a TRAF6- and macrophage-dependent manner, suggesting that MST4 has a crucial role in limiting inflammatory damage and maintaining immune homeostasis upon bacterial infection.
–
During pathogen infection, phagocytes such as neutrophils and macrophages can recognize, engulf, and degrade microorganism [66][67][68]. Phagocytes sense chemical components and physical properties of microorganism and engulf microorganism into phagosomes. Phagosomal NADPH oxidase machinery can generate reactive oxygen species (ROS) to kill pathogens. Mitochondrial ROS (mROS) is required for the optimal clearance of pathogen by macrophages [69] (Figure 1A). Cell surface receptors TLR1/2/4 contributes to the mROS production via recruiting mitochondria to phagosomes. A further study found that MST1/2 are indispensible for this process (Figure 1A). Depletion of MST1/2 in myeloid cells leads to more susceptibility of mice to bacterial infection and severe inflammation [62]. Upon bacterial infection, MST1/2 are activated by TLR1/2/4−MyD88 signaling and in turn phosphorylates PKC-α at Ser226 and Thr228, leading to PKC-α activation. Activated PKC-α interferes with the binding of the Rho-GTP-dissociation inhibitor Ly-GDI (also named Rho-GDI2) to Rac1 via phosphorylating Ly-GDI at Ser31, leading to the release of Rac1 [70]. TRAF6 can further mediate Lys63-linked polyubiquitination of GTP-charged Rac1 at Lys16 that results in its activation. In turn, the polyubiquitination of Rac1 promotes the interaction of TRAF6 with ECSIT. TRAF6 enhances ECSIT ubiquitination and enrichment at the mitochondrial periphery [69]. ECSIT is a regulator of mitochondrial complex I assembly [71]. It associates with mitochondrial complex I assembly chaperone NDUFAF1 to regulate mitochondrial function that is required for the recruitment of mitochondria to phagosomes and the production of ROS in phagocytes. However, the mechanism for the activation of MST1/2 by TLRs is still undefined. Recently, Liu et al. found that Drosophila IRAK homolog Pelle (Pll) acting downstream of Toll (Tl) receptor directly phosphorylates Cka (Drosophila homolog of STRNs) and promotes its degradation [72] (Figure 1B). Because both the kinase Hippo (Hpo, Drosophila homolog of MST1/2) and the phosphatase PP2A B’’’ regulatory subunit Cka are major components of Drosophila STRIPAK complex and Cka is a negative regulator of Hpo activity [28], the degradation of Cka will facilitate the activation of Hpo. Thus mammalian MST1/2 may be activated by TLR1/2/4 in a similar manner.
–
Besides their roles on TLR-mediated ROS production, MST1/2 are also involved in the regulation of canonical TLR-mediated inflammatory response (Figure 1A). Upon the stimulation with poly(I:C), LPS and CpG that activate TLR3/4/9 respectively, the MST1 expression is decreased in mouse PEMs [63]. MST1 deficiency results in increased expression of IL-1β, IL-6 and TNF-α in macrophages in response to LPS and CpG stimulation, but reduced expression of IFN-β in response to LPS and poly(I:C) stimulation [62][63]. IRAK1 is a component of the Myddsome and required for TLR-mediated production of pro-inflammatory cytokines [73]. However, its phosphorylation and degradation promotes IFN-β production [74][75]. MST1 can associate with IRAK1 to induce its phosphorylation and degradation, resulting in impaired TLR4/9-mediated NF-κB activation and pro-inflammatory cytokines production but enhanced TLR3/4-stimulated IRF3 phosphorylation and IFN-β production [63]. Whether IRAK1 is the direct substrate of MST1 needs more biochemical investigation.
–
Numerous studies have revealed the regulatory roles of TLR signaling on cancer development [76][77]. Many TLRs ligands such as poly(I:C), LPS and CpG have potential antitumor activity, indicating the activation of TLRs suppresses tumorigenesis might through stimulating IFN response. However, other reports suggest TLR-mediated chronic inflammation promotes tumor development. For example, ultraviolet irradiation promotes angiotropism and metastasis in melanoma through inducing TLR4/Myd88-driven neutrophilic inflammation [78]. Intestinal microbiota and TLR4 signaling also have potential roles in promoting hepatocellular carcinoma (HCC) proliferation [79]. Li et al. found that the expression of MST1 is significantly decreased in macrophages from HCC patients and inversely correlated with the expression of IRAK1, and the levels of phosphorylated p65 and STAT3 [63]. Consistent with its inhibitory role in pro-inflammatory response and positive role in IFN response, MST1 guards mice from chronic inflammation-driven HCC upon LPS stimulation or bacterial infection.
–
Besides the direct regulation of TLR signaling, the GCKII family kinases also facilitates TLR-mediated antimicrobial response via Hippo signaling (Figure 1B). In Drosophila, like Tl and Myd88 deficiency, depletion of Hpo or Warts (Wts, Drosophila homolog of LATS1/2) in fly fat bodies, as well as overexpression of Yorkie (Yki, Drosophila homolog of YAP), causes higher sensitivity to bacterial and fungal infection [72]. The transcription of Drosophila IκB homolog Cactus (Cact) that inhibits the NF-κB family transcription factors Dorsal (Dl) and Dorsal-related immune factor (Dif) is controlled by the Yki-Scalloped (Sd, Drosophila homolog of TEADs) transcription factor complex. Hpo can suppress the expression of Cact to promote innate immune response via inhibiting Yki activity. Due to the impact of Tl on Hpo activation, microbial infection can trigger Hippo signal transduction, which inhibits Cact expression and subsequently facilitates antimicrobial response.
MST KINASES IN ANTIVIRAL SIGNALING REGULATION
The activation of transcription factor IRF3 is a key step in antiviral response. IRF3 is constituted of an N-terminal DNA binding domain, an IRF association domain and a C-terminal auto-inhibitory region. Upon viral infection, IRF3 is phosphorylated by TBK1 or IKKε at multiple sites in auto-inhibitory region, which leads to its homo-dimerization and nuclear localization [80][81]. In nucleus, IRF3 binds the IFN-stimulated response element (ISRE) sequences within the promoters of IFN genes to activate their transcription. MST1 was reported to suppress RIG-I−MAVS- and cGAS−STING-mediated antiviral response via the regulation of IRF3, as well as its upstream kinase TBK1 [82] (Figure 2). MST1 can interact with and phosphorylate IRF3 at Thr75 and Thr253. MST1 partners SAV1 and RASSF family proteins RASSF1A and RASSF5 may mediate the interaction between MST1 and IRF3 to promote IRF3 phosphorylation. Residues Thr75 and Thr253 localize at DNA binding and IRF association domains of IRF3 respectively. Phosphorylation of these two threonine residues disrupts the dimerization and DNA-binding ability of IRF3 and thus impairs its transcription activity and antiviral signaling. Furthermore, PPM1B was identified as the phosphatase responsible for dephosphorylation of IRF3 at Thr75 and Thr253 [82]. MST1 also suppresses TBK1 activation, further contributing to the attenuation of IRF3 activation. Zebrafish ectopically expressing MST1 are more susceptible to viral infection, while depletion of MST1 protects cells and mice against viral infection, suggesting MST1 physiologically suppresses innate immune response during viral invasion, which may prevent tissue damage caused by excessive IFN response.
–
Recently, two groups reported that YAP/TAZ in the Hippo pathway could negatively regulate antiviral response [83][84] (Figure 2). Zhang et al. found that YAP/TAZ attenuate K63-linked polyuniquitination of TBK1 by TRAFs, and they also associate with TBK1/IKKε to suppress their binding to upstream adaptors STING and MAVS, as well as to the substrate IRF3, both of which result in the inhibition of TBK1 activity and IRF3 phosphorylation [83]. Distinct from this finding, Wang et al. reported that YAP associates with IRF3 to directly block its homo-dimerization and interaction with importins, and therefore to prevent its nuclear translocation, but has no influence on the activity of TBK1/IKKε or the phosphorylation of IRF3 [84]. Both of groups demonstrated that the transcriptional activity of YAP/TAZ is dispensable for their inhibitory role in antiviral response, but the C-terminal transactivation of YAP is required for this function. Overexpressing YAP or TAZ enhances viral infection in human cells and zebrafish, while myeloid YAP deficiency has an opposite effect in mice. When Hippo signal is turned “ON” by cellular nutrition or physical status including serum starvation and high cell confluence, YAP/TAZ are phosphorylated and inactivated by MST1/2 downstream kinases LATS1/2, and thus their suppression on TBK1 is relieved [83]. This finding is different from the aforementioned role of MST1 in antiviral response through directly inhibiting IRF3 that does not depend on Hippo signaling [82]. Furthermore, viral infection induces the phosphorylation of YAP at Ser403 by IKKε, triggering the lysosomal degradation of YAP and also relieving its negative function in antiviral immunity, which is independent of Hippo signaling [84]. However, IKKε was also reported to induce the ubiquitination and proteasomal degradation of LATS1/2 in glioblastoma multiforme cell lines, causing YAP activation [85]. A selective inhibitor of IKKε, amlexanox, reverses the inhibition of IKKε on LATS1/2 and thus declines glioblastoma cell migration and invasion and glioblastoma growth in xenograft mouse model. In this regard, we have recently found that both IRF3 and YAP are unregulated in clinical samples of gastric cancer [86]. Viral infection that activates IRF3 relieves the inhibition of YAP. Mechanistically, phosphorylated IRF3 can bind both YAP and TEAD4 in the nucleus to co-regulate the target genes of the Hippo pathway. Treatment with amlexanox that decreases the activity of IRF3, significantly suppresses YAP-driven gastric cancer growth in mice. These observations further indicate a complex interplay between the Hippo pathway and innate immune regulation.
CONCLUSION AND PERSPECTIVE
MST kinases are emerging as crucial regulators of innate immune response. They regulate antibacterial and antiviral signaling via targeting multiple molecules including kinases IRAK1 and TBK1/IKKε, E3 ligase TRAF6 and transcription factor IRF3. The activity of these signal molecules is important for their functions and must be strictly regulated. Current results suggest that MST kinases can suppress the activity of these molecules via direct phosphorylation, which is required for limiting excessive immune response. MST kinases also have positive roles against pathogen infection through diverse mechanism. Therefore, MST kinases and the Hippo pathway exert their influences on the regulation of innate immune response likely in a context-dependent manner. The type of pathogens and host cells, as well as the specific tissue microenvironment, might determine the positive or negative role of MST kinases in innate immunity. Given that the activation of MST kinases is regulated by cell−cell contact, mechanical cues, GPCR signal and cell stress [87][88], these factors may also influence innate immune response. Furthermore, the activity of MST kinases is negatively regulated by the PP2A module in the STRIPAK complex, so other components of STRIPAK complex are expected to regulate microbial defense programs. Several components of STRIPAK complex have been reported to participate in the regulation of T and B cell development and IFN signaling [89][90][91]. Whether other subunits possess MST-dependent or independent roles on immune regulation need to be investigated.
–
Excessive inflammatory response usually causes tissue damage, while impaired immune response is insufficient to prevent microbial infection. Due to the important function on immune regulation, dysregulation of MST kinases often links to immune diseases. Several mutations of MST1 have been detected in patients with immunodeficiency [92][93]. These patients are susceptive to bacterial and viral infection possibly caused by defective development of T and B cells. This observation is distinct from that in mice where MST1 deficiency resulted in impaired innate antiviral response [82]. Thus MST1 might play distinct roles at different stages of antiviral immunity. In patients with IgG4-related autoimmune pancreatitis and rheumatoid arthritis, the CpG sites in the promoter region of MST1 were hypermethylated and the expression of MST1 was reduced in patient Treg cells [94]. It is possible that the disorders caused by these alterations on MST1 partially result from the dysregulation of innate immune response. The association of MST1 and other MST kinases and immune diseases remains to be elucidated. Until now, studies on the regulatory roles of MST kinases in immune regulation are still limited. Future discoveries will uncover their novel regulatory mechanism and provide new therapeutic strategies for related diseases.
References
- G. Manning, D.B. Whyte, R. Martinez, T. Hunter, and S. Sudarsanam, "The Protein Kinase Complement of the Human Genome", Science, vol. 298, pp. 1912-1934, 2002. http://dx.doi.org/10.1126/science.1075762
- K. Strange, J. Denton, and K. Nehrke, "Ste20-Type Kinases: Evolutionarily Conserved Regulators of Ion Transport and Cell Volume", Physiology, vol. 21, pp. 61-68, 2006. http://dx.doi.org/10.1152/physiol.00139.2005
- F. Yu, B. Zhao, and K. Guan, "Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer", Cell, vol. 163, pp. 811-828, 2015. http://dx.doi.org/10.1016/j.cell.2015.10.044
- Z. Shi, S. Jiao, and Z. Zhou, "Structural dissection of Hippo signaling", Acta Biochimica et Biophysica Sinica, vol. 47, pp. 29-38, 2015. http://dx.doi.org/10.1093/abbs/gmu107
- D. Zhou, C. Conrad, F. Xia, J. Park, B. Payer, Y. Yin, G.Y. Lauwers, W. Thasler, J.T. Lee, J. Avruch, and N. Bardeesy, "Mst1 and Mst2 Maintain Hepatocyte Quiescence and Suppress Hepatocellular Carcinoma Development through Inactivation of the Yap1 Oncogene", Cancer Cell, vol. 16, pp. 425-438, 2009. http://dx.doi.org/10.1016/j.ccr.2009.09.026
- H. Song, K.K. Mak, L. Topol, K. Yun, J. Hu, L. Garrett, Y. Chen, O. Park, J. Chang, R.M. Simpson, C. Wang, B. Gao, J. Jiang, and Y. Yang, "Mammalian Mst1 and Mst2 kinases play essential roles in organ size control and tumor suppression", Proceedings of the National Academy of Sciences, vol. 107, pp. 1431-1436, 2010. http://dx.doi.org/10.1073/pnas.0911409107
- L. Lu, Y. Li, S.M. Kim, W. Bossuyt, P. Liu, Q. Qiu, Y. Wang, G. Halder, M.J. Finegold, J. Lee, and R.L. Johnson, "Hippo signaling is a potent in vivo growth and tumor suppressor pathway in the mammalian liver", Proceedings of the National Academy of Sciences, vol. 107, pp. 1437-1442, 2010. http://dx.doi.org/10.1073/pnas.0911427107
- D. Zhou, Y. Zhang, H. Wu, E. Barry, Y. Yin, E. Lawrence, D. Dawson, J.E. Willis, S.D. Markowitz, F.D. Camargo, and J. Avruch, "Mst1 and Mst2 protein kinases restrain intestinal stem cell proliferation and colonic tumorigenesis by inhibition of Yes-associated protein (Yap) overabundance", Proceedings of the National Academy of Sciences, vol. 108, 2011. http://dx.doi.org/10.1073/pnas.1110428108
- T. Moroishi, C.G. Hansen, and K. Guan, "The emerging roles of YAP and TAZ in cancer", Nature Reviews Cancer, vol. 15, pp. 73-79, 2015. http://dx.doi.org/10.1038/nrc3876
- F. Zanconato, M. Cordenonsi, and S. Piccolo, "YAP/TAZ at the Roots of Cancer", Cancer Cell, vol. 29, pp. 783-803, 2016. http://dx.doi.org/10.1016/j.ccell.2016.05.005
- C.F. Huang, Y. Wu, C. Hsu, W. Lee, M. Lai, T. Lu, C. Huang, T. Leu, H. Shih, H. Fang, D.R. Robinson, H. Kung, and C. Yuan, "Caspase Activation of Mammalian Sterile 20-like Kinase 3 (Mst3)", Journal of Biological Chemistry, vol. 277, pp. 34367-34374, 2002. http://dx.doi.org/10.1074/jbc.M202468200
- H. Cornils, R.S. Kohler, A. Hergovich, and B.A. Hemmings, "Human NDR Kinases Control G1/S Cell Cycle Transition by Directly Regulating p21 Stability", Molecular and Cellular Biology, vol. 31, pp. 1382-1395, 2011. http://dx.doi.org/10.1128/MCB.01216-10
- M.R. Stegert, A. Hergovich, R. Tamaskovic, S.J. Bichsel, and B.A. Hemmings, "Regulation of NDR Protein Kinase by Hydrophobic Motif Phosphorylation Mediated by the Mammalian Ste20-Like Kinase MST3", Molecular and Cellular Biology, vol. 25, pp. 11019-11029, 2005. http://dx.doi.org/10.1128/MCB.25.24.11019-11029.2005
- A. Vichalkovski, E. Gresko, H. Cornils, A. Hergovich, D. Schmitz, and B.A. Hemmings, "NDR Kinase Is Activated by RASSF1A/MST1 in Response to Fas Receptor Stimulation and Promotes Apoptosis", Current Biology, vol. 18, pp. 1889-1895, 2008. http://dx.doi.org/10.1016/j.cub.2008.10.060
- S. Ultanir, S. Yadav, N. Hertz, J. Oses-Prieto, S. Claxton, A. Burlingame, K. Shokat, L. Jan, and Y. Jan, "MST3 Kinase Phosphorylates TAO1/2 to Enable Myosin Va Function in Promoting Spine Synapse Development", Neuron, vol. 84, pp. 968-982, 2014. http://dx.doi.org/10.1016/j.neuron.2014.10.025
- B. Lorber, M.L. Howe, L.I. Benowitz, and N. Irwin, "Mst3b, an Ste20-like kinase, regulates axon regeneration in mature CNS and PNS pathways", Nature Neuroscience, vol. 12, pp. 1407-1414, 2009. http://dx.doi.org/10.1038/nn.2414
- J.P. ten Klooster, M. Jansen, J. Yuan, V. Oorschot, H. Begthel, V. Di Giacomo, F. Colland, J. de Koning, M.M. Maurice, P. Hornbeck, and H. Clevers, "Mst4 and Ezrin Induce Brush Borders Downstream of the Lkb1/Strad/Mo25 Polarization Complex", Developmental Cell, vol. 16, pp. 551-562, 2009. http://dx.doi.org/10.1016/j.devcel.2009.01.016
- T. Matsuki, R.T. Matthews, J.A. Cooper, M.P. van der Brug, M.R. Cookson, J.A. Hardy, E.C. Olson, and B.W. Howell, "Reelin and Stk25 Have Opposing Roles in Neuronal Polarization and Dendritic Golgi Deployment", Cell, vol. 143, pp. 826-836, 2010. http://dx.doi.org/10.1016/j.cell.2010.10.029
- C. Preisinger, B. Short, V. De Corte, E. Bruyneel, A. Haas, R. Kopajtich, J. Gettemans, and F.A. Barr, "YSK1 is activated by the Golgi matrix protein GM130 and plays a role in cell migration through its substrate 14-3-3ζ", The Journal of Cell Biology, vol. 164, pp. 1009-1020, 2004. http://dx.doi.org/10.1083/jcb.200310061
- Z. Shi, S. Jiao, Z. Zhang, M. Ma, Z. Zhang, C. Chen, K. Wang, H. Wang, W. Wang, L. Zhang, Y. Zhao, and Z. Zhou, "Structure of the MST4 in Complex with MO25 Provides Insights into Its Activation Mechanism", Structure, vol. 21, pp. 449-461, 2013. http://dx.doi.org/10.1016/j.str.2013.01.007
- B.M. Filippi, P. de los Heros, Y. Mehellou, I. Navratilova, R. Gourlay, M. Deak, L. Plater, R. Toth, E. Zeqiraj, and D.R. Alessi, "MO25 is a master regulator of SPAK/OSR1 and MST3/MST4/YSK1 protein kinases", The EMBO Journal, vol. 30, pp. 1730-1741, 2011. http://dx.doi.org/10.1038/emboj.2011.78
- Q. Hao, M. Feng, Z. Shi, C. Li, M. Chen, W. Wang, M. Zhang, S. Jiao, and Z. Zhou, "Structural insights into regulatory mechanisms of MO25-mediated kinase activation", Journal of Structural Biology, vol. 186, pp. 224-233, 2014. http://dx.doi.org/10.1016/j.jsb.2014.04.005
- C.D. Madsen, S. Hooper, M. Tozluoglu, A. Bruckbauer, G. Fletcher, J.T. Erler, P.A. Bates, B. Thompson, and E. Sahai, "STRIPAK components determine mode of cancer cell migration and metastasis", Nature Cell Biology, vol. 17, pp. 68-80, 2014. http://dx.doi.org/10.1038/ncb3083
- X. Ma, H. Zhao, J. Shan, F. Long, Y. Chen, Y. Chen, Y. Zhang, X. Han, and D. Ma, "PDCD10 Interacts with Ste20-related Kinase MST4 to Promote Cell Growth and Transformation via Modulation of the ERK Pathway", Molecular Biology of the Cell, vol. 18, pp. 1965-1978, 2007. http://dx.doi.org/10.1091/mbc.E06-07-0608
- M. Zhang, L. Dong, Z. Shi, S. Jiao, Z. Zhang, W. Zhang, G. Liu, C. Chen, M. Feng, Q. Hao, W. Wang, M. Yin, Y. Zhao, L. Zhang, and Z. Zhou, "Structural Mechanism of CCM3 Heterodimerization with GCKIII Kinases", Structure, vol. 21, pp. 680-688, 2013. http://dx.doi.org/10.1016/j.str.2013.02.015
- M. Fidalgo, M. Fraile, A. Pires, T. Force, C. Pombo, and J. Zalvide, "CCM3/PDCD10 stabilizes GCKIII proteins to promote Golgi assembly and cell orientation", Journal of Cell Science, vol. 123, pp. 1274-1284, 2010. http://dx.doi.org/10.1242/jcs.061341
- Z. Shi, S. Jiao, and Z. Zhou, "STRIPAK complexes in cell signaling and cancer", Oncogene, vol. 35, pp. 4549-4557, 2016. http://dx.doi.org/10.1038/onc.2016.9
- P.S. Ribeiro, F. Josué, A. Wepf, M.C. Wehr, O. Rinner, G. Kelly, N. Tapon, and M. Gstaiger, "Combined Functional Genomic and Proteomic Approaches Identify a PP2A Complex as a Negative Regulator of Hippo Signaling", Molecular Cell, vol. 39, pp. 521-534, 2010. http://dx.doi.org/10.1016/j.molcel.2010.08.002
- X. Du, A. Yu, and W. Tao, "The non-canonical Hippo/Mst pathway in lymphocyte development and functions", Acta Biochimica et Biophysica Sinica, vol. 47, pp. 60-64, 2015. http://dx.doi.org/10.1093/abbs/gmu112
- D. Zhou, B.D. Medoff, L. Chen, L. Li, X. Zhang, M. Praskova, M. Liu, A. Landry, R.S. Blumberg, V.A. Boussiotis, R. Xavier, and J. Avruch, "The Nore1B/Mst1 complex restrains antigen receptor-induced proliferation of naïve T cells", Proceedings of the National Academy of Sciences, vol. 105, pp. 20321-20326, 2008. http://dx.doi.org/10.1073/pnas.0810773105
- J. Choi, S. Oh, D. Lee, H.J. Oh, J.Y. Park, S.B. Lee, and D. Lim, "Mst1-FoxO Signaling Protects Naïve T Lymphocytes from Cellular Oxidative Stress in Mice", PLoS ONE, vol. 4, pp. e8011, 2009. http://dx.doi.org/10.1371/journal.pone.0008011
- M.K. Lehtinen, Z. Yuan, P.R. Boag, Y. Yang, J. Villén, E.B. Becker, S. DiBacco, N. de la Iglesia, S. Gygi, T.K. Blackwell, and A. Bonni, "A Conserved MST-FOXO Signaling Pathway Mediates Oxidative-Stress Responses and Extends Life Span", Cell, vol. 125, pp. 987-1001, 2006. http://dx.doi.org/10.1016/j.cell.2006.03.046
- T. Kinashi, "Integrin Regulation of Lymphocyte Trafficking: Lessons from Structural and Signaling Studies", Advances in Immunology, pp. 185-227, 2007. http://dx.doi.org/10.1016/S0065-2776(06)93005-3
- F. Mou, M. Praskova, F. Xia, D. Van Buren, H. Hock, J. Avruch, and D. Zhou, "The Mst1 and Mst2 kinases control activation of rho family GTPases and thymic egress of mature thymocytes", Journal of Experimental Medicine, vol. 209, pp. 741-759, 2012. http://dx.doi.org/10.1084/jem.20111692
- Y. Dong, X. Du, J. Ye, M. Han, T. Xu, Y. Zhuang, and W. Tao, "A Cell-Intrinsic Role for Mst1 in Regulating Thymocyte Egress", The Journal of Immunology, vol. 183, pp. 3865-3872, 2009. http://dx.doi.org/10.4049/jimmunol.0900678
- K. Katagiri, T. Katakai, Y. Ebisuno, Y. Ueda, T. Okada, and T. Kinashi, "Mst1 controls lymphocyte trafficking and interstitial motility within lymph nodes", The EMBO Journal, vol. 28, pp. 1319-1331, 2009. http://dx.doi.org/10.1038/emboj.2009.82
- Y. Ueda, K. Katagiri, T. Tomiyama, K. Yasuda, K. Habiro, T. Katakai, S. Ikehara, M. Matsumoto, and T. Kinashi, "Mst1 regulates integrin-dependent thymocyte trafficking and antigen recognition in the thymus", Nature Communications, vol. 3, 2012. http://dx.doi.org/10.1038/ncomms2105
- K. Katagiri, M. Imamura, and T. Kinashi, "Spatiotemporal regulation of the kinase Mst1 by binding protein RAPL is critical for lymphocyte polarity and adhesion", Nature Immunology, vol. 7, pp. 919-928, 2006. http://dx.doi.org/10.1038/ni1374
- K. Katagiri, A. Maeda, M. Shimonaka, and T. Kinashi, "RAPL, a Rap1-binding molecule that mediates Rap1-induced adhesion through spatial regulation of LFA-1", Nature Immunology, vol. 4, pp. 741-748, 2003. http://dx.doi.org/10.1038/ni950
- F. Tang, J. Gill, X. Ficht, T. Barthlott, H. Cornils, D. Schmitz-Rohmer, D. Hynx, D. Zhou, L. Zhang, G. Xue, M. Grzmil, Z. Yang, A. Hergovich, G.A. Hollaender, J.V. Stein, B.A. Hemmings, and P. Matthias, "The kinases NDR1/2 act downstream of the Hippo homolog MST1 to mediate both egress of thymocytes from the thymus and lymphocyte motility", Science Signaling, vol. 8, 2015. http://dx.doi.org/10.1126/scisignal.aab2425
- X. Xu, X. Wang, E.M. Todd, E.R. Jaeger, J.L. Vella, O.L. Mooren, Y. Feng, J. Hu, J.A. Cooper, S.C. Morley, and Y.H. Huang, "Mst1 Kinase Regulates the Actin-Bundling Protein L-Plastin To Promote T Cell Migration", The Journal of Immunology, vol. 197, pp. 1683-1691, 2016. http://dx.doi.org/10.4049/jimmunol.1600874
- N. Kondo, Y. Ueda, T. Kita, M. Ozawa, T. Tomiyama, K. Yasuda, D. Lim, and T. Kinashi, "NDR1-Dependent Regulation of Kindlin-3 Controls High-Affinity LFA-1 Binding and Immune Synapse Organization", Molecular and Cellular Biology, vol. 37, 2017. http://dx.doi.org/10.1128/MCB.00424-16
- S.Z. Josefowicz, L. Lu, and A.Y. Rudensky, "Regulatory T Cells: Mechanisms of Differentiation and Function", Annual Review of Immunology, vol. 30, pp. 531-564, 2012. http://dx.doi.org/10.1146/annurev.immunol.25.022106.141623
- T. Tomiyama, Y. Ueda, T. Katakai, N. Kondo, K. Okazaki, and T. Kinashi, "Antigen-Specific Suppression and Immunological Synapse Formation by Regulatory T Cells Require the Mst1 Kinase", PLoS ONE, vol. 8, pp. e73874, 2013. http://dx.doi.org/10.1371/journal.pone.0073874
- X. Du, H. Shi, J. Li, Y. Dong, J. Liang, J. Ye, S. Kong, S. Zhang, T. Zhong, Z. Yuan, T. Xu, Y. Zhuang, B. Zheng, J. Geng, and W. Tao, "Mst1/Mst2 Regulate Development and Function of Regulatory T Cells through Modulation of Foxo1/Foxo3 Stability in Autoimmune Disease", The Journal of Immunology, vol. 192, pp. 1525-1535, 2014. http://dx.doi.org/10.4049/jimmunol.1301060
- F. Ramsdell, and S.F. Ziegler, "FOXP3 and scurfy: how it all began", Nature Reviews Immunology, vol. 14, pp. 343-349, 2014. http://dx.doi.org/10.1038/nri3650
- J. van Loosdregt, and P.J. Coffer, "Post-translational modification networks regulating FOXP3 function", Trends in Immunology, vol. 35, pp. 368-378, 2014. http://dx.doi.org/10.1016/j.it.2014.06.005
- J. Li, X. Du, H. Shi, K. Deng, H. Chi, and W. Tao, "Mammalian Sterile 20-like Kinase 1 (Mst1) Enhances the Stability of Forkhead Box P3 (Foxp3) and the Function of Regulatory T Cells by Modulating Foxp3 Acetylation", Journal of Biological Chemistry, vol. 290, pp. 30762-30770, 2015. http://dx.doi.org/10.1074/jbc.M115.668442
- J. Geng, S. Yu, H. Zhao, X. Sun, X. Li, P. Wang, X. Xiong, L. Hong, C. Xie, J. Gao, Y. Shi, J. Peng, R.L. Johnson, N. Xiao, L. Lu, J. Han, D. Zhou, and L. Chen, "The transcriptional coactivator TAZ regulates reciprocal differentiation of TH17 cells and Treg cells", Nature Immunology, vol. 18, pp. 800-812, 2017. http://dx.doi.org/10.1038/ni.3748
- B. Amulic, C. Cazalet, G.L. Hayes, K.D. Metzler, and A. Zychlinsky, "Neutrophil Function: From Mechanisms to Disease", Annual Review of Immunology, vol. 30, pp. 459-489, 2012. http://dx.doi.org/10.1146/annurev-immunol-020711-074942
- Y. Zhang, W. Tang, H. Zhang, X. Niu, Y. Xu, J. Zhang, K. Gao, W. Pan, T. Boggon, D. Toomre, W. Min, and D. Wu, "A Network of Interactions Enables CCM3 and STK24 to Coordinate UNC13D-Driven Vesicle Exocytosis in Neutrophils", Developmental Cell, vol. 27, pp. 215-226, 2013. http://dx.doi.org/10.1016/j.devcel.2013.09.021
- A.R. Kurz, M. Pruenster, I. Rohwedder, M. Ramadass, K. Schäfer, U. Harrison, G. Gouveia, C. Nussbaum, R. Immler, J.R. Wiessner, A. Margraf, D. Lim, B. Walzog, S. Dietzel, M. Moser, C. Klein, D. Vestweber, R. Haas, S.D. Catz, and M. Sperandio, "MST1-dependent vesicle trafficking regulates neutrophil transmigration through the vascular basement membrane", Journal of Clinical Investigation, vol. 126, pp. 4125-4139, 2016. http://dx.doi.org/10.1172/JCI87043
- S.W. Brubaker, K.S. Bonham, I. Zanoni, and J.C. Kagan, "Innate Immune Pattern Recognition: A Cell Biological Perspective", Annual Review of Immunology, vol. 33, pp. 257-290, 2015. http://dx.doi.org/10.1146/annurev-immunol-032414-112240
- S. Akira, S. Uematsu, and O. Takeuchi, "Pathogen Recognition and Innate Immunity", Cell, vol. 124, pp. 783-801, 2006. http://dx.doi.org/10.1016/j.cell.2006.02.015
- S. Pandey, T. Kawai, and S. Akira, "Microbial Sensing by Toll-Like Receptors and Intracellular Nucleic Acid Sensors", Cold Spring Harbor Perspectives in Biology, vol. 7, pp. a016246, 2014. http://dx.doi.org/10.1101/cshperspect.a016246
- T. Kawai, and S. Akira, "The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors", Nature Immunology, vol. 11, pp. 373-384, 2010. http://dx.doi.org/10.1038/ni.1863
- Y. Loo, and M. Gale, "Immune Signaling by RIG-I-like Receptors", Immunity, vol. 34, pp. 680-692, 2011. http://dx.doi.org/10.1016/j.immuni.2011.05.003
- B. Wu, and S. Hur, "How RIG-I like receptors activate MAVS", Current Opinion in Virology, vol. 12, pp. 91-98, 2015. http://dx.doi.org/10.1016/j.coviro.2015.04.004
- D. Goubau, S. Deddouche, and C. Reis e Sousa, "Cytosolic Sensing of Viruses", Immunity, vol. 38, pp. 855-869, 2013. http://dx.doi.org/10.1016/j.immuni.2013.05.007
- Z. Ma, and B. Damania, "The cGAS-STING Defense Pathway and Its Counteraction by Viruses", Cell Host & Microbe, vol. 19, pp. 150-158, 2016. http://dx.doi.org/10.1016/j.chom.2016.01.010
- S. Jiao, Z. Zhang, C. Li, M. Huang, Z. Shi, Y. Wang, X. Song, H. Liu, C. Li, M. Chen, W. Wang, Y. Zhao, Z. Jiang, H. Wang, C.C.L. Wong, C. Wang, and Z. Zhou, "The kinase MST4 limits inflammatory responses through direct phosphorylation of the adaptor TRAF6", Nature Immunology, vol. 16, pp. 246-257, 2015. http://dx.doi.org/10.1038/ni.3097
- J. Geng, X. Sun, P. Wang, S. Zhang, X. Wang, H. Wu, L. Hong, C. Xie, X. Li, H. Zhao, Q. Liu, M. Jiang, Q. Chen, J. Zhang, Y. Li, S. Song, H. Wang, R. Zhou, R.L. Johnson, K. Chien, S. Lin, J. Han, J. Avruch, L. Chen, and D. Zhou, "Kinases Mst1 and Mst2 positively regulate phagocytic induction of reactive oxygen species and bactericidal activity", Nature Immunology, vol. 16, pp. 1142-1152, 2015. http://dx.doi.org/10.1038/ni.3268
- W. Li, J. Xiao, X. Zhou, M. Xu, C. Hu, X. Xu, Y. Lu, C. Liu, S. Xue, L. Nie, H. Zhang, Z. Li, Y. Zhang, F. Ji, L. Hui, W. Tao, B. Wei, and H. Wang, "STK4 regulates TLR pathways and protects against chronic inflammation–related hepatocellular carcinoma", Journal of Clinical Investigation, vol. 125, pp. 4239-4254, 2015. http://dx.doi.org/10.1172/JCI81203
- C. Ea, L. Sun, J. Inoue, and Z.J. Chen, "TIFA activates IκB kinase (IKK) by promoting oligomerization and ubiquitination of TRAF6", Proceedings of the National Academy of Sciences, vol. 101, pp. 15318-15323, 2004. http://dx.doi.org/10.1073/pnas.0404132101
- C. Wang, L. Deng, M. Hong, G.R. Akkaraju, J. Inoue, and Z.J. Chen, "TAK1 is a ubiquitin-dependent kinase of MKK and IKK", Nature, vol. 412, pp. 346-351, 2001. http://dx.doi.org/10.1038/35085597
- S.H. Kaufmann, and A. Dorhoi, "Molecular Determinants in Phagocyte-Bacteria Interactions", Immunity, vol. 44, pp. 476-491, 2016. http://dx.doi.org/10.1016/j.immuni.2016.02.014
- S. Gordon, "Phagocytosis: An Immunobiologic Process", Immunity, vol. 44, pp. 463-475, 2016. http://dx.doi.org/10.1016/j.immuni.2016.02.026
- D.M. Underhill, and H.S. Goodridge, "Information processing during phagocytosis", Nature Reviews Immunology, vol. 12, pp. 492-502, 2012. http://dx.doi.org/10.1038/nri3244
- A.P. West, I.E. Brodsky, C. Rahner, D.K. Woo, H. Erdjument-Bromage, P. Tempst, M.C. Walsh, Y. Choi, G.S. Shadel, and S. Ghosh, "TLR signalling augments macrophage bactericidal activity through mitochondrial ROS", Nature, vol. 472, pp. 476-480, 2011. http://dx.doi.org/10.1038/nature09973
- D. Mehta, A. Rahman, and A.B. Malik, "Protein Kinase C-α Signals Rho-Guanine Nucleotide Dissociation Inhibitor Phosphorylation and Rho Activation and Regulates the Endothelial Cell Barrier Function", Journal of Biological Chemistry, vol. 276, pp. 22614-22620, 2001. http://dx.doi.org/10.1074/jbc.M101927200
- R.O. Vogel, R.J. Janssen, M.A. van den Brand, C.E. Dieteren, S. Verkaart, W.J. Koopman, P.H. Willems, W. Pluk, L.P. van den Heuvel, J.A. Smeitink, and L.G. Nijtmans, "Cytosolic signaling protein Ecsit also localizes to mitochondria where it interacts with chaperone NDUFAF1 and functions in complex I assembly", Genes & Development, vol. 21, pp. 615-624, 2007. http://dx.doi.org/10.1101/gad.408407
- B. Liu, Y. Zheng, F. Yin, J. Yu, N. Silverman, and D. Pan, "Toll Receptor-Mediated Hippo Signaling Controls Innate Immunity in Drosophila", Cell, vol. 164, pp. 406-419, 2016. http://dx.doi.org/10.1016/j.cell.2015.12.029
- N.J. Gay, M.F. Symmons, M. Gangloff, and C.E. Bryant, "Assembly and localization of Toll-like receptor signalling complexes", Nature Reviews Immunology, vol. 14, pp. 546-558, 2014. http://dx.doi.org/10.1038/nri3713
- H. An, J. Hou, J. Zhou, W. Zhao, H. Xu, Y. Zheng, Y. Yu, S. Liu, and X. Cao, "Phosphatase SHP-1 promotes TLR- and RIG-I-activated production of type I interferon by inhibiting the kinase IRAK1", Nature Immunology, vol. 9, pp. 542-550, 2008. http://dx.doi.org/10.1038/ni.1604
- T.T. Yamin, and D.K. Miller, "The interleukin-1 receptor-associated kinase is degraded by proteasomes following its phosphorylation.", The Journal of biological chemistry, 1997. http://www.ncbi.nlm.nih.gov/pubmed/9261174
- S. Rakoff-Nahoum, and R. Medzhitov, "Toll-like receptors and cancer", Nature Reviews Cancer, vol. 9, pp. 57-63, 2008. http://dx.doi.org/10.1038/nrc2541
- J. Pradere, D.H. Dapito, and R.F. Schwabe, "The Yin and Yang of Toll-like receptors in cancer", Oncogene, vol. 33, pp. 3485-3495, 2013. http://dx.doi.org/10.1038/onc.2013.302
- T. Bald, T. Quast, J. Landsberg, M. Rogava, N. Glodde, D. Lopez-Ramos, J. Kohlmeyer, S. Riesenberg, D. van den Boorn-Konijnenberg, C. Hömig-Hölzel, R. Reuten, B. Schadow, H. Weighardt, D. Wenzel, I. Helfrich, D. Schadendorf, W. Bloch, M.E. Bianchi, C. Lugassy, R.L. Barnhill, M. Koch, B.K. Fleischmann, I. Förster, W. Kastenmüller, W. Kolanus, M. Hölzel, E. Gaffal, and T. Tüting, "Ultraviolet-radiation-induced inflammation promotes angiotropism and metastasis in melanoma", Nature, vol. 507, pp. 109-113, 2014. http://dx.doi.org/10.1038/nature13111
- D. Dapito, A. Mencin, G. Gwak, J. Pradere, M. Jang, I. Mederacke, J. Caviglia, H. Khiabanian, A. Adeyemi, R. Bataller, J. Lefkowitch, M. Bower, R. Friedman, R. Sartor, R. Rabadan, and R. Schwabe, "Promotion of Hepatocellular Carcinoma by the Intestinal Microbiota and TLR4", Cancer Cell, vol. 21, pp. 504-516, 2012. http://dx.doi.org/10.1016/j.ccr.2012.02.007
- T. Tamura, H. Yanai, D. Savitsky, and T. Taniguchi, "The IRF Family Transcription Factors in Immunity and Oncogenesis", Annual Review of Immunology, vol. 26, pp. 535-584, 2008. http://dx.doi.org/10.1146/annurev.immunol.26.021607.090400
- H. Ikushima, H. Negishi, and T. Taniguchi, "The IRF Family Transcription Factors at the Interface of Innate and Adaptive Immune Responses", Cold Spring Harbor Symposia on Quantitative Biology, vol. 78, pp. 105-116, 2013. http://dx.doi.org/10.1101/sqb.2013.78.020321
- F. Meng, R. Zhou, S. Wu, Q. Zhang, Q. Jin, Y. Zhou, S.W. Plouffe, S. Liu, H. Song, Z. Xia, B. Zhao, S. Ye, X. Feng, K. Guan, J. Zou, and P. Xu, "Mst1 shuts off cytosolic antiviral defense through IRF3 phosphorylation", Genes & Development, vol. 30, pp. 1086-1100, 2016. http://dx.doi.org/10.1101/gad.277533.116
- Q. Zhang, F. Meng, S. Chen, S.W. Plouffe, S. Wu, S. Liu, X. Li, R. Zhou, J. Wang, B. Zhao, J. Liu, J. Qin, J. Zou, X. Feng, K. Guan, and P. Xu, "Hippo signalling governs cytosolic nucleic acid sensing through YAP/TAZ-mediated TBK1 blockade", Nature Cell Biology, vol. 19, pp. 362-374, 2017. http://dx.doi.org/10.1038/ncb3496
- S. Wang, F. Xie, F. Chu, Z. Zhang, B. Yang, T. Dai, L. Gao, L. Wang, L. Ling, J. Jia, H. van Dam, J. Jin, L. Zhang, and F. Zhou, "YAP antagonizes innate antiviral immunity and is targeted for lysosomal degradation through IKKɛ-mediated phosphorylation", Nature Immunology, vol. 18, pp. 733-743, 2017. http://dx.doi.org/10.1038/ni.3744
- Y. Liu, J. Lu, Z. Zhang, L. Zhu, S. Dong, G. Guo, R. Li, Y. Nan, K. Yu, Y. Zhong, and Q. Huang, "Amlexanox, a selective inhibitor of IKBKE, generates anti-tumoral effects by disrupting the Hippo pathway in human glioblastoma cell lines", Cell Death & Disease, vol. 8, pp. e3022-e3022, 2017. http://dx.doi.org/10.1038/cddis.2017.396
- S. Jia, J. Guan, M. Chen, W. Wang, C. Li, Y. Wang, Y. Cheng, and Z. Zhou, " Targeting IRF3 as a YAP agonist therapy against gastric cancer.", J Exp Med in press., 2017.
- Z. Meng, T. Moroishi, and K. Guan, "Mechanisms of Hippo pathway regulation", Genes & Development, vol. 30, pp. 1-17, 2016. http://dx.doi.org/10.1101/gad.274027.115
- B.J. Thompson, and E. Sahai, "MST kinases in development and disease", Journal of Cell Biology, vol. 210, pp. 871-882, 2015. http://dx.doi.org/10.1083/jcb.201507005
- Q. Zou, J. Jin, Y. Xiao, H. Hu, X. Zhou, Z. Jie, X. Xie, J.Y. Li, X. Cheng, and S. Sun, "T cell development involves TRAF3IP3-mediated ERK signaling in the Golgi", Journal of Experimental Medicine, vol. 212, pp. 1323-1336, 2015. http://dx.doi.org/10.1084/jem.20150110
- I. Hoebeke, M. De Smedt, F. Stolz, K. Pike-Overzet, F.J.T. Staal, J. Plum, and G. Leclercq, "T-, B- and NK-lymphoid, but not myeloid cells arise from human CD34+CD38−CD7+ common lymphoid progenitors expressing lymphoid-specific genes", Leukemia, vol. 21, pp. 311-319, 2006. http://dx.doi.org/10.1038/sj.leu.2404488
- J. Huang, T. Liu, L. Xu, D. Chen, Z. Zhai, and H. Shu, "SIKE is an IKKε/TBK1-associated suppressor of TLR3- and virus-triggered IRF-3 activation pathways", The EMBO Journal, vol. 24, pp. 4018-4028, 2005. http://dx.doi.org/10.1038/sj.emboj.7600863
- H. Abdollahpour, G. Appaswamy, D. Kotlarz, J. Diestelhorst, R. Beier, A.A. Schäffer, E.M. Gertz, A. Schambach, H.H. Kreipe, D. Pfeifer, K.R. Engelhardt, N. Rezaei, B. Grimbacher, S. Lohrmann, R. Sherkat, and C. Klein, "The phenotype of human STK4 deficiency", Blood, vol. 119, pp. 3450-3457, 2012. http://dx.doi.org/10.1182/blood-2011-09-378158
- N.T. Nehme, J.P. Schmid, F. Debeurme, I. André-Schmutz, A. Lim, P. Nitschke, F. Rieux-Laucat, P. Lutz, C. Picard, N. Mahlaoui, A. Fischer, and G. de Saint Basile, "MST1 mutations in autosomal recessive primary immunodeficiency characterized by defective naive T-cell survival", Blood, vol. 119, pp. 3458-3468, 2012. http://dx.doi.org/10.1182/blood-2011-09-378364
- T. Fukuhara, T. Tomiyama, K. Yasuda, Y. Ueda, Y. Ozaki, Y. Son, S. Nomura, K. Uchida, K. Okazaki, and T. Kinashi, "Hypermethylation of MST1 in IgG4-related autoimmune pancreatitis and rheumatoid arthritis", Biochemical and Biophysical Research Communications, vol. 463, pp. 968-974, 2015. http://dx.doi.org/10.1016/j.bbrc.2015.06.043
ACKNOWLEDGMENTS
This work was supported by National Key R&D Program of China (2017YFA0504504), the National Natu-ral Science Foundation of China (91442125, 31470736, 31470868, 91542125, 31600731, 31700679, 81773212, 81725014), the 'Strategic Priority Research Program' (XDB19020202, XDA12020342), Shanghai Sailing Program (17YF1422200) and the Natural Science Foundation of Shang-hai (17ZR1435400).
COPYRIGHT
© 2017

MST kinases in innate immune signaling by Shi and Zhou is licensed under a Creative Commons Attribution 4.0 International License.


