Reviews:
Cell Stress, Vol. 3, No. 7, pp. 221 - 235; doi: 10.15698/cst2019.07.192
Stress and catecholamines modulate the bone marrow microenvironment to promote tumorigenesis

1 Department of Biomedicine, University Hospital and University of Basel, Basel, Switzerland.
2 Division of Clinical Hematology, University Hospital Basel, Basel, Switzerland.
Keywords: bone marrow, cancer, metastasis, leukemia, catecholamines, CXCL12/CXCR4, angiogenesis, stress.
Abbreviatons:
ALL – acute lymphoblastic leukemia,
AML – acute myeloid leukemia,
AR – adrenergic receptor,
BCL-2 – B-cell lymphoma 2,
bFGF – basic fibroblast growth factor,
BM – bone marrow,
cAMP – cyclic adenosine monophosphate,
COX2 – cyclooxygenase 2,
DA – dopamine,
DTC – disseminated tumor cells,
EPI – epinephrine,
FAK – focal adhesion kinase,
GM-CSF – granulocyte-macrophage colony-stimulating factor,
HSC – hematopoietic stem cell,
HSPC – hematopoietic stem and progenitor cell,
IL-6 – interleukin 6,
LSC – leukemic stem cell,
MDS – myelodysplastic syndrome,
MSC – mesenchymal stromal cell,
NK – natural killer,
NOR – norepinephrine,
PGE2 – prostaglandin E2,
PKA – protein kinase A,
RANK – receptor activator of NF-κB,
RANKL – RANK ligand,
SNS – sympathetic nervous system,
TAM – tumor associated macrophage,
TNF – tumor necrosis factor,
VCAM-1 – vascular cell adhesion molecule 1,
VEGF – vascular endothelial growth factor,
VEGF-R – VEGF receptor.
Received originally: 20/12/2018 Received in revised form: 30/04/2019
Accepted: 02/05/2019
Published: 04/06/2019
Correspondence:
Claudia Lengerke, MD, University Hospital Basel, Petersgraben 4, 4031 Basel, Switzerland; Tel.: +41 61 2652525; Fax: +41 61 2653380; claudia.lengerke@unibas.ch
Conflict of interest statement: The authors declare no conflict of financial interest.
Please cite this article as: Pauline Hanns, Anna M. Paczulla, Michael Medinger, Martina Konantz and Claudia Lengerke (2019). Stress and cate-cholamines modulate the bone marrow microenvironment to promote tumorigenesis. Cell Stress 3(7): 221-235. doi: 10.15698/cst2019.07.192
Abstract
High vascularization and locally secreted factors make the bone marrow (BM) microenvironment particularly hospitable for tumor cells and bones to a preferred metastatic site for disseminated cancer cells of different origins. Cancer cell homing and proliferation in the BM are amongst other regulated by complex interactions with BM niche cells (e.g. osteoblasts, endothelial cells and mesenchymal stromal cells (MSCs)), resident hematopoietic stem and progenitor cells (HSPCs) and pro-angiogenic cytokines leading to enhanced BM microvessel densities during malignant progression. Stress and catecholamine neurotransmitters released in response to activation of the sympathetic nervous system (SNS) reportedly modulate various BM cells and may thereby influence cancer progression. Here we review the role of catecholamines during tumorigenesis with particular focus on pro-tumorigenic effects mediated by the BM niche.
STRESS AND CATECHOLAMINE SIGNALING
Stress is defined as the relationship between a person and her/his environment when latter is perceived as endangering to her/his well-being. While the stimulus-based definition understands stress as the sum of effects that emerge after exerting acute or a chronic external discomfort on a subject, the response-based definition proposes stress as part of the physiological alert reaction activated by the body to better master a dangerous situation [1]. On the physiological and biochemical levels, stress involves the sympathetic nervous system (SNS) and the release of catecholamine neurotransmitters (epinephrine (EPI), norepinephrine (NOR), dopamine (DA)) from SNS fibers. Catecholamine release may differ in acute compared to chronic stress: whereas acute and chronic stress both induce EPI as well as NOR [2][3][4][5], brain concentrations of DA were instead found elevated in acute but however reduced in chronic stress [2][6][7]. The downstream adrenergic signaling responses activated by stress are furthermore dependent on the expression of specific adrenergic receptors (ARs) on various organs and tissues participating in the alert reaction [8][9]. ARs belong to the class of G protein-coupled receptors and are subdivided into α- and β-ARs. Activation of α1-ARs (comprising α1a, α1b, α1d-ARs) increases intracellular calcium levels and induces vasoconstriction, while α2-AR (comprising α2a, α2b, α2c-ARs) activation inhibits intracellular cyclic adenosine monophosphate (cAMP), insulin, acetylcholine and NOR release [10]. Stimulation of β-ARs (comprising β1, β2, β3-ARs) elevates cytosolic cAMP levels and activates protein kinase A (PKA) leading to smooth muscle relaxation and lipolysis [11][12][13]. While β3-ARs bind EPI and NOR with the same affinity, α2 and β2-ARs are more potently stimulated by EPI and α1 and β1-ARs by NOR [14][15][16]. ARs can be pharmacologically modulated by drugs with selectivity for certain receptors, e.g. “selective” blockers targeting α1-ARs (e.g. prazosin), α2-ARs, (e.g. yohimbine), β1-ARs (e.g. acebutolol) or by so-called “unselective” blockers targeting for example all types of α-ARs (e.g. phenoxybenzamine), or all types of β-ARs (e.g. propranolol). AR-blockers are in routine clinical use especially for the treatment of patients with hypertension and cardiovascular disorders.
–
Interestingly, cancer cells may also express ARs, and enhanced levels of EPI and NOR were detected in fluids and tissues of patients with cancer [17][18][19][20][21], suggesting that catecholamines and adrenergic signaling are involved in cancer pathogenesis.
EPIDEMIOLOGIC STUDIES
The relationships between stress, catecholamine levels, AR-blocker use and cancer incidence or outcome were investigated by several epidemiological studies with in part controversial results. Enhanced perceived stress (measured by self-assessment via a questionnaire) was identified as a risk factor for rectal cancer in a prospective study involving 61,563 Japanese men and women followed-up for 21 years [22]. Furthermore, elevated intra-tumor NOR levels determined by high performance liquid chromatography were associated with advanced stage and high-grade histology in ovarian carcinoma [23]. Consistently, retrospective studies indicated treatment with β-blocker to associate with increased overall and progression-free survival in patients with prostate [24][25], breast [26][27][28][29][30][31], ovarian cancer [32] or melanoma [33][34], and treatment with propranolol to reduce the incidence of head and neck, esophagus, stomach, colon, and prostate carcinoma in an analysis of 24,000 people followed-up for twelve years [35]. Finally, a meta-analysis of 20,898 patients with cancer (including patients from twelve studies published between 1993 and 2013, among which the studies mentioned above) indicated that β-blocker usage may associate with prolonged survival in early stage cancer patients undergoing surgical resection [36].
–
In contrast, other reports – also involving patients with a variety of different tumors (e.g. breast cancer [37], renal carcinoma [38], acute myeloid leukemia (AML) [39], melanoma [40][41] as well as a retrospective meta-analysis with more than 88,000 cancer patients from approximately thirty studies failed to reproduce these associations [42]. We hypothesize that these controversial results may derive from the heterogeneity among the analyzed patient groups and tumor types, stages and therapies, as well as the differences in employed β-blockers. Different types of stress may induce different catecholamine compositions (e.g. acute or chronic stress, versus exercise-induced, see also below) that influence results. Selected tumor subtypes or phases during tumorigenesis might be more or less responsive to pro-tumorigenic effects mediated by catecholamines and large meta-analyses may not sufficiently capture such effects. Prospective randomized trials focusing on defined patient subgroups, disease stages and medications are required to obtain conclusive results. Epidemiologic studies investigating β-blocker treatments in patients with cancer were recently comprehensively reviewed by Yap et al. [43].
STRESS AND CATECHOLAMINES PROMOTE TUMORIGENESIS VIA CELL AUTONOMOUS AND NON-AUTONOMOUS MECHANISMS
Experimental data indicate that stress and catecholamines promote tumor growth and metastasis via both cell autonomous and non-autonomous mechanisms [17][44][45][46][47][48][49][50][51][52][53][54][55][56] (see also the recent review by Qiao et al. [57]). In an ovarian cancer animal model, restraint stress enhanced NOR and EPI levels and thereby promoted malignant cell growth by suppressing anoikis and enhancing phosphorylation of focal adhesion kinases (FAK) [58]. Elevated housing temperature enhanced NOR levels in an orthotopic pancreatic carcinoma model thereby up-regulating the expression of anti-apoptotic B-cell lymphoma 2 (BCL-2), B-cell lymphoma-extra large (BCL-xL) and induced myeloid leukemia cell differentiation (MCL) proteins, suppressing the pro-apoptotic Bcl-2-associated death promoter (BAD) protein and inducing apoptosis resistance [59]. Similarly, in a prostate cancer xenograft model, behavioral stress increased EPI levels, induced β2-AR signaling activation and accelerated tumor progression by enhancing anti-apoptotic responses in tumor cells [60]. Finally, recent work has pointed out that catecholamines induce cytoskeleton alterations and expression of genes mediating invasive properties thereby enhancing the aggressiveness of tumor cells [45]. Molecularly, β-AR activation by catecholamines activated cAMP and downstream PKA signaling, resulting in higher Ca2+ efflux from the endoplasmic reticulum and finally modulation of cadherins and actin [45].
–
Consistent with these data, expression of different ARs has been documented on different cancer cell types and linked to cancer progression (β1: [61][62], β2 : [17][61][62], β3 : [61][63][64][65][66], and targeting of catecholamine signaling by treatment with specific β-AR inhibitors has been proposed as a potential therapeutic approach for cancer [61][67]. In fact, treatment with specific β3-AR antagonists was shown to reduce proliferation and activate cell death in tumor cells thereby inhibiting melanoma progression in a mouse model [65].
–
Non-cell autonomous catecholamine-mediated pro-tumorigenic mechanisms include effects on blood and lymphatic vessels, fibroblasts, immune cells as well as different subtypes of bone marrow (BM) cells and are thus even more complex. For example, daily restraint stress was shown to activate cancer-associated fibroblasts to produce extracellular matrix components favoring ovarian cancer growth [68]. Chronic psychological stress (induced by different types of stressors) furthermore facilitated breast cancer cell metastasis to the lungs by modulating macrophage responses and the pre-metastatic niche [56]. Additionally, chronic restraint stress promoted angio- and lymphangiogenesis [49][69] and the reorganisation of lymphatic networks within and around the primary tumor via induction of tumor-derived vascular endothelial growth factor C (VEGF-C), which in turn was found to depend on cyclooxygenase-2 (COX-2) mediated inflammatory signaling from macrophages [69]. Furthermore, in a prostate cancer mouse model NOR release in the stroma was shown to activate an angiogenic switch fueling tumor growth via the endothelial β-AR signaling pathway [70]. Consistently, β-adrenergic-mediated chronic restraint stress also enhanced leukemic burden in an acute lymphoblastic leukemia (ALL) mouse xenograft model. Interestingly, the pro-leukemogenic effect of catecholamines in this setting was not mediated by adrenergic signaling in leukemic cells themselves but rather by pro-leukemogenic modulation of host cells that interact with human ALL cells. The effects could potentially be mediated by SNS regulation of anti-tumor immune response (e.g. involving natural killer (NK) cell-mediated killing of leukemia cells) and of BM stromal cells, including osteoblasts that play a key role in the maintenance of healthy hematopoietic cells [71]. In response to stress, tumor cells furthermore showed increased release of pro-inflammatory prostaglandin E2 (PGE2) [72]. Further studies demonstrated that NOR induced activation of β3-ARs in both melanoma cells and cells of the tumor microenvironment enhanced the response of stromal macrophages and fibroblasts by inducing pro-inflammatory cytokine secretion and de novo angiogenesis in the tumor, thus sustaining tumor growth and aggressiveness [64]. Interestingly, pharmaceutical blockade of β3-AR could significantly decrease the tumor vasculature by activating apoptosis signaling pathways of endothelial cells in tumor blood vessels, thus reducing melanoma malignancy [65].
–
The pro-tumorigenic effects of stress summarized above were obtained in models studying the effects of catecholamine release associated with physical or psychological stress, or a combination of both. Interestingly, different results were observed when catecholamines released upon exercise were analyzed. Mice given the voluntary opportunity to run in an environmentally enriched cage also showed enhanced catecholamine levels, but these did not promote tumor growth [73]. Interestingly, suppressive effects on tumor growth were instead observed upon such exercise in different murine cancer models (e.g. of breast [74], pancreas [75], lung and melanoma [76]). There is no definitive molecular explanation for the different impact of stress- versus exercise-induced catecholamine increase on tumorgenesis. Possible explanations include (a) that different types of catecholamine are released in exercise compared to stress (e.g. higher induction of NOR and DA in exercise [77] and of EPI in stress conditions, perhaps due to the fact that exercise preferentially induces a response of the SNS, while stress primarily triggers an adrenal response [78]), (b) different catecholamine dynamics (intensive peaks upon exercise versus more constant enrichment under “chronic” stress with latter perhaps being only permissive for modeling of a pro-tumorgenic environment [79] and (c) confounding non-catecholamine related physiological and biochemical processes associated with exercise versus stress. Exercise-induced cancer protection could also be linked to the activation of the immune system. For example, high EPI levels induced by voluntary wheel running mobilized NK cells to the tumor site thereby reducing incidence and growth of melanoma, liver and lung tumors [76]. Catecholamine effects on the immune system are reviewed in detail elsewhere [57][80][81][82].
CANCER AND THE BM MICROENVIRONMENT
Bones and the BM are also preferred metastatic sites for different solid tumors. According to Suva et al., the post mortem incidence of BM metastases is highest in patients with breast carcinoma (73%), followed by prostate (68%), thyroid (42%), lung and renal carcinoma and melanoma [83][84]. Detectable bone metastases are a strong predictor of poor outcome and associate with 5-year survival rates of only 10% in patients with breast carcinoma [84]. In order to seed the BM, cancer cells first have to leave the primary tumor by extravasation into the circulation as disseminated tumor cells (DTCs). Next, they have to survive in the circulation until they reach their final destination, probably via a process similar to the well-described “homing” of healthy hematopoietic stem and progenitor cells (HSPCs). Upon arrival in the BM, DTCs adhere to components of this new microenvironment, which is considered a “fertile soil” for tumor cells because of its high vascularization and enhanced concentrations of pro-tumorigenic growth factors, cytokines and chemokines [85][86]. Importantly, the BM microenvironment may facilitate tumor cell dormancy and confer chemotherapy resistance to tumor cells, thus enabling their long-term persistence despite treatments, and facilitating subsequent relapses and progression. As for example shown by Carlson et al. for breast carcinoma cells, DTCs occupy perivascular niches through integrin-mediated interactions driven by endothelial-derived von Willebrand factor and vascular cell adhesion molecule 1 (VCAM-1) and thereby receive selective protection against chemotherapies [87]. Interestingly, as shown for hematologic neoplasia, solid tumor cells also appear to actively modulate the BM microenvironment to facilitate its colonization. For example, high levels of soluble intercellular adhesion molecule 1 (ICAM-1), VCAM-1 and platelet-derived growth factor (PDGF) detectable in the BM plasma of untreated advanced breast cancer patients possibly contribute to DTCs escape out of the blood vessels into the BM [88]. When entering the bone to cause osteolytic or osteoblastic lesions, cancer cells initiate a cellular crosstalk that further supports tumor growth and invasion [89], often triggering a destructive auto-regulatory feedback loop promoting tumor growth. Taken together, these data indicate that cancer cells actively modulate the BM niche to facilitate its occupation. Whether vice versa a perturbed niche by itself can trigger malignancy is currently under heavy debate [89].
THE BM NICHE IN HEMATOLOGIC MALIGNANCIES
Next to the healthy situation, the BM niche is best studied in hematologic malignancies involving malignant cells residing at this site. BM microenvironment changes are commonly observed in patients with hematologic diseases, and experimental models demonstrate an intensive crosstalk between malignant and healthy cells, suggesting that the BM niche plays important pathophysiological roles (reviewed in more detail in [90][91][92][93]). When compared to healthy controls, patients suffering from myelodysplastic syndromes (MDS) show higher CXCL12 (also named stromal cell-derived factor 1, SDF-1) levels and enhanced tumor necrosis factor α (TNFα), interleukin 6 (IL-6) and tumor necrosis factor β (TNF-β) expression in fibroblasts and macrophages, which likely contribute to disease pathogenesis [94]. However, it often remains unclear whether the observed microenvironment changes are cause or consequence of the disturbed hematopoiesis.
–
Leukemic cells are hypothesized to actively modulate the BM microenvironment to allow specific subpopulations of leukemia-initiating cells (so-called leukemic stem cells, LSCs) to colonize BM niches granting their long-term survival and perhaps protection against anti-neoplastic therapies (reviewed in more details in [95][96][97][98]). For example, in murine models of myeloproliferative neoplasia, malignant cells were shown to stimulate mesenchymal stromal cell (MSCs) to produce higher numbers of functionally altered osteoblasts, which then accumulate as inflammatory myelofibrotic cells to induce fibrosis and ultimately promote LSC persistence [99].
BM ANGIOGENESIS
Many studies have suggested a role for angiogenesis not only in the pathogenesis of solid tumors but also in hematological malignancies [100][101][102][103][104][105][106]. The first link between leukemia progression and increased BM vascularization [107] was provided 1997, when an increased blood vessel content was demonstrated in the BM of ALL patients compared to healthy controls. A detailed analysis of BM sections from ALL patients showed irregular, albeit abundant, BM vasculature. Moreover, urine and peripheral blood samples from ALL patients exhibited elevated levels of proangiogenic basic fibroblast growth factor (bFGF) and vascular endothelial growth factor (VEGF), which correlated with the increase in BM angiogenesis [108]. In AML, increased levels of plasma VEGF were associated with worse outcome [109] and higher numbers of circulating blasts [110]. In patients with MDS, enhanced BM VEGF expression was detected in high compared to low risk patients and showed to predict evolution to transfusion dependence [111]. Increased BM microvessel density and VEGF expression were furthermore observed in patients with myeloproliferative neoplasms or lymphoma [102][112], where enhanced BM angiogenic activity also associated with disease aggressiveness and worse outcome [113][114].
–
Cells, cytokines and growth factors that maintain physiological angiogenesis are unbalanced in the neoplastic BM. Tumor cells themselves secrete cytokines (e.g. IL-6, granulocyte-macrophage colony-stimulating factor (GM-CSF), VEGF, bFGF) [115][116][117] to stimulate angiogenesis and promote tumor growth and dissemination [100]. In fact, the VEGF gene was first cloned from an AML cell line (HL-60) [118]. Malignant cells themselves furthermore may express cytokine receptors (e.g. VEGF receptors (VEGF-R)) [119][120], through which they stimulate their own survival, proliferation and migration in an autocrine manner [115][121].
BM REGULATION BY CATECHOLAMINES
The BM naturally harbors hematopoietic stem cells (HSCs) responsible for sustaining the blood production. In order to fulfill this function over the whole life-span of an organism, HSCs balance dormancy and self-renewal activity with basal or demand-oriented proliferation and differentiation. HSCs reside in so-called BM niches, which are embedded in complex cellular networks that intensively communicate via molecular, biophysical (e.g. oxygen levels, blood pressure) and structural (e.g. extracellular matrix) signals [122][123][124]. Different BM niches have been described and reported to support the unique requirements of HSCs (as elegantly reviewed in [125][126]). Osteoblasts producing among others osteopontin [127][128] were initially considered major regulators of HSCs shown to reside in proximity of the endosteum [129][130][131][132]. More recent studies have questioned the importance of molecular signals deriving from osteoblasts for the regulation of HSC quiescence and rather pointed out roles of osteoblasts in the maintenance of more committed hematopoietic progenitors, and particularly in B-cell lymphopoiesis [95][133][134][135][136][137][138][139][140]. More recently there is instead increasing evidence for the so-called vascular HSC niche. HSCs were shown to localize in proximity of sinusoids enriched for MSC activity [140][141] and endothelial cells lining the BM vasculature and MSCs to secrete factors sustaining the maintenance and activation of HSCs and derived progenitors [133][143][144][145]. The influence of the vascular niche on HSCs fate is nicely summarized in [126][146].
–
Sympathetic nerve fibers are a further critical component of the BM niche. Already back in 1925 they were described by De Castro to enter the bone with blood vessels and branch to form rings around osteoblasts and osteocytes, as described in [147]. Next to a baseline routine secretion, these fibers release catecholamines to the BM space [148] in response to circadian rhythm oscillations with especially NOR levels peaking during night and EPI release instead less dependent on circadian oscillations [149]. As shown by Heidt et al., chronic stress applied to mouse models induces a surplus release of NOR, which then reduces CXCL12 levels in the BM through activation of β3-ARs. Chronic stress as a consequence activates HSCs and increased their proliferation and differentiation, thereby causing increased output of inflammatory cells and inducing functional decline of HSCs [150]. Neural regulation of the BM as well as the interplay of the nerve system with the bone, BM and immunity has been recently reviewed in [151]. Several other cell types residing in the BM (e.g. immune cells [152], mast cells [153], HSPCs [154]) were also shown to produce catecholamines, which adds another layer of complexity to the regulation of adrenergic signaling in the BM.
–
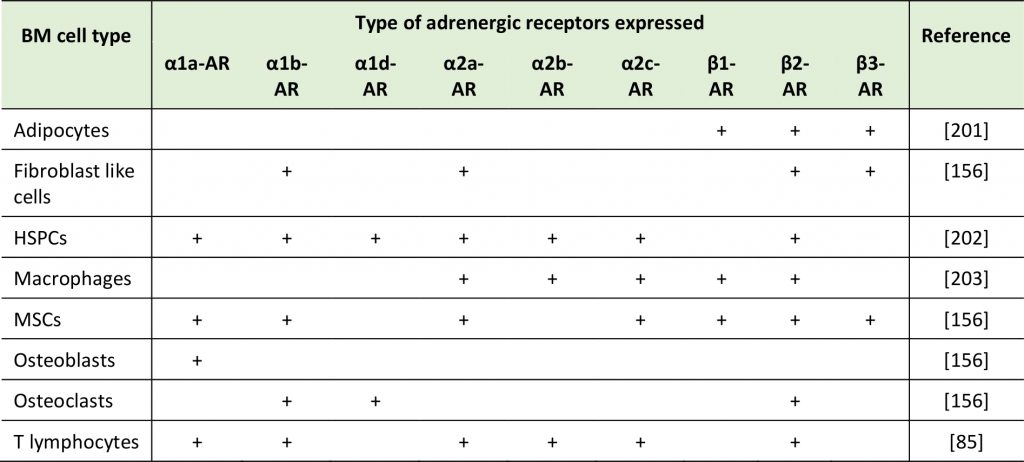
Various cell types in the BM – among which niche cells as well as HSPCs themselves – are known to express ARs and respond to catecholamines as part of their baseline regulatory program or of demand reactions (Table 1) [155][156][157][158]. The circadian rhythm influences the release of HSPCs from the BM into circulation (with a maximum of mobilized HSPCs at five hours after light onset and another five hours after the onset of darkness), in part via catecholamine secretion. This cyclic release of HSPCs is in antiphase with the expression of the chemokine CXCL12 responsive for HSPCs homing and retention to the BM [156]. Interestingly, latter are regulated by core genes of the molecular clock through circadian NOR secretion by the SNS. Nerve fibers locally deliver these adrenergic signals to the BM where β3-ARs expressing stromal cells respond with CXCL12 downregulation [156]. Furthermore, granulocyte colony-stimulating factor (G-CSF) produced in response to systemic bacterial infections, mobilizes HSPCs by suppressing CXCL12 secretion from osteoblasts via NOR/EPI release [95][156][158][159]. NOR release also reinforces the egress of HSPCs from the BM by acting on CAR (CXCL12 abundant reticular) cells expressing β3-ARs, in which exposure to NOR leads to degradation of specificity protein 1 (Sp1), a protein required for CXCL12 expression [156][160][161]. Consistently, low catecholamine levels associate with enhanced CXCL12 levels and enhanced homing and retention of CXCR4 expressing HSPCs in BM niches [161]. Thus, one major role of catecholamines in the BM is to regulate the HSPC pool via controlling their egress [161] (reviewed in more detail in [144][162][163][164][165][166][167]). Furthermore, adrenergic signals were associated with circadian leukocyte recruitment to the BM. Perivascular SNS fibers acting on β-ARs that are expressed on non-hematopoietic cells lead to differential circadian oscillations in the expression of adhesion cell molecules and chemokines, thus governing CXCR4-independent leukocyte recruitment to the BM [168]. The influence of catecholamines on cancer cells and the roles of such processes on the BM colonization by cancer cells are still understudied.
–
TABLE 1: Expression of ARs on the surface of different BM cell type in rodents. |
 |
| [85][156][201][202]203 |
HOW DO STRESS AND CATECHOLAMINES FACILITATE BM METASTASIS?
HSPC BM homing and retention is regulated by the CXCL12/CXCR4 molecular axis [169][170]. As discussed above, this pathway is regulated by adrenergic signals, which in part are released under the influence of circadian rhythms [95][156][158]. While these molecular cues are best characterized for healthy HSPCs, some data suggest that they are co-used by cancer cells. As such, cancer cells may also express the CXCR4 receptor and migrate towards BM osteoblasts releasing CXCL12 (Figure 1) [171]. Consistently, BM areas showing metastasis also display enhanced CXCL12 expression [172]. In experimental mouse models, inhibition of CXCL12 was furthermore shown to reduce BM homing of injected multiple myeloma cells and thereby to impair disease progression [171][172]. Another molecular axis promoting the colonization of bones by cancer cells is the receptor activator of NF-κB (RANK)/ receptor activator of NF-κB ligand (RANKL) pathway [171][173]. RANKL released by osteoblasts was shown to promote BM colonization and retention of metastatic cancer cells expressing the RANK receptor. RANK expression on tumor cells furthermore promotes their migration to the bones, while inhibition of RANKL/RANK signaling resulted in reduced bone metastasis in an experimental breast cancer model (Figure 1) [171][173]. Notably, RANKL producing osteoblasts are the main source responsive to sympathetic nerves in bones because of their very high expression of β2-AR. The stimulation of these receptors by NOR was indeed shown to induce RANKL synthesis [174] and thereby promoting BM metastasis [170]. Another less prominently studied pathway is the CXCR6/CXCL16 molecular axis recently involved in homing of prostate cancer cells to the bones. While mainly expressed by antigen-presenting cells, CXCL16 is also produced by bone tissues including osteocytes and was shown to be involved in migration of CXCR6 expressing prostate cancer cells to this site (Figure 1) [85][171][175].
–
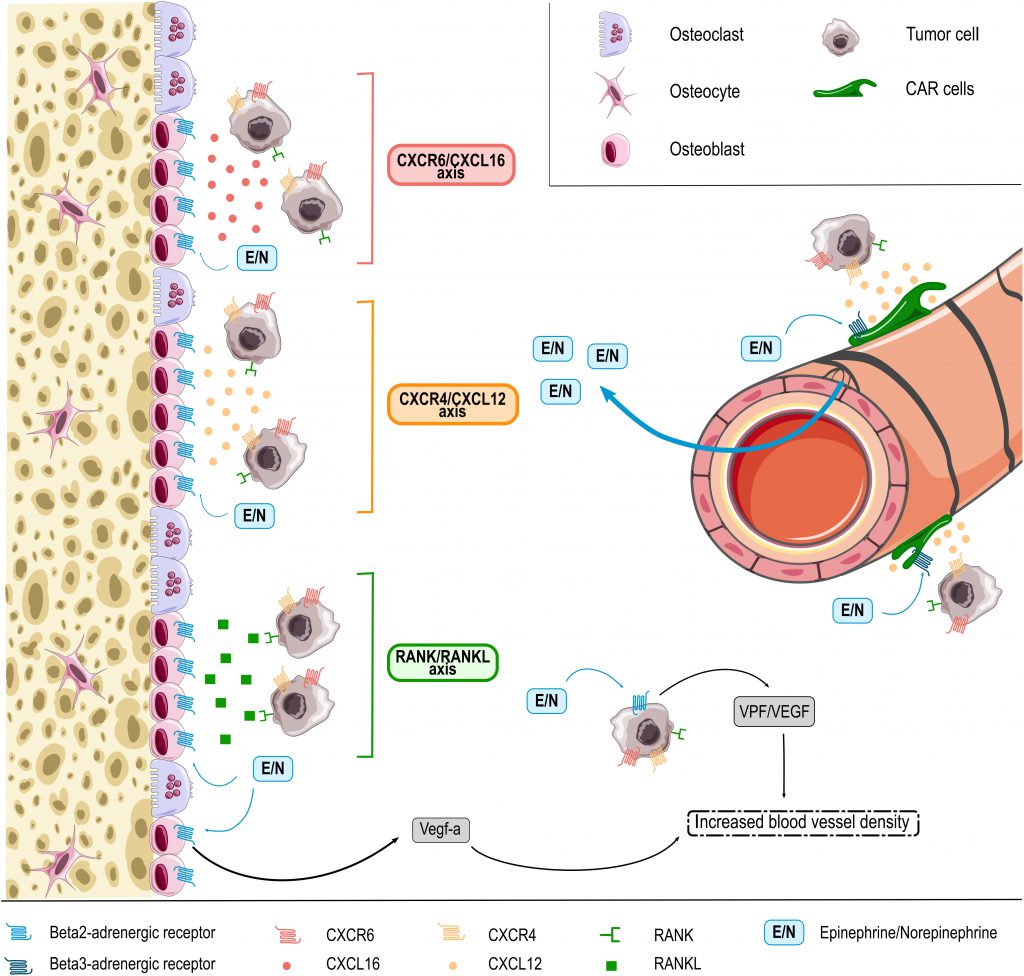 | FIGURE 1: EPI and NOR (E/N, blue square) are released in the BM microenvironment from SNS fibers entering the bone with blood vessels. EPI and NOR influence interaction of tumor cells with b-ARs -expressing BM niche cells, e.g. CXCL12 abundant reticular (CAR) cells, osteoblasts and osteoclasts through different axis. In response to adrenergic signaling niche cells release (1) CXCL16 chemokine (red) that interacts with CXCR6 expressed on the surface of several tumor cells types, (2) CXCL12 (orange) that chemoattracts CXCR4 expressing cancer cells and (3) RANKL (green) protein that binds RANK-expressing malignant cells. In addition, adrenergic signaling in osteoblasts and also directly in tumor cells themselves can promote release of angiogenic factors thus promoting BM colonization by tumor cells through increased blood vessel density. |
–
Cancer cells may also reach the BM without specific cues, as DTCs that are part of the circulating blood flow. Enhanced sympathetic activity in the bone microenvironment increases the density of blood vessels, which may contribute to BM colonization in patients with circulating DTCs [176]. In line, EPI and NOR were shown to increase the synthesis of pro-angiogenic factors (e.g. VEGF) thereby stimulating angiogenesis and the formation of abnormal vessels with higher permeability (Figure 1) (as reviewed by Chakroborty et al. [10]). Mulcrone et al. for example recently also showed that stimulation of β2-AR-expressing osteoblasts using the non-selective β-adrenergic agonist isoproterenol effectively induced VEGF-a expression thereby increasing the vascular density in the mouse BM and promoting BM metastasis (Figure 1) [176]. Importantly, specific blockade of the VEGF-a/VEGF-R axis abrogated the stimulatory effect of isoproterenol on tumor seeding in bones [176], suggesting direct involvement of this molecular axis in the pro-metastatic effect of catecholamines at this site.
–
Beyond the mechanisms discussed above, cancer cell retention in BM niche structures may be promoted by the expression of specific adhesion molecules such as cadherins, integrins or annexins that promote tumor cell binding to BM stromal cells and the bone matrix [86]. For example, osteoblasts and endothelial cells were shown to produce annexin II for which the receptor is widely expressed in cancer cells [169][177]. Adhesion can also be mediated by E-cadherin, which was found to be expressed by cancer cells and to form adherent junctions with N-cadherin from osteogenic cells. Furthermore, αVβ3 and αVβ5 integrins expressed by tumor cells mediate binding to bone extracellular matrix proteins such as fibronectin, vitronectin or osteopontin as nicely reviewed in [171]. Interestingly, stress behavior and associated increased levels of catecholamines have been described to regulate the expression of adhesion molecules in cancer cells. For example, high levels of NOR induced a β1-integrin-mediated increase of the adhesion of human breast carcinoma cells with the vascular endothelium in an in vitro model of human breast cancer. Importantly, this effect was mediated by β-ARs and could be abrogated by β-blockers [178]. Furthermore, restraint stress and the associated increases in catecholamines induced increased levels of FAK in an orthotopic mouse model of human ovarian cancer, thereby affecting adhesion of tumor cells to the extracellular matrix, which contributed to cancer progression [58]. Very recently, Obradovic et al. showed that increase in stress hormones levels during breast cancer progression mediated activation of glucocorticoid receptors in tumor cells promoted breast cancer metastasis through induction of signaling networks and protein kinases known to facilitate breast cancer progression [179]. Whether catecholamines regulate adhesion of DTCs to the BM matrix is still under-investigated.
COMPETITION OF NEOPLASTIC STEM CELLS WITH HEALTHY BM HSCs
As discussed above, healthy HSCs reside in specific BM niches, which however neoplastic cells modify to better serve their own requirements. As shown by imaging experiments, leukemic cells specifically engraft in microvascular BM domains showing high E-selectin and CXCL12 expression levels, where HSCs are also known to localize, indicating a possible competition between malignant and healthy cells [180]. Furthermore, transplantation of MLL-AF9 AML cells in immunodeficient mice transformed the HSC niche by reducing the density of the SNS nerve network and remodeled the BM microenvironment by depleting niche cells required for the maintenance of healthy HSCs (e.g. arteriole associated stromal cells) and expanding leukemia-supportive cells (e.g. more differentiated mesenchymal progenitors). Thus, manipulation of the adrenergic system could provide a strategy to re-install conditions favoring healthy HSCs over LSCs [181]. This notion is further supported by the work from Arranz et al. who showed a disturbed niche consisting in reduced numbers of sympathetic nerve fibers, supporting nestin+ MSCs and Schwann cells in the BM of myeloproliferative neoplasia patients as well as in mouse models. Sympathetic regulation of nestin+ MSCs was restored by pharmacological treatment with a β3-adrenergic agonist leading to improvement in BM fibrosis and restoration of healthy over malignant hematopoiesis [182].
–
Neoplastic cells from solid tumors may also outcompete healthy HSPCs from niches via selected molecular cues. Both the endosteal zone and the perivascular niche, known to harbor healthy HSPCs, are also colonization sites for tumor cells [89][177]. This is perhaps due to the fact that neoplastic cells co-use molecular signals regulating healthy HSPCs BM homing and retention, as mentioned above. Metastatic prostate cancer cells for example were shown to use CXCR4/CXL12 [183][184], Annexin II/Annexin II receptor [185] as well as CXCR7 [186] pathways to establish themselves in the bone [187][188][189]. As an example, prostate cancer cells were shown to co-localize with HSCs in the BM niche, both with a preferred binding to annexin-2 expressing osteoblasts [190]. However, prostate cancer cells showed superior ability to bind to common receptors providing them with an advantage over HSCs [191]. Interestingly, there is no direct link between the size of the primary tumor and the prevalence of DTCs in the BM of cancer patients. A limited number of available niche sites in the BM was discussed as possible cause for this phenomenon [192]. Of note, HSCs derived from animals with disseminated prostate carcinoma were found to express lower levels of niche adhesion molecules and receptors (e.g. NOTCH, angiopoietin-1 receptor (TIE2)) and transcription factors regulating HSC self-renewal and proliferation (B-cell specific Moloney murine virus integration site 1 (BMI1) and inhibitors of CDK4 A (INK4A)) [190], suggesting that these aggressive prostate carcinoma cells actively alter HSCs to vacate the niche. On the other side, as described above, DTCs take advantage of the RANKL/RANK signaling pathway induced by sympathetic activation to migrate to the BM and liberation of a few niche spaces by mechanisms described above would further give the cancer cells an advantage to settle in their new microenvironment.
INFLUENCE ON TUMOR ASSOCIATED MACROPHAGES (TAMs) AND ANTI-TUMOR IMMUNITY
The immune system is a major player during tumor development and progression. Catecholamines, which are known to profoundly impact immune cells, may thus also exert pro-tumorigenic effects via immune modulation. For example, TAMs, which have been linked to cancer progression, metastasis and resistance to therapy [193], were shown to express ARs and respond to NOR by secretion of proangiogenic factors and matrix metalloproteinases (MMPs) promoting tumor-angiogenesis [194][195]. Adrenergic stimulation furthermore increases cAMP and PGE2 levels within the tumor, which further mediates immune-suppressing effects [196]. More recently, EPI was shown to cooperate with inflammatory cytokines (e.g. TNFα) in the regulation of several immunosuppressive factors in both cancer cells and macrophages via COX2 activity. Consistently, EPI-dependent immune suppression was reversed by treatment with the COX2-inhibitor celecoxib [197]. Moreover, in a rat model of highly malignant syngeneic CRNK-16 induced leukemia, psychological stress or injection of stress hormones accelerated leukemia-induced death. Treatment with the β-AR blocker nadolol reversed this pro-leukemogenic effect and increased baseline survival rates. Since reduced NK cell activity was observed in animals exposed to stress hormones, impaired anti-tumor immunity was interpreted to cause the accelerated leukemia progression [198]. The influences of catecholamines on the immune system and possible links to anti-tumor immunity are elegantly reviewed elsewhere [8][199][200].
CONCLUSION
Different types of stress and catecholamine release influence cancer pathogenesis [2] via regulation of the BM environment. Cancer cells of hematopoietic as well as solid tumor origin use BM niches as “safe harbor” promoting their growth and therapy resistance. They modulate the BM niche by secretion of proteins and cytokines and disrupt normal hematopoiesis by altering its adrenergic innervation. Catecholamines regulate the BM at multiple levels and further exert pro-tumorigenic effects by directly acting on AR-expressing tumor cells. Treatment with beta-blockers might be beneficial for the disease evolution in cancer patients, but the benefit is probably limited to certain tumor subtypes and stages. A better understanding of the precise molecular cues and cellular mechanisms underlying pro-tumorigenic effects of catecholamines requires further investigation but could eventually provide a rationale for more selected pharmacological intervention studies that may benefit patients with cancer.
REFERENCES
- Butler G (1993). Definitions of stress. Occas Pap R Coll Gen Pract (61): 1–5. PMID: 8199583
- Moreno-Smith M, Lutgendorf SK, and Sood AK (2010). Impact of stress on cancer metastasis. Future Oncol 6(12): 1863–1881. doi: 10.2217/fon.10.142
- Schmidt C, and Kraft K (1996). Beta-endorphin and catecholamine concentrations during chronic and acute stress in intensive care patients. Eur J Med Res 1(11):528-32. PMID: 9438155
- Rupp H, Dhallas KS, and Dhallas NS (1994). Mechanisms of cardiac cell damage due to catecholamine significance of drugs regulating central sympathetic outflow. J Cardiovasc Pharmacol 24 (Suppl 1):S16-24. doi: 10.1097/00005344-199424001-00004
- Rupp H, and Jacob R (1995). Excess catecholamine and the metabolic syndrome: should central imidazoline receptors be a therapeutic target? Med Hypotheses 44(3):217-25. doi: 10.1016/0306-9877(95)90139-6
- Puglisi-Allegra S, Imperato A, Angelucci L, and Cabib S (1991). Acute stress induces time-dependent response in dopamine mesolimbic system. Brain Res 554(1-2):217-22. doi: 10.1016/0006-8993(91)90192-x
- Imperato A, Angelucci L, Casolini P, Zocchi A, Puglisi-Allegra S (1992) Repeated stressful experiences differently affect limbic dopamine release during and following stress. Brain Res 577(2):194-9. doi: 10.1016/0006-8993(92)90274-d
- Elenkov IJ, Wilder RL, Chrousos GP, and Vizi ES (2000). The sympathetic nerve–an integrative interface between two supersystems: the brain and the immune system. Pharmacol Rev 52(4): 595–638. PMID: 11121511.
- Esler M, Eikelis N, Schlaich M, Lambert G, Alvarenga M, Kaye D, El-Osta A, Guo L, Barton D, Pier C, Brenchley C, Dawood T, Jennings G, and Lambert E (2008). Human sympathetic nerve biology: parallel influences of stress and epigenetics in essential hypertension and panic disorder. Ann N Y Acad Sci 1148: 338–348. doi: 10.1196/annals.1410.064
- Chakroborty D, Sarkar C, Basu B, Dasgupta PS, and Basu S (2009). Catecholamines regulate tumor angiogenesis. Cancer Res 69(9): 3727–3730. doi: 10.1158/0008-5472.can-08-4289
- Hieble JP, Bondinell WE, and Ruffolo Jr. RR (1995). Alpha- and beta-adrenoceptors: from the gene to the clinic. 1. Molecular biology and adrenoceptor subclassification. J Med Chem 38(18): 3415–3444. doi: 10.1021/jm00018a001
- Hein L, and Kobilka BK (1995). Adrenergic receptor signal transduction and regulation. Neuropharmacology 34(4): 357–366. doi: 10.1016/0028-3908(95)00018-2
- Molinoff PB (1984). Alpha- and beta-adrenergic receptor subtypes properties, distribution and regulation. Drugs 28 (Suppl 2): 1–15. doi: 10.2165/00003495-198400282-00002
- Rang H, Dale M, Ritter J, and Flower R Rang & Dale's Pharmacology E-Book – 6th Edition.
- Orenberg EK, Pfendt EA, and Wilkinson DI (1983). Characterization of α- and β-Adrenergic Agonist Stimulation of Adenylate Cyclase Activity in Human Epidermal Keratinocytes In Vitro. J Invest Dermatol 80(6): 503–507. doi: 10.1111/1523-1747.ep12535068
- Ariëns EJ, and Simonis AM (1983). Physiological and pharmacological aspects of adrenergic receptor classification. Biochem Pharmacol 32(10): 1539–1545. doi: 10.1016/0006-2952(83)90324-6
- Chang A, Le CP, Walker AK, Creed SJ, Pon CK, Albold S, Caroll D, Halls ML, Lane R, Ferrari D, Sloan EK, Carroll D, Halls ML, Lane JR, Riedel B, Ferrari D, and Sloan EK (2016). β2-Adrenoceptors on tumor cells play a critical role in stress-enhanced metastasis in a mouse model of breast cancer. Brain, Behav Immunol 57: 106–115. doi: 10.1016/j.bbi.2016.06.011
- Tang J, Li Z, Lu L, and Cho CH (2013). β-Adrenergic system, a backstage manipulator regulating tumour progression and drug target in cancer therapy. Semin Cancer Biol 23(6): 533–542. doi: 10.1016/j.semcancer.2013.08.009
- Pon CK, Lane JR, Sloan EK, and Halls ML (2016). The β2-adrenoceptor activates a positive cAMP-calcium feedforward loop to drive breast cancer cell invasion. FASEB J 30(3): 1144–1154. doi: 10.1096/fj.15-277798
- Reeder A, Attar M, Nazario L, Bathula C, Zhang A, Hochbaum D, Roy E, Cooper KL, Oesterreich S, Davidson NE, Neumann CA, and Flint MS (2015). Stress hormones reduce the efficacy of paclitaxel in triple negative breast cancer through induction of DNA damage. Br J Cancer 112(9): 1461–1470. doi: 10.1038/bjc.2015.133
- James GD, Berge-Landry Hv H, Valdimarsdottir HB, Montgomery GH, and Bovbjerg DH (2004). Urinary catecholamine levels in daily life are elevated in women at familial risk of breast cancer. Psychoneuroendocrinology 29(7): 831–838. doi: 10.1016/s0306-4530(03)00150-1
- Kikuchi N, Nishiyama T, Sawada T, Wang C, Lin Y, Watanabe Y, Tamakoshi A, and Kikuchi S (2017). Perceived Stress and Colorectal Cancer Incidence: The Japan Collaborative Cohort Study. Sci Rep 7: 40363. doi: 10.1038/srep40363
- Lutgendorf SK, DeGeest K, Dahmoush L, Farley D, Penedo F, Bender D, Goodheart M, Buekers TE, Mendez L, Krueger G, Clevenger L, Lubaroff DM, Sood AK, and Cole SW (2011). Social isolation is associated with elevated tumor norepinephrine in ovarian carcinoma patients. Brain Behav Immun 25(2): 250–255. doi: 10.1016/j.bbi.2010.10.012
- Grytli HH, Fagerland MW, Fossa SD, and Tasken KA (2014). Association between use of beta-blockers and prostate cancer-specific survival: a cohort study of 3561 prostate cancer patients with high-risk or metastatic disease. Eur Urol 65(3): 635–641. doi: 10.1016/j.eururo.2013.01.007
- Kaapu KJ, Murtola TJ, Maattanen L, Talala K, Taari K, Tammela TL, and Auvinen A (2016). Prostate cancer risk among users of digoxin and other antiarrhythmic drugs in the Finnish Prostate Cancer Screening Trial. Cancer Causes Control 27(2): 157–164. doi: 10.1007/s10552-015-0693-2
- Botteri E, Munzone E, Rotmensz N, Cipolla C, De Giorgi V, Santillo B, Zanelotti A, Adamoli L, Colleoni M, Viale G, Goldhirsch A, and Gandini S (2013). Therapeutic effect of beta-blockers in triple-negative breast cancer postmenopausal women. Breast Cancer Res Treat 140(3): 567–575. doi: 10.1007/s10549-013-2654-3
- Melhem-Bertrandt A, Chavez-MacGregor M, Lei X, Brown EN, Lee RT, Meric-Bernstam F, Sood AK, Conzen SD, Hortobagyi GN, and Gonzalez-Angulo A-M (2011). Beta-Blocker Use Is Associated With Improved Relapse-Free Survival in Patients With Triple-Negative Breast Cancer. J Clin Oncol 29(19): 2645–2652. doi: 10.1200/JCO.2010.33.4441
- Powe DG, Voss MJ, Zänker KS, Habashy HO, Green AR, Ellis IO, and Entschladen F (2010). Beta-blocker drug therapy reduces secondary cancer formation in breast cancer and improves cancer specific survival. Oncotarget 1(7): 628–38. doi: 10.18632/oncotarget.101009
- Choy C, Raytis JL, Smith DD, Duenas M, Neman J, Jandial R, and Lew MW (2016). Inhibition of beta2-adrenergic receptor reduces triple-negative breast cancer brain metastases: The potential benefit of perioperative beta-blockade. Oncol Rep 35(6): 3135–3142. doi: 10.3892/or.2016.4710
- Barron TI, Connolly RM, Sharp L, Bennett K, and Visvanathan K (2011). Beta Blockers and Breast Cancer Mortality: A Population- Based Study. J Clin Oncol 29(19): 2635–2644. doi: 10.1200/JCO.2010.33.5422
- Montoya A, Amaya CN, Belmont A, Diab N, Trevino R, Villanueva G, Rains S, Sanchez LA, Badri N, Otoukesh S, Khammanivong A, Liss D, Baca ST, Aguilera RJ, Dickerson EB, Torabi A, Dwivedi AK, Abbas A, Chambers K, Bryan BA, and Nahleh Z (2017). Use of non-selective β-blockers is associated with decreased tumor proliferative indices in early stage breast cancer. Oncotarget 8: 6446–6460. doi: 10.18632/oncotarget.14119
- Diaz ES, Karlan BY, and Li AJ (2012). Impact of beta blockers on epithelial ovarian cancer survival. Gynecol Oncol 127(2): 375–378. doi: 10.1016/j.ygyno.2012.07.102
- De Giorgi V, Grazzini M, Gandini S, Benemei S, Lotti T, Marchionni N, and Geppetti P (2011). Treatment With β-Blockers and Reduced Disease Progression in Patients With Thick Melanoma. Arch Intern Med 171(8): 779–81. doi: 10.1001/archinternmed.2011.131
- Lemeshow S, Sorensen HT, Phillips G, Yang E V, Antonsen S, Riis AH, Lesinski GB, Jackson R, and Glaser R (2011). beta-Blockers and survival among Danish patients with malignant melanoma: a population-based cohort study. Cancer Epidemiol Biomarkers Prev 20(10): 2273–2279. doi: 10.1158/1055-9965.epi-11-0249
- Chang PY, Huang WY, Lin CL, Huang TC, Wu YY, Chen JH, and Kao CH (2015). Propranolol Reduces Cancer Risk: A Population-Based Cohort Study. Med 94(27): e1097. doi: 10.1097/md.0000000000001097
- Choi CH, Song T, Kim TH, Choi JK, Park JY, Yoon A, Lee YY, Kim TJ, Bae DS, Lee JW, Kim BG (2014). Meta-analysis of the effect of beta-blocker on survival time in cancer patients. J Cancer Res Clin Oncol 140(7):1179-88. doi: 10.1007/s00432-014-1658-7
- Cardwell CR, Coleman HG, Murray LJ, Entschladen F, and Powe DG (2013). Beta-blocker usage and breast cancer survival: a nested case-control study within a UK clinical practice research datalink cohort. Int J Epidemiol 42(6): 1852–1861. doi: 10.1093/ije/dyt196
- Parker WP, Lohse CM, Zaid HB, Cheville JC, Boorjian SA, Leibovich BC, and Thompson RH (2017). Evaluation of beta-blockers and survival among hypertensive patients with renal cell carcinoma. Urol Oncol 35(1): 36.e1-36.e6. doi: 10.1016/j.urolonc.2016.08.013
- Chae YK, Dimou A, Pierce S, Kantarjian H, and Andreeff M (2014). The effect of calcium channel blockers on the outcome of acute myeloid leukemia. Leuk Lymphoma 55(12): 2822–2829. doi: 10.3109/10428194.2014.901513
- Livingstone E, Hollestein LM, van Herk-Sukel MP, van de Poll-Franse L, Nijsten T, Schadendorf D, and de Vries E (2013). beta-Blocker use and all-cause mortality of melanoma patients: results from a population-based Dutch cohort study. Eur J Cancer 49(18): 3863–3871. doi: 10.1016/j.ejca.2013.07.141
- McCourt C, Coleman HG, Murray LJ, Cantwell MM, Dolan O, Powe DG, and Cardwell CR (2014). Beta-blocker usage after malignant melanoma diagnosis and survival: a population-based nested case-control study. Br J Dermatol 170(4): 930–938. doi: 10.1111/bjd.12894
- Weberpals J, Jansen L, Carr PR, Hoffmeister M, and Brenner H (2016). Beta blockers and cancer prognosis – The role of immortal time bias: A systematic review and meta-analysis. Cancer Treat Rev 47: 1–11. doi: 10.1016/j.ctrv.2016.04.004
- Yap A, Lopez-Olivo MA, Dubowitz J, Pratt G, Hiller J, Gottumukkala V, Sloan E, Riedel B, and Schier R (2018). Effect of beta-blockers on cancer recurrence and survival: a meta-analysis of epidemiological and perioperative studies. Br J Anaesth 121(1): 45–57. doi: 10.1016/j.bja.2018.03.024
- Kim-Fuchs C, Le CP, Pimentel MA, Shackleford D, Ferrari D, Angst E, Hollande F, and Sloan EK (2014). Chronic stress accelerates pancreatic cancer growth and invasion: a critical role for beta-adrenergic signaling in the pancreatic microenvironment. Brain Behav Immun 40: 40–47. doi: 10.1016/j.bbi.2014.02.019
- Kim TH, Gill NK, Nyberg KD, Nguyen A V, Hohlbauch S V, Geisse NA, Nowell CJ, Sloan EK, and Rowat AC (2016). Cancer cells become less deformable and more invasive with activation of beta-adrenergic signaling. J Cell Sci 129(24): 4563–4575. doi: 10.1242/jcs.194803
- Behrenbruch C, Shembrey C, Paquet-Fifield S, Molck C, Cho HJ, Michael M, Thomson BNJ, Heriot AG, and Hollande F (2018). Surgical stress response and promotion of metastasis in colorectal cancer: a complex and heterogeneous process. Clin Exp Metastasis doi: 10.1007/s10585-018-9873-2
- Magnon C, Hall SJ, Lin J, Xue X, Gerber L, Freedland SJ, and Frenette PS (2013). Autonomic nerve development contributes to prostate cancer progression. Science 341(6142): 1236361. doi: 10.1126/science.1236361
- Zhang J, Deng Y, Liu J, Wang Y, Yi T, Huang B, He S, Zheng B, and Jiang Y (2016). Norepinephrine induced epithelial–mesenchymal transition in HT-29 and A549 cells in vitro. J Cancer Res Clin Oncol 142(2): 423–435. doi: 10.1007/s00432-015-2044-9
- Hulsurkar M, Li Z, Zhang Y, Li X, Zheng D, and Li W (2017). Beta-adrenergic signaling promotes tumor angiogenesis and prostate cancer progression through HDAC2-mediated suppression of thrombospondin-1. Oncogene 36(11): 1525–1536. doi: 10.1038/onc.2016.319
- Lin Q, Wang F, Yang R, Zheng X, Gao H, and Zhang P (2013). Effect of chronic restraint stress on human colorectal carcinoma growth in mice. PLoS One 8(4): e61435. doi: 10.1371/journal.pone.0061435
- Madden KS, Szpunar MJ, and Brown EB (2013). Early impact of social isolation and breast tumor progression in mice. Brain Behav Immun 30 (Suppl): S135-41. doi: 10.1016/j.bbi.2012.05.003
- Partecke LI, Speerforck S, Kading A, Seubert F, Kuhn S, Lorenz E, Schwandke S, Sendler M, Kessler W, Trung DN, Oswald S, Weiss FU, Mayerle J, Henkel C, Menges P, Beyer K, Lerch MM, Heidecke CD, and von Bernstorff W (2016). Chronic stress increases experimental pancreatic cancer growth, reduces survival and can be antagonised by beta-adrenergic receptor blockade. Pancreatology 16(3): 423–433. doi: 10.1016/j.pan.2016.03.005
- Qin J, Jin F, Li N, Guan H, Lan L, Ni H, and Wang Y (2015). Adrenergic receptor β2 activation by stress promotes breast cancer progression through macrophages M2 polarization in tumor microenvironment. BMB Rep 48(5): 295–300. doi: 10.5483/bmbrep.2015.48.5.008
- Sloan EK, Priceman SJ, Cox BF, Yu S, Pimentel MA, Tangkanangnukul V, Arevalo JMG, Morizono K, Karanikolas BDW, Wu L, Sood AK, and Cole SW (2010). The sympathetic nervous system induces a metastatic switch in primary breast cancer. Cancer Res 70(18): 7042–7052. doi: 10.1158/0008-5472.CAN-10-0522
- Thaker PH, Han LY, Kamat AA, Arevalo JM, Takahashi R, Lu C, Jennings NB, Armaiz-Pena G, Bankson JA, Ravoori M, Merritt WM, Lin YG, Mangala LS, Kim TJ, Coleman RL, Landen CN, Li Y, Felix E, Sanguino AM, Newman RA, Lloyd M, Gershenson DM, Kundra V, Lopez-Berestein G, Lutgendorf SK, Cole SW, and Sood AK (2006). Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat Med 12(8): 939–944. doi: 10.1038/nm1447
- Chen H, Liu D, Guo L, Cheng X, Guo N, and Shi M (2018). Chronic psychological stress promotes lung metastatic colonization of circulating breast cancer cells by decorating a pre-metastatic niche through activating beta-adrenergic signaling. J Pathol 244(1): 49–60. doi: 10.1002/path.4988
- Qiao G, Chen M, Bucsek MJ, Repasky EA, and Hylander BL (2018). Adrenergic Signaling: A Targetable Checkpoint Limiting Development of the Antitumor Immune Response. Front Immunol 9: 164. doi: 10.3389/fimmu.2018.00164
- Sood AK, Armaiz-Pena GN, Halder J, Nick AM, Stone RL, Hu W, Carroll AR, Spannuth WA, Deavers MT, Allen JK, Han LY, Kamat AA, Shahzad MM, McIntyre BW, Diaz-Montero CM, Jennings NB, Lin YG, Merritt WM, DeGeest K, Vivas-Mejia PE, Lopez-Berestein G, Schaller MD, Cole SW, and Lutgendorf SK (2010). Adrenergic modulation of focal adhesion kinase protects human ovarian cancer cells from anoikis. J Clin Invest 120(5): 1515–1523. doi: 10.1172/jci40802
- Eng JW, Reed CB, Kokolus KM, Pitoniak R, Utley A, Bucsek MJ, Ma WW, Repasky EA, and Hylander BL (2015). Housing temperature-induced stress drives therapeutic resistance in murine tumour models through beta2-adrenergic receptor activation. Nat Commun 6: 6426. doi: 10.1038/ncomms7426
- Hassan S, Karpova Y, Baiz D, Yancey D, Pullikuth A, Flores A, Register T, Cline JM, D'Agostino Jr. R, Danial N, Datta SR, and Kulik G (2013). Behavioral stress accelerates prostate cancer development in mice. J Clin Invest 123(2): 874–886. doi: 10.1172/jci63324
- Rains SL, Amaya CN, and Bryan BA (2017). Beta-adrenergic receptors are expressed across diverse cancers. Oncoscience 4(7–8): 95–105. doi: 10.18632/oncoscience.357.
- Hu P, He J, Liu S, Wang M, Pan B, Zhang W (2016) Beta2-adrenergic receptor activation promotes the proliferation of A549 lung cancer cells via the ERK1/2/CREB pathway. Onc Rep 36(3): 1757-1763. doi: 10.3892/or.2016.4966
- Perrone MG, Notarnicola M, Caruso MG, Tutino V, Scilimati A (2008) Upregulation of beta3-adrenergic receptor mRNA in human colon cancer: a preliminary study. Oncology 75(3-4):224-9. doi: 10.1159/000163851
- Calvani M, Pelon F, Comito G, Taddei ML, Moretti S, Innocenti S, Nassini R, Gerlini G, Borgognoni L, Bambi F, Giannoni E, Filippi L, Chiarugi P (2015). Norepinephrine promotes tumor microenvironment reactivity through beta3-adrenoreceptor during melanoma progression. Oncotarget 6(7): 4615-4632. doi: 10.18632/oncotarget.2652
- Dal Monte M, Casini G, Filippi L, Nicchia GP, Svelto M, Bagnoli P (2013). Functional involvment of beta3-adrenergic receptors in melanoma growth and vascularization. J Mol Med 91(12):1407-19. doi: 10.1007/s00109-013-1073-6
- Calvani M, Cavallini L, Tondo A, Spinelli V, Ricci L, Pasha A, Bruno G, Buonvivino D, Bigagli E, Vignoli M, Bianchimi F, Sartiani L, Lodovici M, Semeraro R, Fontani F, De Logu F, Dal Monte M, Chiarugi P, Favre C, Filippi L (2018) Beta3-adrenoreceptors control mitochondrial dormancy in melanoma and embryonic stem cells. Oxid Med Cell Longev 2018: 6816508. doi: 10.1155/2018/6816508
- Cole SW, Sood AK (2012). Molecular Pathways: beta-adrenergic signaling in cancer. Clin Cancer Res 18(5):1201-6. doi: 10.1158/1078-0432.CCR-11-0641
- Nagaraja AS, Dood RL, Armaiz-Pena G, Kang Y, Wu SY, Allen JK, Jennings NB, Mangala LS, Pradeep S, Lyons Y, Haemmerle M, Gharpure KM, Sadaoui NC, Rodriguez-Aguayo C, Ivan C, Wang Y, Baggerly K, Ram P, Lopez-Berestein G, Liu J, Mok SC, Cohen L, Lutgendorf SK, Cole SW, and Sood AK (2017). Adrenergic-mediated increases in INHBA drive CAF phenotype and collagens. JCI Insight 2(16). doi: 10.1172/jci.insight.93076
- Le CP, Nowell CJ, Kim-Fuchs C, Botteri E, Hiller JG, Ismail H, Pimentel MA, Chai MG, Karnezis T, Rotmensz N, Renne G, Gandini S, Pouton CW, Ferrari D, Moller A, Stacker SA, and Sloan EK (2016). Chronic stress in mice remodels lymph vasculature to promote tumour cell dissemination. Nat Commun 7: 10634. doi: 10.1038/ncomms10634
- Zahalka AH, Arnal-Estape A, Maryanovich M, Nakahara F, Cruz CD, Finley LWS, and Frenette PS (2017). Adrenergic nerves activate an angio-metabolic switch in prostate cancer. Science 358(6361): 321–326. doi: 10.1126/science.aah5072
- Lamkin DM, Sloan EK, Patel AJ, Chiang BS, Pimentel MA, Ma JCY, Arevalo JM, Morizono K, and Cole SW (2012). Chronic stress enhances progression of acute lymphoblastic leukemia via β-adrenergic signaling. Brain Behav Immun 26(4): 635–641. doi: 10.1016/j.bbi.2012.01.013
- Nagaraja AS, Dorniak PL, Sadaoui NC, Kang Y, Lin T, Armaiz-Pena G, Wu SY, Rupaimoole R, Allen JK, Gharpure KM, Pradeep S, Zand B, Previs RA, Hansen JM, Ivan C, Rodriguez-Aguayo C, Yang P, Lopez-Berestein G, Lutgendorf SK, Cole SW, and Sood AK (2016). Sustained adrenergic signaling leads to increased metastasis in ovarian cancer via increased PGE2 synthesis. Oncogene 35(18): 2390–2397. doi: 10.1038/onc.2015.302
- Westwood JA, Darcy PK, and Kershaw MH (2013). Environmental enrichment does not impact on tumor growth in mice. F1000Res 2: 140. doi: 10.12688/f1000research.2-140.v1
- Dethlefsen C, Hansen LS, Lillelund C, Andersen C, Gehl J, Christensen JF, Pedersen BK, and Hojman P (2017). Exercise-Induced Catecholamines Activate the Hippo Tumor Suppressor Pathway to Reduce Risks of Breast Cancer Development. Cancer Res 77(18): 4894–4904. doi: 10.1158/0008-5472.can-16-3125
- Song Y, Gan Y, Wang Q, Meng Z, Li G, Shen Y, Wu Y, Li P, Yao M, Gu J, and Tu H (2017). Enriching the Housing Environment for Mice Enhances Their NK Cell Antitumor Immunity via Sympathetic Nerve-Dependent Regulation of NKG2D and CCR5. Cancer Res 77(7): 1611–1622. doi: 10.1158/0008-5472.can-16-2143
- Pedersen L, Idorn M, Olofsson GH, Lauenborg B, Nookaew I, Hansen RH, Johannesen HH, Becker JC, Pedersen KS, Dethlefsen C, Nielsen J, Gehl J, Pedersen BK, Thor Straten P, and Hojman P (2016). Voluntary Running Suppresses Tumor Growth through Epinephrine- and IL-6-Dependent NK Cell Mobilization and Redistribution. Cell Metab 23(3): 554–562. doi: 10.1016/j.cmet.2016.01.011
- Devalon ML, Miller TD, Squires RW, Rogers PJ, Bove AA, and Tyce GM (1989). Dopa in plasma increases during acute exercise and after exercise training. J Lab Clin Med 114(3): 321–327. PMID: 2504856
- Dimsdale JE, and Moss J (2018). Plasma Catecholamines in Stress and Exercise. JAMA 243(4): 340–342. doi: 10.1001/jama.1980.03300300018017
- Hanson ED, Sakkal S, Evans WS, Violet JA, Battaglini CL, McConell GK, and Hayes A (2018). Altered stress hormone response following acute exercise during prostate cancer treatment. Scand J Med Sci Sport 28(8): 1925–1933. doi: 10.1111/sms.13199
- Scanzano A, and Cosentino M (2015). Adrenergic regulation of innate immunity: a review. Front Pharmacol 6: 171. doi: 10.3389/fphar.2015.00171
- Kohm AP, and Sanders VM (2000). Norepinephrine: a messenger from the brain to the immune system. Immunol Today 21(11): 539–542. doi: 10.1016/S0167-5699(00)01747-3
- Sanders VM, and Kohm AP (2002). Sympathetic nervous system interaction with the immune system. In: Int. Rev. Neurobiol. Int Rev Neurobiol 53: 17–41. doi: 10.1016/s0074-7742(02)52004-3
- Suva LJ, Washam C, Nicholas RW, and Griffin RJ (2011). Bone metastasis: mechanisms and therapeutic opportunities. Nat Rev Endocrinol 7(4): 208–218. doi: 10.1038/nrendo.2010.227
- Coleman RE (2006). Clinical Features of Metastatic Bone Disease and Risk of Skeletal Morbidity. Clin Cancer Res 12(20): 6243s–6249s. doi: 10.1158/1078-0432.CCR-06-0931
- Bussard KM, Gay C V., and Mastro AM (2008). The bone microenvironment in metastasis; what is special about bone? Cancer Metastasis Rev 27(1): 41–55. doi: 10.1007/s10555-007-9109-4
- Roodman GD (2004). Mechanisms of Bone Metastasis. N Engl J Med 350(16): 1655–1664. doi: 10.1056/NEJMra030831
- Carlson P, Dasgupta A, Grzelak CA, Kim J, Barrett A, Coleman IM, Shor RE, Goddard ET, Dai J, Schweitzer EM, Lim AR, Crist SB, Cheresh DA, Nelson PS, Hansen KC, Ghajar CM (2019). Targeting the perivascular niche sensitizes disseminated tumour cells to chemotherapy. Nat Cell Biol 21(2):238-250. doi: 10.1038/s41556-018-0267-0
- Martinez LM, Vallone VB, Labovsky V, Choi H, Hofer EL, Feldman L, Bordenave RH, Batagelj E, Dimase F, Villafane AR, and Chasseing NA (2014). Changes in the peripheral blood and bone marrow from untreated advanced breast cancer patients that are associated with the establishment of bone metastases. Clin Exp Metastasis 31(2): 213–232. doi: 10.1007/s10585-013-9622-5
- Reagan MR, and Rosen CJ (2016). Navigating the bone marrow niche: translational insights and cancer-driven dysfunction. Nat Rev Rheumatol 12(3): 154–168. doi: 10.1038/nrrheum.2015.160
- Agarwal P, and Bhatia R (2015). Influence of Bone Marrow Microenvironment on Leukemic Stem Cells: Breaking Up an Intimate Relationship. Adv Cancer Res 127: 227–252. doi: 10.1016/bs.acr.2015.04.007
- Krause DS, and Scadden DT (2015). A hostel for the hostile: the bone marrow niche in hematologic neoplasms. Haematologica 100(11): 1376–1387. doi: 10.3324/haematol.2014.113852
- Schepers K, Campbell TB, and Passegue E (2015). Normal and leukemic stem cell niches: insights and therapeutic opportunities. Cell Stem Cell 16(3): 254–267. doi: 10.1016/j.stem.2015.02.014
- Manier S, Sacco A, Leleu X, Ghobrial IM, and Roccaro AM (2012). Bone marrow microenvironment in multiple myeloma progression. J Biomed Biotechnol 2012: 157496. doi: 10.1155/2012/157496
- Cogle CR, Saki N, Khodadi E, Li J, Shahjahani M, and Azizidoost S (2015). Bone marrow niche in the myelodysplastic syndromes. Leuk Res 39(10): 1020–1027. doi: 10.1016/j.leukres.2015.06.017
- Asada N, Takeishi S, and Frenette PS (2017). Complexity of bone marrow hematopoietic stem cell niche. Int J Hematol 106: 45–54. doi: 10.1007/s12185-017-2262-9
- Zhou HS, Carter BZ, and Andreeff M (2016). Bone marrow niche-mediated survival of leukemia stem cells in acute myeloid leukemia: Yin and Yang. Cancer Biol Med 13(2): 248–259. doi: 10.20892/j.issn.2095-3941.2016.0023
- Andreeff M et al. (2013). Proteomic, Gene Expression, and Micro-RNA Analysis Of Bone Marrow Mesenchymal Stromal Cells In Acute Myeloid Leukemia Identifies Pro-Inflammatory, Pro-Survival Signatures In Vitro and In Vivo. Blood 122:3685.
- Tabe Y, and Konopleva M (2015). Role of microenvironment in resistance to therapy in AML. Curr Hematol Malig Rep 10(2): 96–103. doi: 10.1007/s11899-015-0253-6
- Schepers K, Pietras EM, Reynaud D, Flach J, Binnewies M, Garg T, Wagers AJ, Hsiao EC, and Passegué E (2013). Myeloproliferative Neoplasia Remodels the Endosteal Bone Marrow Niche into a Self-Reinforcing Leukemic Niche. Cell Stem Cell 13(3): 285–299. doi: 10.1016/j.stem.2013.06.009
- Bellamy WT, Richter L, Frutiger Y, and Grogan TM (1999). Expression of vascular endothelial growth factor and its receptors in hematopoietic malignancies. Cancer Res 59(3): 728–733. PMID: 9973224
- Aguayo A (2004). The role of angiogenesis in the biology and therapy of myelodysplastic syndromes. Curr Hematol Rep 3(3): 184–191. PMID: 15087066
- Medinger M, Skoda R, Gratwohl A, Theocharides A, Buser A, Heim D, Dirnhofer S, Tichelli A, and Tzankov A (2009). Angiogenesis and vascular endothelial growth factor-/receptor expression in myeloproliferative neoplasms: correlation with clinical parameters and JAK2-V617F mutational status. Br J Haematol 146(2): 150–157. doi: 10.1111/j.1365-2141.2009.07726.x
- Padro T, Ruiz S, Bieker R, Burger H, Steins M, Kienast J, Buchner T, Berdel WE, and Mesters RM (2000). Increased angiogenesis in the bone marrow of patients with acute myeloid leukemia. Blood 95(8): 2637–2644. PMID: 10753845
- Pruneri G, Bertolini F, Soligo D, Carboni N, Cortelezzi A, Ferrucci PF, Buffa R, Lambertenghi-Deliliers G, and Pezzella F (1999). Angiogenesis in myelodysplastic syndromes. Br J Cancer 81: 1398–1401. doi: 10.1038/sj.bjc.6693515
- Vacca A, Ribatti D, Roncali L, Ranieri G, Serio G, Silvestris F, and Dammacco F (1994). Bone marrow angiogenesis and progression in multiple myeloma. Br J Haematol 87(3): 503–508. doi: 10.1111/j.1365-2141.1994.tb08304.x
- Medinger M, and Passweg J (2014). Role of tumour angiogenesis in haematological malignancies. Swiss Med Wkly 144: w14050. doi: 10.4414/smw.2014.14050
- Perez-Atayde AR, Sallan SE, Tedrow U, Connors S, Allred E, and Folkman J (1997). Spectrum of tumor angiogenesis in the bone marrow of children with acute lymphoblastic leukemia. Am J Pathol 150(3): 815–821. PMID: 9060819
- Yetgin S, Yenicesu I, Cetin M, and Tuncer M (2001). Clinical importance of serum vascular endothelial and basic fibroblast growth factors in children with acute lymphoblastic leukemia. Leuk Lymphoma 42(1–2): 83–88. doi: 10.3109/10428190109097679
- Aguayo A, Kantarjian HM, Estey EH, Giles FJ, Verstovsek S, Manshouri T, Gidel C, O'Brien S, Keating MJ, and Albitar M (2002). Plasma vascular endothelial growth factor levels have prognostic significance in patients with acute myeloid leukemia but not in patients with myelodysplastic syndromes. Cancer 95(9): 1923–1930. doi: 10.1002/cncr.10900
- Aguayo A, Estey E, Kantarjian H, Mansouri T, Gidel C, Keating M, Giles F, Estrov Z, Barlogie B, and Albitar M (1999). Cellular vascular endothelial growth factor is a predictor of outcome in patients with acute myeloid leukemia. Blood 94(11): 3717–3721. PMID: 10572084
- Gianelli U, Fracchiolla NS, Bucciarelli P, Ferla V, Boiocchi L, Savi F, Moro A, Reda G, Bosari S, and Cortelezzi A (2013). High levels of vascular endothelial growth factor protein expression are associated with an increased risk of transfusion dependence in myelodysplastic syndromes. Am J Clin Pathol 139(3): 380–387. doi: 10.1309/ajcp5o3ybktggwnq
- Tzankov A, Heiss S, Ebner S, Sterlacci W, Schaefer G, Augustin F, Fiegl M, and Dirnhofer S (2007). Angiogenesis in nodal B cell lymphomas: a high throughput study. J Clin Pathol 60(5): 476–482. doi: 10.1136/jcp.2006.038661
- Loges S, Heil G, Bruweleit M, Schoder V, Butzal M, Fischer U, Gehling UM, Schuch G, Hossfeld DK, and Fiedler W (2005). Analysis of concerted expression of angiogenic growth factors in acute myeloid leukemia: expression of angiopoietin-2 represents an independent prognostic factor for overall survival. J Clin Oncol 23(6): 1109–1117. doi: 10.1200/jco.2005.05.058
- Vacca A, Ribatti D, Presta M, Minischetti M, Iurlaro M, Ria R, Albini A, Bussolino F, and Dammacco F (1999). Bone marrow neovascularization, plasma cell angiogenic potential, and matrix metalloproteinase-2 secretion parallel progression of human multiple myeloma. Blood 93(9): 3064–3073. PMID: 10216103
- Dias S, Hattori K, Zhu Z, Heissig B, Choy M, Lane W, Wu Y, Chadburn A, Hyjek E, Gill M, Hicklin DJ, Witte L, Moore MA, and Rafii S (2000). Autocrine stimulation of VEGFR-2 activates human leukemic cell growth and migration. J Clin Invest 106(4): 511–521. doi: 10.1172/jci8978
- Fiedler W, Graeven U, Ergun S, Verago S, Kilic N, Stockschlader M, and Hossfeld DK (1997). Vascular endothelial growth factor, a possible paracrine growth factor in human acute myeloid leukemia. Blood 89(6): 1870–1875. PMID: 9058706
- Chen H, Treweeke AT, West DC, Till KJ, Cawley JC, Zuzel M, and Toh CH (2000). In vitro and in vivo production of vascular endothelial growth factor by chronic lymphocytic leukemia cells. Blood 96(9): 3181–3187. PMID: 11050001
- Leung DW, Cachianes G, Kuang WJ, Goeddel D V, and Ferrara N (1989). Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 246(4935): 1306–1309. doi: 10.1126/science.2479986
- Dias S, Shmelkov S V, Lam G, and Rafii S (2002). VEGF(165) promotes survival of leukemic cells by Hsp90-mediated induction of Bcl-2 expression and apoptosis inhibition. Blood 99(7): 2532–2540. doi: 10.1182/blood.V99.7.2532
- Fragoso R, Elias AP, and Dias S (2007). Autocrine VEGF loops, signaling pathways, and acute leukemia regulation. Leuk Lymphoma 48(3): 481–488. doi: 10.1080/10428190601064720
- Santos SC, and Dias S (2004). Internal and external autocrine VEGF/KDR loops regulate survival of subsets of acute leukemia through distinct signaling pathways. Blood 103(10): 3883–3889. doi: 10.1182/blood-2003-05-1634
- Rieger MA, Hoppe PS, Smejkal BM, Eitelhuber AC, and Schroeder T (2009). Hematopoietic cytokines can instruct lineage choice. Science 325(5937): 217–218. doi: 10.1126/science.1171461
- Zhang CC, and Lodish HF (2008). Cytokines regulating hematopoietic stem cell function. Curr Opin Hematol 15(4): 307–311. doi: 10.1097/MOH.0b013e3283007db5
- Guilak F, Cohen DM, Estes BT, Gimble JM, Liedtke W, and Chen CS (2009). Control of stem cell fate by physical interactions with the extracellular matrix. Cell Stem Cell 5(1): 17–26. doi: 10.1016/j.stem.2009.06.016
- Birbrair A, and Frenette PS (2016). Niche heterogeneity in the bone marrow. Ann N Y Acad Sci 1370(1): 82–96. doi: 10.1111/nyas.13016
- Pinho S, Frenette PS (2019). Haematopoietic stem cell activity and interactions with the niche. Nat Rev Mol Cell Biol 20(5): 303-320. doi: 10.1038/s41580-019-0103-9
- Stier S, Ko Y, Forkert R, Lutz C, Neuhaus T, Grünewald E, Cheng T, Dombkowski D, Calvi LM, Rittling SR, Scadden DT (2005). Osteopontin is a hematopoietic stem cell niche component that negatively regulates stem cell pool size. J Exp Med 201(11):1781-91. doi: 10.1084/jem.20041992
- Nilsson SK, Johnston HM, Whitty GA, Williams B, Webb RJ, Denhardt DT, Bertoncello I, Bendall LJ, Simmons PJ, Haylock DN (2005). Osteopontin, a key component of the hematopoietic stem cell niche and regulator of primitive hematopoietic progenitor cells. Blood 106(4):1232-9. doi: 10.1182/blood-2004-11-4422
- Lord BI, Tesla NG, Hendry JH (1975). The relative spatial distribution of CFUs and CFUc in the normal mouse femur. Blood 46(1):65-72. PMID: 1131427
- Gong JK (1978). Endosteal marrow: a rich source of hematopoietic stem cells. Science 199(4336):1443-5. doi: 10.1126/science.75570
- Nilsson SK, Johnston HM, Coverdale JA (2001). Spatial localization of transplanted hemopoietic stem cells: inferences for the localization of stem cell niches. Blood 97(8):2293-9. doi: 10.1182/blood.V97.8.2293
- Taichman RS, Reilly MJ, Emerson SG (1996). Human osteoblasts support human hematopoietic progenitor cells in vitro bone marrow culture. Blood 87(2):518-24. PMID: 8555473
- Greenbaum A, Hsu YMS, Day RB, Schuettpelz LG, Christopher MJ, Borgerding JN, Nagasawa T, and Link DC (2013). CXCL12 Production by Early Mesenchymal Progenitors is Required for Hematopoietic Stem Cell Maintenance. Nature 495(7440): 227–230. doi: 10.1038/nature11926
- Nilsson SK, Johnston HM, Whitty GA, Williams B, Webb RJ, Denhardt DT, Bertoncello I, Bendall LJ, Simmons PJ, and Haylock DN (2005). Osteopontin, a key component of the hematopoietic stem cell niche and regulator of primitive hematopoietic progenitor cells. Blood 106(4): 1232–1239. doi: 10.1182/blood-2004-11-4422
- Visnjic D, Kalajzic Z, Rowe DW, Katavic V, Lorenzo J, Aguila HL (2004). Hematopoiesis is severely altered in mice with an induced osteoblast deficiency. Blood 103(9):3258-64. doi: 10.1182/blood-2003-11-4011
- Kunisaki Y, Bruns I, Scheiermann C, Ahmed J, Pinho S, Zhang D, Mizoguchi T, Wei Q, Lucas D, Ito K, Mar JC, Bergman A, Frenette PS (2013). Arteriolar niches maintan haematopoietic stem cell quiescence. Nature 502(7473):637-43. doi: 10.1038/nature12612
- Nombela-Arrieta C, Pivarnik G, Winkel B, Canty KJ, Harley B, Mahoney JE, Park SY, Lu J, Protopopov A, Silberstein LE (2013). Quantitative imaging of haematopoietic stem and progenitor cell localization and hypoxic status in the bone marrow microenvironment. Nat Cell Biol 15(5):533-43. doi: 10.1038/ncb2730
- Ding L, Morrison SJ (2013). Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature 495(7440):231-5. doi: 10.1038/nature11885
- Zhu J, Garrett R, Jung Y, Zhang Y, Kim N, Wang J, Joe GJ, Hexner E, Choi Y, Taichman TS, Emerson SG (2007). Osteoblasts support B-lymphocyte commitment and differentiation from hematopoietic stem cells. Blood 109(9):3706-12. doi: 10.1182/blood-2006-08-041384
- Yu VW, Saez B, Cook C, Lotinun S, Pardo-Saganda A, Wang YH, Lymperi S, Ferraro F, Raaijmakers MH, Wu JY, Zhou L, Rajagopal J, Kronenberg HM, Baron R, Scadden DT (2015). Specific bone cells produced DLL4 to generate thymus-seeding progenitors from bone marrow. J Exp Med 212(5):759-74. doi: 10.1084/jem.20141843
- Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ (2005). SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 127(7):1109-21. doi: 10.1016/j.cell.2005.05.026
- Sacchetti B, Funari A, Michienzi S, Di Cesare S, Piersanti S, Saggio I, Tagliafico E, Ferrari S, Robey PG, Riminucci M, Bianco P (2007). Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell 131(2):324-36. doi: 10.1016/j.cell.2007.08.025
- Ding L, Saunders TL, Enikolopov G, and Morrison SJ (2012). Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 481(7382): 457–462. doi: 10.1038/nature10783
- Morrison SJ, and Scadden DT (2014). The bone marrow niche for haematopoietic stem cells. Nature 505(7483): 327. doi: 10.1038/nature12984
- Asada N, Kunisaki Y, Pierce H, Wang Z, Fernandez NF, Birbrair A, Ma'ayan A, and Frenette PS (2017). Differential cytokine contributions of perivascular haematopoietic stem cell niches. Nat Cell Biol 19(3): 214–223. doi: 10.1038/ncb3475
- Kunisaki Y, and Frenette PS (2014). Influences of vascular niches on hematopoietic stem cell fate. Int J Hematol 99(6): 699–705. doi: 10.1007/s12185-014-1580-4
- Mach DB, Rogers SD, Sabino MC, Luger NM, Schwei MJ, Pomonis JD, Keyser CP, Clohisy DR, Adams DJ, O'Leary P, and Mantyh PW (2002). Origins of skeletal pain: sensory and sympathetic innervation of the mouse femur. Neuroscience 113(1): 155–66. doi: 10.1016/S0306-4522(02)00165-3
- Nadri S, Soleimani M, Mobarra Z, and Amini S (2008). Expression of dopamine-associated genes on conjunctiva stromal-derived human mesenchymal stem cells. Biochem Biophys Res Commun 377(2): 423–428. doi: 10.1016/j.bbrc.2008.09.148
- Maestroni GJ, Cosentino M, Marino F, Togni M, Conti A, Lecchini S, and Frigo G (1998). Neural and endogenous catecholamines in the bone marrow. Circadian association of norepinephrine with hematopoiesis? Exp Hematol 26(12): 1172–1177. PMID: 9808057
- Heidt T, Sager HB, Courties G, Dutta P, Iwamodo Y, Zaltsman A, von zur Muhlen C, Bode C, Fricchione GL, Denninger J, Lin CP, Vinegoni C, Libby P, Swirski FK, Weissleder R, Nahrendorf M (2015). Chronic variable stress activates hematopoietic stem cells. Nat Med 20(7): 754-758. doi: 10.1038/nm.3589
- Maryanovich M, Takeishi S, and Frenette PS (2018). Neural Regulation of Bone and Bone Marrow. Cold Spring Harb Perspect Med 8(9): a031344. doi: 10.1101/cshperspect.a031344
- Cosentino M, Kustrimovic N, and Marino F (2013). Endogenous Catecholamines in Immune Cells: An Overview. Available at http://www.brainimmune.com/endogenous-catecholamines-in-immune-cells-discovery-functions-and-clinical-potential-as-pharmacotherapeutic-targets-3/ [Accessed 30.09.18]
- Freeman JG, Ryan JH, Shelburne CP, Bailey DP, Bouton A, Narasimhachari N, Domen J, Siméon N, Courderc F, and Stewart JK (2001). Catecholamines in murine bone marrow derived mast cells. J Neuroimmunol 119(2): 231–238. doi: 10.1016/S0165-5728(01)00384-8
- Kuci Z, Seitz G, Kuci S, Kreyenberg H, Schumm M, Lang P, Niethammer D, Handgretinger R, and Bruchelt G (2006). Pitfalls in detection of contaminating neuroblastoma cells by tyrosine hydroxylase RT-PCR due to catecholamine-producing hematopoietic cells. Anticancer Res 26(3a): 2075–2080. PMID: 16827147
- Seidel J, Niggemann B, Punzel M, Fischer J, Zanker KS, and Dittmar T (2007). The neurotransmitter GABA is a potent inhibitor of the stromal cell-derived factor-1alpha induced migration of adult CD133+ hematopoietic stem and progenitor cells. Stem Cells Dev 16(5): 827–836. doi: 10.1089/scd.2007.0004
- Méndez-Ferrer S, Lucas D, Battista M, and Frenette PS (2008). Haematopoietic stem cell release is regulated by circadian oscillations. Nature 452(7186): 442–447. doi: 10.1038/nature06685
- Spiegel A, Shivtiel S, Kalinkovich A, Ludin A, Netzer N, Goichberg P, Azaria Y, Resnick I, Hardan I, Ben-Hur H, Nagler A, Rubinstein M, and Lapidot T (2007). Catecholaminergic neurotransmitters regulate migration and repopulation of immature human CD34+ cells through Wnt signaling. Nat Immunol 8(10): 1123–1131. doi: 10.1038/ni1509
- Hoggatt J, and Scadden DT (2012). The stem cell niche: tissue physiology at a single cell level. J Clin Invest 122(9): 3029–3034. doi: 10.1172/JCI60238
- Kropfl JM, Stelzer I, Mangge H, Pekovits K, Fuchs R, Allard N, Schinagl L, Hofmann P, Dohr G, Wallner-Liebmann S, Domej W, and Muller W (2014). Exercise-induced norepinephrine decreases circulating hematopoietic stem and progenitor cell colony-forming capacity. PLoS One 9(9): e106120. doi: 10.1371/journal.pone.0106120
- Méndez-Ferrer S, Battista M, and Frenette PS (2010). Cooperation of β2- and β3-adrenergic receptors in hematopoietic progenitor cell mobilization. Ann N Y Acad Sci 1192(1): 139–144. doi: 10.1111/j.1749-6632.2010.05390.x
- Dar A, Schajnovitz A, Lapid K, Kalinkovich A, Itkin T, Ludin A, Kao W-MM, Battista M, Tesio M, Kollet O, Cohen NN, Margalit R, Buss EC, Baleux F, Oishi S, Fujii N, Larochelle A, Dunbar CE, Broxmeyer HE, Frenette PS, and Lapidot T (2011). Rapid mobilization of hematopoietic progenitors by AMD3100 and catecholamines is mediated by CXCR4-dependent SDF-1 release from bone marrow stromal cells. Leukemia 25(8): 1286–1296. doi: 10.1038/leu.2011.62
- Wei Q, and Frenette PS (2018). Niches for Hematopoietic Stem Cells and Their Progeny. Immunity 48(4): 632–648. doi: 10.1016/j.immuni.2018.03.024
- Asada N (2018). Regulation of Malignant Hematopoiesis by Bone Marrow Microenvironment. Front Oncol doi: 10.3389/fonc.2018.00119
- Sarkaria SM, Decker M, and Ding L (2018). Bone Marrow Micro-Environment in Normal and Deranged Hematopoiesis: Opportunities for Regenerative Medicine and Therapies. Bioessays 40(3). doi: 10.1002/bies.201700190
- Doron B, Handu M, and Kurre P (2018). Concise Review: Adaptation of the Bone Marrow Stroma in Hematopoietic Malignancies: Current Concepts and Models. Stem Cells 36(3): 304–312. doi: 10.1002/stem.2761
- Costa MHG, de Soure AM, Cabral JMS, Ferreira FC, and da Silva CL (2018). Hematopoietic Niche – Exploring Biomimetic Cues to Improve the Functionality of Hematopoietic Stem/Progenitor Cells. Biotechnol J 13(2): 1700088. doi: 10.1002/biot.201700088
- Shafat MS, Gnaneswaran B, Bowles KM, and Rushworth SA (2017). The bone marrow microenvironment – Home of the leukemic blasts. Blood Rev 31(5): 277–286. doi: 10.1016/j.blre.2017.03.004
- Scheiermann C, Kunisaki Y, Lucas D, Chow A, Jang JE, Zhang D, Hashimoto D, Merad M, and Frenette PS (2012). Adrenergic nerves govern circadian leukocyte recruitment to tissues. Immunity 37(2): 290–301. doi: 10.1016/j.immuni.2012.05.021
- Shiozawa Y, Eber MR, Berry JE, and Taichman RS (2015). Bone marrow as a metastatic niche for disseminated tumor cells from solid tumors. Bonekey Rep 4: 689. doi: 10.1038/bonekey.2015.57
- Campbell JP, Merkel AR, Masood-Campbell SK, Elefteriou F, and Sterling JA (2012). Models of Bone Metastasis. J Vis Exp 67: e4260. doi: 10.3791/4260
- Kan C, Vargas G, Pape F Le, and Clézardin P (2016). Cancer Cell Colonisation in the Bone Microenvironment. Int J Mol Sci 17(10): 1674. doi: 10.3390/ijms17101674
- Roccaro AM, Sacco A, Purschke WG, Moschetta M, Buchner K, Maasch C, Zboralski D, Zöllner S, Vonhoff S, Mishima Y, Maiso P, Reagan MR, Lonardi S, Ungari M, Facchetti F, Eulberg D, Kruschinski A, Vater A, Rossi G, Klussmann S, and Ghobrial IM (2014). SDF-1 inhibition targets the bone marrow niche for cancer therapy. Cell Rep 9(1): 118–128. doi: 10.1016/j.celrep.2014.08.042
- Jones DH, Nakashima T, Sanchez OH, Kozieradzki I, Komarova S V, Sarosi I, Morony S, Rubin E, Sarao R, Hojilla C V, Komnenovic V, Kong Y-Y, Schreiber M, Dixon SJ, Sims SM, Khokha R, Wada T, and Penninger JM (2006). Regulation of cancer cell migration and bone metastasis by RANKL. Nature 440(7084): 692–696. doi: 10.1038/nature04524
- Elefteriou F (2016). Role of sympathetic nerves in the establishment of metastatic breast cancer cells in bone. J bone Oncol 5(3): 132–134. doi: 10.1016/j.jbo.2016.03.003
- Hu W, Zhen X, Xiong B, Wang B, Zhang W, and Zhou W (2008). CXCR6 is expressed in human prostate cancer in vivo and is involved in the in vitro invasion of PC3 and LNCap cells. Cancer Sci 99(7): 1362–1369. doi: 10.1111/j.1349-7006.2008.00833.x
- Mulcrone PL, Campbell JP, Clément-Demange L, Anbinder AL, Merkel AR, Brekken RA, Sterling JA, and Elefteriou F (2017). Skeletal Colonization by Breast Cancer Cells Is Stimulated by an Osteoblast and β2AR-Dependent Neo-Angiogenic Switch. J Bone Miner Res 32(7): 1442–1454. doi: 10.1002/jbmr.3133
- Zhang W, Ching Lo H, Xiang, and Zhang HF (2017). Mapping bone marrow niches of disseminated tumor cells. Sci China Life Sci 60(10): 1125–1132. doi: 10.1007/s11427-017-9180-5
- Strell C, Niggemann B, Voss MJ, Powe DG, Zanker KS, and Entschladen F (2012). Norepinephrine promotes the beta1-integrin-mediated adhesion of MDA-MB-231 cells to vascular endothelium by the induction of a GROalpha release. Mol Cancer Res 10(2): 197–207. doi: 10.1158/1541-7786.mcr-11-0130
- Obradovic MMS, Hamelin B, Manevski N, Pinto Couto J, Sethi A, Coissieux MM, Münst S, Okamoto R, Kohler H, Schmidt A, Bentires-Alj M (2019). Glucocorticoids promote breast cancer metastasis. Nature 567(7749):540-544. doi: 10.1038/s41586-019-1019-4
- Sipkins DA, Wei X, Wu JW, Runnels JM, Côté D, Means TK, Luster AD, Scadden DT, and Lin CP (2005). In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nature 435(7044): 969–973. doi: 10.1038/nature03703
- Hanoun M, Zhang D, Mizoguchi T, Pinho S, Pierce H, Kunisaki Y, Lacombe J, Armstrong SA, Dührsen U, and Frenette PS (2014). Acute myelogenous leukemia-induced sympathetic neuropathy promotes malignancy in an altered hematopoietic stem cell niche. Cell Stem Cell 15(3): 365–375. doi: 10.1016/j.stem.2014.06.020
- Arranz L, Sánchez-Aguilera A, Martín-Pérez D, Isern J, Langa X, Tzankov A, Lundberg P, Muntión S, Tzeng Y-S, Lai D-M, Schwaller J, C. Skoda R, Méndez-Ferrer S, Skoda RC, and Méndez-Ferrer S (2014). Neuropathy of haematopoietic stem cell niche is essential for myeloproliferative neoplasms. Nature 512(7512): 78. doi: 10.1038/nature13383
- Sun YX, Schneider A, Jung Y, Wang J, Dai J, Cook K, Osman NI, Koh-Paige AJ, Shim H, Pienta KJ, Keller ET, McCauley LK, and Taichman RS (2005). Skeletal localization and neutralization of the SDF-1(CXCL12)/CXCR4 axis blocks prostate cancer metastasis and growth in osseous sites in vivo. J Bone Min Res 20(2): 318–329. doi: 10.1359/jbmr.041109
- Sun YX, Fang M, Wang J, Cooper CR, Pienta KJ, and Taichman RS (2007). Expression and activation of alpha v beta 3 integrins by SDF-1/CXC12 increases the aggressiveness of prostate cancer cells. Prostate 67(1): 61–73. doi: 10.1002/pros.20500
- Shiozawa Y, Havens AM, Jung Y, Ziegler AM, Pedersen EA, Wang J, Lu G, Roodman GD, Loberg RD, Pienta KJ, and Taichman RS (2008). Annexin II/annexin II receptor axis regulates adhesion, migration, homing, and growth of prostate cancer. J Cell Biochem 105(2): 370–380. doi: 10.1002/jcb.21835
- Wang J, Shiozawa Y, Wang J, Wang Y, Jung Y, Pienta KJ, Mehra R, Loberg R, and Taichman RS (2008). The Role of CXCR7/RDC1 as a Chemokine Receptor for CXCL12/SDF-1 in Prostate Cancer. J Biol Chem 283(7):4283-94. doi: 10.1074/jbc.M707465200
- Asri A, Sabour J, Atashi A, and Soleimani M (2016). Homing in hematopoietic stem cells: focus on regulatory role of CXCR7 on SDF1a/CXCR4 axis. EXCLI J 15:134-43. doi: 10.17179/excli2014-585
- Sharma M, Afrin F, Satija N, Tripathi RP, and Gangenahalli GU (2011). Stromal-derived factor-1/CXCR4 signaling: indispensable role in homing and engraftment of hematopoietic stem cells in bone marrow. Stem Cells Dev 20(6): 933–946. doi: 10.1089/scd.2010.0263
- Jung Y, Wang J, Song J, Shiozawa Y, Havens A, Wang Z, Sun YX, Emerson SG, Krebsbach PH, and Taichman RS (2007). Annexin II expressed by osteoblasts and endothelial cells regulates stem cell adhesion, homing, and engraftment following transplantation. Blood 110(1): 82–90. doi: 10.1182/blood-2006-05-021352
- Shiozawa Y, Pedersen EA, Havens AM, Jung Y, Mishra A, Joseph J, Kim JK, Patel LR, Ying C, Ziegler AM, Pienta MJ, Song J, Wang J, Loberg RD, Krebsbach PH, Pienta KJ, and Taichman RS (2011). Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J Clin Invest 121: 1298–1312. doi: 10.1172/jci43414
- Shiozawa Y, Pedersen EA, Patel LR, Ziegler AM, Havens AM, Jung Y, Wang J, Zalucha S, Loberg RD, Pienta KJ, and Taichman RS (2010). GAS6/AXL axis regulates prostate cancer invasion, proliferation, and survival in the bone marrow niche. Neoplasia 12(2): 116–27. doi: 10.1593/neo.91384
- Shiozawa Y, Havens AM, Pienta KJ, and Taichman RS (2008). The bone marrow niche: habitat to hematopoietic and mesenchymal stem cells and unwitting host to molecular parasites. Leukemia 22(5): 941–950. doi: 10.1038/leu.2008.48
- Cook J, and Hagemann T (2013). Tumour-associated macrophages and cancer. Curr Opin Pharmacol 13(4): 595–601. doi: 10.1016/j.coph.2013.05.017
- Lutgendorf SK, Lamkin DM, Jennings NB, Arevalo JM, Penedo F, DeGeest K, Langley RR, Lucci 3rd JA, Cole SW, Lubaroff DM, and Sood AK (2008). Biobehavioral influences on matrix metalloproteinase expression in ovarian carcinoma. Clin Cancer Res 14(21): 6839–6846. doi: 10.1158/1078-0432.ccr-08-0230
- Pollard JW (2004). Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer 4: 71–78. doi: 10.1038/nrc1256
- Kalinski P (2012). Regulation of immune responses by prostaglandin E2. J Immunol 188(1): 21–28. doi: 10.4049/jimmunol.1101029
- Muthuswamy R, Okada NJ, Jenkins FJ, McGuire K, McAuliffe PF, Zeh HJ, Bartlett DL, Wallace C, Watkins S, Henning JD, Bovbjerg DH, and Kalinski P (2017). Epinephrine promotes COX-2-dependent immune suppression in myeloid cells and cancer tissues. Brain Behav Immun 62: 78–86. doi: 10.1016/j.bbi.2017.02.008
- Inbar S, Neeman E, Avraham R, Benish M, Rosenne E, and Ben-Eliyahu S (2011). Do Stress Responses Promote Leukemia Progression? An Animal Study Suggesting a Role for Epinephrine and Prostaglandin-E2 through Reduced NK Activity. PLoS One 6(4): e19246–e19246. doi: 10.1371/journal.pone.0019246
- Marino F, and Cosentino M (2013). Adrenergic modulation of immune cells: an update. Amino Acids 45(1): 55–71. doi: 10.1007/s00726-011-1186-6
- Eng JW, Kokolus KM, Reed CB, Hylander BL, Ma WW, and Repasky EA (2014). A nervous tumor microenvironment: the impact of adrenergic stress on cancer cells, immunosuppression, and immunotherapeutic response. Cancer Immunol Immunother 63(11): 1115–1128. doi: 10.1007/s00262-014-1617-9
- Scheller EL, Khandaker S, Learman BS, Cawthorn WP, Anderson LM, Pham HA, Robles H, Wang Z, Li Z, Parlee SD, Simon BR, Mori H, Bree AJ, Craft CS, and MacDougald OA (2018). Bone marrow adipocytes resist lipolysis and remodeling in response to beta-adrenergic stimulation. Bone 118:32-41. doi: 10.1016/j.bone.2018.01.016
- Muthu K, Iyer S, He LK, Szilagyi A, Gamelli RL, Shankar R, and Jones SB (2007). Murine Hematopoietic Stem cells and Progenitors Express Adrenergic Receptors. J Neuroimmunol 186(1–2): 27–36. doi: 10.1016/j.jneuroim.2007.02.007
- Shen HM, Sha LX, Kennedy JL, and Ou DW (1994). Adrenergic receptors regulate macrophage secretion. Int J Immunopharmacol.16(11): 905–910. doi: 10.1016/0192-0561(94)90045-0
ACKNOWLEDGMENTS
This study was supported by grants from the Krebsliga Schweiz (KFS-4013-08-2016-R) to C.L.
COPYRIGHT
© 2019

Stress and catecholamines modulate the bone marrow microenvironment to promote tumorigenesis by Hanns et al. is licensed under a Creative Commons Attribution 4.0 International License.


