Reviews:
Cell Stress, Vol. 5, No. 4, pp. 40 - 51; doi: 10.15698/cst2021.04.246
Roles of phosphatidyl inositol 3 kinase gamma (PI3Kγ) in respiratory diseases

1 Department of Molecular Biotechnology and Health Sciences, University of Torino, Via Nizza 52, 10126, Torino, Italy.
2 Kither Biotech S.r.l. Via Nizza 52, 10126, Torino, Italy.
# Equal contribution to senior authorship
Keywords: PI3K signaling, chronic respiratory disease, restrictive airway disease, obstructive airway disease, inflammation.
Abbreviatons:
AHR – airway hyperresponsiveness;
ASM – airway smooth muscle;
cAMP – cyclic adenosine monophosphate;
CF – cystic fibrosis;
CFTR – CF transmembrane conductance regulator;
GPCR – G-protein coupled receptor;
COPD – chronic obstructive pulmonary disease;
IAV – influenza A;
IFN – interferon;
IL – interleukin;
IPF – idiopathic pulmonary fibrosis;
KD – knockdown;
KO – knockout;
LPS – lipopolysaccharide;
MIF – migration inhibitory factor;
NK – natural killer;
PI3K – phosphatidyl inositol 3 kinase;
PLY – pneumolysin;
SNP - single-nucleotide polymorphisms;
TLR – Toll-like receptor;
VILI – ventilation-induced lung injury.
Received originally: 05/11/2020 Received in revised form: 15/02/2021
Accepted: 23/02/2021
Published: 08/03/2021
Correspondence:
Emilio Hirsch, via Nizza 52, 10126 Torino, Italy, Tel. +390116706225, Fax: +390116706432; emilio.hirsch@unito.it
Valentina Sala, via Nizza 52, 10126 Torino, Italy, Tel. +390116706335, Fax: +390116706432; valentina.sala@unito.it
Conflict of interest statement: Alessandra Ghigo and Emilio Hirsch are founders of Kither Biotech S.r.l. Valentina Sala received a contract fee from Kither Biotech S.r.l.
Please cite this article as: Valentina Sala, Angela Della Sala, Alessandra Ghigo and Emilio Hirsch (2021). Roles of phosphatidyl inositol 3 kinase gamma (PI3Kγ) in respiratory diseases. Cell Stress 5(4): 40-51. doi: 10.15698/cst2021.04.246
Abstract
Phosphatidyl inositol 3 kinase gamma (PI3Kγ) is expressed in all the cell types that are involved in airway inflammation and disease, including not only leukocytes, but also structural cells, where it is expressed at very low levels under physiological conditions, while is significantly upregulated after stress. In the airways, PI3Kγ behaves as a trigger or a controller, depending on the pathological context. In this review, the contribution of PI3Kγ in a plethora of respiratory diseases, spanning from acute lung injury, pulmonary fibrosis, asthma, cystic fibrosis and response to both bacterial and viral pathogens, will be commented.
INTRODUCTION
Phosphatidyl inositol 3 kinases (PI3Ks) are a family of lipid kinases that play key roles in a plethora of processes, including cell growth, proliferation and differentiation, tissue morphogenesis, metabolism, and immune function. The PI3K family is divided into three classes with distinct functions, among which the best characterized is class I, which phosphorylates phosphatidylinositol 4,5 bisphosphate in the third position to generate the second messenger phosphatidylinositide 3,4,5 trisphosphate. Class I PI3K subfamily is further divided into two classes: class IA, which is composed of α, β and δ isoforms, and class IB, whose only member is PI3Kγ [1]. Class I PI3K isoforms display different expression patterns: while PI3Kα and PI3Kβ are ubiquitously expressed, PI3Kδ and PI3Kγ have a more restricted expression pattern. Accordingly, deficiency in PI3Kα or PI3Kβ is embryonic lethal in murine models, whereas PI3Kγ or PI3Kδ knockout (KO) mice are viable and fertile [2].
–
In particular, PI3Kγ is expressed, at very low levels under physiological conditions, in cell types including cardiomyocytes [3][4][5][6][7][8][9], vascular smooth muscle cells [10], and the microglia [11], where it is significantly upregulated after stress.
–
On the contrary, PI3Kγ is constitutively enriched in leukocytes (neutrophils, eosinophils, macrophages, T cells and mast cells) [12]. Consistently, PI3Kγ KO mice exposed to natural pathogens/microbiota display altered immune traits that closely mirror the human Inactivated PI3Kγ Syndrome (IPGS) [13]. Intriguingly, clinical signs related to loss of PIK3γ include autoimmune cytopenia and infections, as well as pathological infiltration of T cells in barrier organs, including the lungs, that are hyper-responsive to microbial products [13].
–
Of utmost relevance for respiratory homeostasis and disease, PI3Kγ is also expressed in all the other cell types that are involved in airway disease, like endothelial cells [14], fibroblasts [15], and epithelial cells [16][17].
–
Besides such diverse expression patterns, class I PI3Ks own non-redundant roles in the response to a variety of stimuli. Class IA PI3Ks, exception done for PI3Kβ that can be also activated by G-protein Coupled Receptors (GPCRs) [18], are recruited to receptor tyrosine kinases through the SH2 domains of p85-like regulatory subunits. Class IB PI3Kγ is composed of the p110γ catalytic subunit, and of the p101 and p84/p87 subunits. These two adapter companions have important non-redundant roles in coupling PI3Kγ to upstream Ras/GPCRs signaling pathways [19]. While p84 is a component of a constitutively-expressed PI3K complex, p101 is part of an inducible PI3K complex [20]. Moreover, the p110γ/p84 heterodimer is less sensitive to the activation promoted by Gβγ subunits and depends on Ras partnership, while activation of the p110γ/p101 variant by Gβγ subunits is more favorable and Ras-independent [21]. Importantly, p110γ acts as an A-kinase anchoring protein (AKAP), being engaged in a functional and physical interaction with PKA that does not involve its kinase activity [7][8]. Thus, PI3Kγ is not only a kinase but also a scaffold protein for PKA in a complex containing type 3 and 4 phosphodiesterases (PDEs). This complex acts in a negative feedback loop, suppressing cyclic adenosine monophosphate (cAMP) levels in the vicinity of the β2-adrenergic receptor, through PKA-mediated activation of PDEs [7][8].
–
Therefore, acting at the crossroads of multiple pathways [1][22], PI3Kγ is a hub of intracellular signaling. As an example, PI3Kγ is activated downstream of GPCRs by both metabolic signals acting on β-adrenergic receptors, and immune signals like chemokines and complement fragments. Moreover, PI3Kγ can be activated by pathogen- and damage-associated molecular patterns downstream of Toll-like receptors (TLRs) in myeloid cells [23][24][25] and cardiomyocytes [5], functioning as a master regulator at the interface between metabolic and immune homeostasis. The relevance of the PI3Kγ hub as a regulator and amplifier for diverse and converging signaling pathways is evident in mast cells, where the FcεRI receptor mediates PI3Kγ activation. Yet, the FcεRI receptor has no direct link to GPCRs, but degranulation relies on PI3Kγ [26]. Intriguingly, the combinatorial regulation of PI3Kγ heterodimer variants can lead to a remarkable level of signaling specificity, which depends on both the tissue and the physio-pathological context [27].
–
TABLE 1. Differential responses of PI3Kγ KO mouse models of respiratory diseases. |
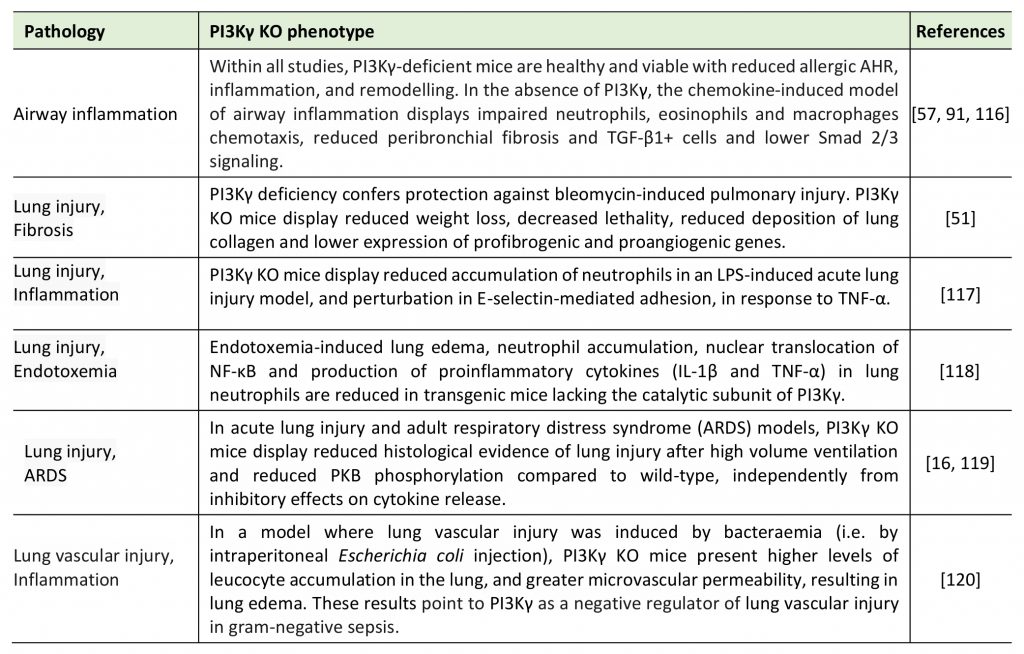 |
| [16][51][57][91][116][117][118][119][120] |
–
Moreover, studies demonstrating the effects of knocking out PI3Kγ in murine disease models (Table 1) led to great interest in the immunological functions and in the potential of PI3Kγ as a therapeutic target in inflammatory-driven diseases [15], including those affecting the airways. Within this review, we intend to highlight the relevance of PI3Kγ as a trigger or target in a plethora of respiratory diseases, spanning from acute lung injury, pulmonary fibrosis, asthma, cystic fibrosis and response to both bacterial and viral pathogens (Figure 1).
–
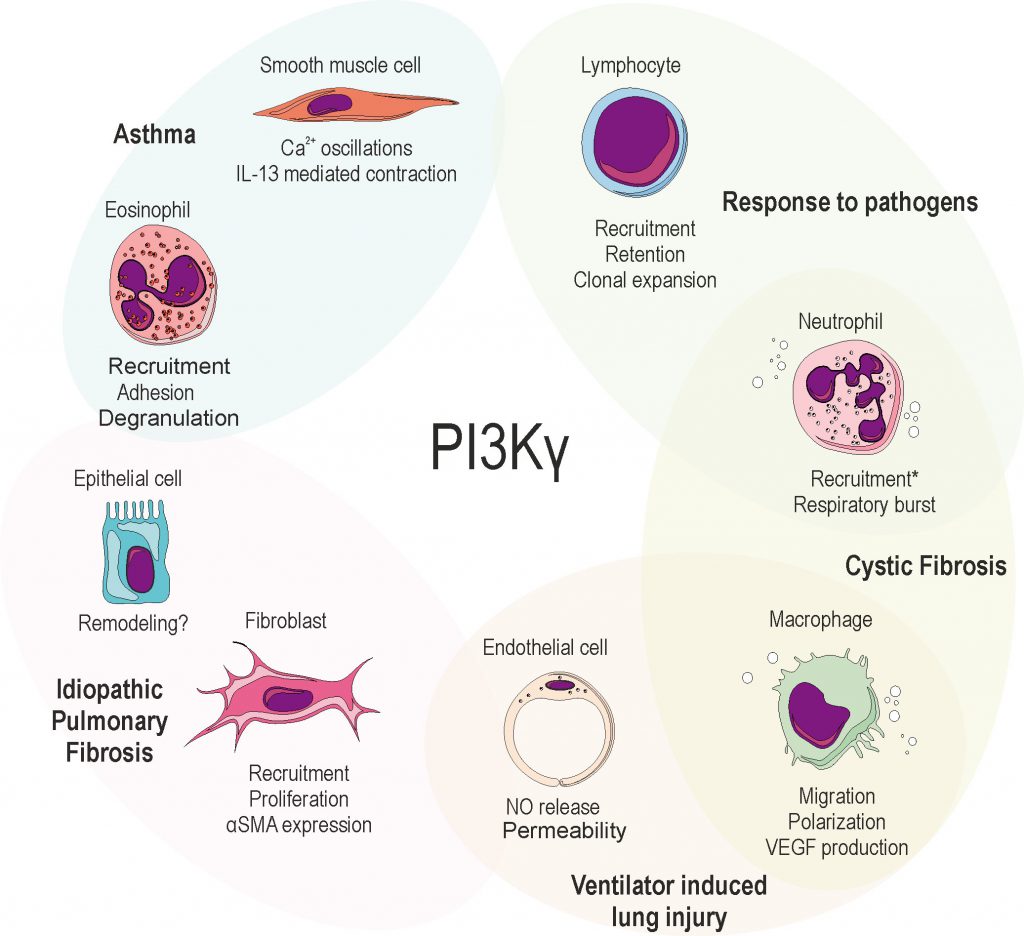 | FIGURE 1: Schematic representation of the biological processes induced or mediated by PI3Kγ in the cell types that are relevant for respiratory disease. PI3Kγ has been involved in almost all target cells shown in the figure using either isoform-selective PI3K inhibitors or genetic engineering. *Neutrophilic recruitment may be enhanced by either activation or inhibition of PI3Kγ, depending on the context. NO: Nitric Oxide; αSMA: α-smooth muscle actin; VEGF: Vascular endothelial growth factor; IL-13: Interleukin-13. |
PI3Kγ ROLE IN RESPIRATORY DISEASES
Ventilator-induced damage
Mechanical ventilation is a life-saving therapy but can contribute to the progression or even initiate lung injury per se. Ventilation-induced lung injury (VILI) clinically displays signs of alveolar edema, including increased vascular permeability and accumulation of fluids in the alveoli [28].
–
In VILI, the mechanical stress induced by ventilation activates the inflammasome in macrophages and endothelial cells, leading to enhanced nitric oxide (NO), oxygen radicals, and peroxynitrite production, which contributes to the increase of alveolar and vascular permeability [28] and impairs alveolar fluid clearance [29]. In line with these findings, the inhibition of PI3Kγ kinase activity specifically in resident lung cells attenuates VILI through the reduction of NO release [30].
–
Besides NO, the intracellular level of cAMP is critical for the modulation of endothelial permeability [31]. Since PI3Kγ can act as a scaffold, independently of its kinase activity, to modulate cAMP levels [7][8], its role in the formation of edema during VILI has been investigated. Accordingly, PI3Kγ knockout lungs are protected from VILI [16]. Moreover, pharmacological combined regimens aimed at blocking PI3Kγ kinase activity while increasing cAMP levels attenuate VILI in PI3Kγ wild-type lungs deprived of circulating leukocytes [30], pointing to the central role of PI3Kγ in lung cell types other than the immune cells.
–
Nevertheless, PI3Kγ is largely expressed in leukocytes, whose contribution to Vascular endothelial growth factor (VEGF) production, inflammation and injury in VILI is recognized [32]. A major player in the control of local and systemic immune response is TLR4 [33][34]. Notably, it has been shown that TLR4 is overexpressed [35], and has a key role in experimental models of VILI [36][37][38][39]. In macrophages, upon TLR4 receptor activation, PI3Kγ is recruited by Rab8, and is required to activate the Akt/mTOR pathway to bias the cytokine response towards an anti-inflammatory scenario [24]. Therefore, these findings suggest that the contribution of the TLR4/PI3Kγ axis to VILI pathogenesis deserves further investigation.
–
PI3Kγ emerged therefore as a possible therapeutic target in the treatment and/or prevention of VILI and edema. However, strategies aimed at blocking PI3Kγ kinase dependent and independent activities only in resident inflammatory and parenchymal lung cells, such as airway epithelial cells, should be addressed. This could enable to mitigate potential systemic side effects on the innate immune system on one side, and on different cAMP-responsive cells, like cardiomyocytes, on the other.
–
Idiopathic pulmonary fibrosis (IPF)
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive, fibrotic interstitial lung disease of unknown etiology, which occurs primarily in older adults (median age at diagnosis is about 65 years) [40]. Although it is classified as a rare disease (occurring in less than 5/10,000 persons per year), IPF is the most common type of idiopathic interstitial pneumonia, occurring with a frequency comparable to that of stomach, brain and testis cancers [41]. Moreover, the global burden of IPF is extremely high, due to the poor prognosis, with a median survival time of two to four years from diagnosis [42].
–
Historically, IPF was considered a chronic and progressive inflammatory disorder, which gradually leads to established fibrosis. However, the failure of anti-inflammatory therapies [43] caused a profound revision of this concept [44]. IPF is now thought to result from the concomitance of repetitive local micro-injuries to the ageing alveolar epithelium, genetic factors [45], and environmental risk factors (such as cigarette smoke, drugs, lung microbiome, infections or environmental pollutants) [42][46]. In turn, intracellular signaling initiated by micro-injuries gives rise to an aberrant communication between epithelial cells and fibroblasts, leading to increased extracellular matrix accumulation, and, ultimately, to lung interstitial remodeling and loss of function.
–
Within this context, PI3K signaling emerged as a crucial pathway in models of pulmonary fibrosis [47][48]. In particular, class I PI3Ks play key roles in the homeostasis of all the cell types that are involved in the pathogenesis of IPF. Consistently, an inhaled pan-Class I PI3K inhibitor has been demonstrated to have a protective effect against the rapid, progressive pulmonary fibrosis induced by instillation of bleomycin in vivo[48], by reducing the expression of pro-fibrotic genes, including transforming growth factor-β (TGF-β) and connective tissue growth factor (CTGF) [49]. Among class I isoforms, PI3Kγ is overexpressed in myofibroblasts and bronchiolar basal cells in the lungs of IPF patients, and, ex vivo, in human IPF primary fibroblasts [50]. Both genetic and pharmacological inhibition of PI3Kγ are able to inhibit proliferation as well as α-smooth muscle actin (αSMA) expression in IPF fibroblasts in vitro[50]. Accordingly, mice lacking PI3Kγ are protected from the accumulation of matrix and leukocytes in the lungs after bleomycin injury [51], pointing to PI3Kγ as a promising therapeutic target for IPF.
–
Recently, the need of pathway-specific biomarkers and genetic phenotyping has emerged in order to identify patient subtypes for new combinatorial trials [52]. In fact, due to its intrinsic complexity, the natural history of IPF is highly variable and the course of the disease in an individual patient is somewhat unpredictable, as some patients experience a rapid lung decline, while others progress much more slowly. Of note, a rapidly progressive disease has been associated with the upregulation of several genes, including TLR9 [53], downstream of which PI3Kγ is activated, at least in cardiomyocytes [5]. Moreover, in the past two decades, metabolic dysregulation, impaired mitochondrial autophagy, and mitochondrial dysfunction have been observed in cells of IPF lungs [54].
–
Overall, these results suggest the intriguing hypothesis that the activation level of PI3Kγ might act as a master controller in the different processes that converge on IPF pathogenesis and influence the fate of the lung environment. Whether PI3Kγ will be a suitable biomarker or therapeutic target in IPF patients, however, still has to be investigated.
–
Asthma
The role of PI3K family members in asthma is well documented and pan-class I PI3K topical inhibition is effective against acute and, more importantly, glucocorticoid resistant asthma [49]. Focusing on the specific contribution of PI3Kγ to asthma pathogenesis, KO of PI3Kγ or treatment with an aerosolized dual inhibitor of PI3Kγ and δ (TG100-115), is able to reduce eosinophilic airway hyper-responsiveness (AHR) and inflammation in experimental models [54][55][56][57].
–
Moreover, the PI3Kγ-specific inhibitor AS605240 dampens eosinophilic inflammation induced by the CC chemokine eotaxin (CCL11), by suppressing signaling pathways downstream of CC chemokine receptor 3 (CCR3) [58]. In detail, AS605240 inhibits eotaxin-induced chemotaxis, adhesion to Intercellular Adhesion Molecule 1 (ICAM-1), and degranulation of human peripheral blood eosinophils by inhibiting ERK1/2 phosphorylation, without down-regulation of surface CCR3 expression [58].
–
Mechanistically, the pathological role of PI3Kγ in asthma implicates the release of inflammatory cell mediators, including macrophage migration inhibitory factor (MIF) and the T-helper type II cytokine Interleukin-13 (IL-13).
–
MIF participates as a proinflammatory cytokine in both innate and adaptive immune responses, contributing to the pathogenesis of inflammatory, metabolic, autoimmune, and allergic diseases. Of note, MIF plays a pivotal role in activating the expression of PI3Kγ regulatory (p101) and catalytic subunits (p110) [59]. In turn, increased PI3Kγ activity is responsible for IL-13-mediated contraction of airway smooth muscle (ASM) cells, the underlying mechanism of AHR induced by allergen sensitization or cytokines in asthma [59][60][61][62][63]. IL-13 receptor and PI3Kγ are both expressed in ASM cells, in which they control contractility by regulating Ca2+ oscillations [64]. Notably, IL-13, which is increased in the airways of asthmatic patients and correlates with AHR [65], is sufficient [66] and required [67] for the development of allergen-induced AHR. In a translational perspective, targeting PI3Kγ, either pharmacologically or by RNA interference, suppresses IL-13-dependent contractility of ASM cells, and, more importantly, intranasal administration of a PI3Kγ inhibitor attenuates IL-13-induced AHR in mice [64]. Therefore, dampening IL-13 levels by targeting the upstream PI3Kγ signaling might be a feasible and efficient strategy to reduce Ca2+ oscillations and contraction in ASMs.
–
Overall, these data underline the promising therapeutic potential of PI3Kγ inhibition in asthma [68].
–
Cystic Fibrosis
Cystic fibrosis (CF) is the most common genetic disease in the Caucasian population, affecting ~1 in 3,500 persons. The basic defect of CF results from mutations in a single gene encoding for the CF transmembrane conductance regulator (CFTR), a 1,480 residues transmembrane glycoprotein that regulates cAMP-mediated chloride (Cl–) conductance at the apical surface of secretory epithelia. Impaired secretion of Cl– and bicarbonate triggers dehydration of the airway surface liquid, resulting in increased mucus viscosity and impaired mucociliary clearance. The accumulated mucus ultimately favors colonization by pathogens and resistance to treatments [69]. In turn, airway mucus obstruction and recurrent/persistent bacterial infections trigger a chronic neutrophilic inflammation, which are responsible for the release of neutrophilic elastases and for the ensuing, progressive lung damage and decline of function in CF patients [70].
–
In this context, the inflammatory response in CF lungs is non-resolving and self-perpetuating. In fact, the vicious cycle of neutrophilic burden and release of noxious mediators, further fuels inflammation and infection, and further contributes to disease progression towards irreversible lung damage. Notably, albeit chronic bacterial infections play a prominent role in the progression of CF lung disease, inflammation was observed in the lungs of asymptomatic CF infants without any apparent established bacterial infection [71], suggesting that sterile inflammation can precede, and possibly promote, infection in early-stage CF lung disease, by favoring the expansion of more pathogenic strains among the lung microbiota. Consistently, recent studies suggest that, upon migration to CF airways, neutrophils undergo a phenotypic reprogramming, leading to dysregulated lifespan, metabolism and effector function, ultimately contributing, together with the epithelium and resident microbiota, to the evolution of a pathological microenvironment [72].
–
Therefore, anti-inflammatory therapy, eventually combined with antibiotics, is crucial to prevent lung damage. However, currently used therapeutic strategies show limited clinical benefit. With the aim of filling this gap, the possibility to interfere with leukocyte trafficking into CF airways has been explored. PI3Kγ has a key role in this process, triggering signaling pathways evoked by binding of chemotactic factors to GPCRs. Among these, IL-8 represents the principal neutrophil chemoattractant and its elevated concentration characterizes CF lung inflammation. In the CF context, the biological efficacy of both genetic deletion and pharmacological inhibition of PI3Kγ in reducing chronic neutrophilic inflammation in the lungs has been demonstrated in β-ENaC overexpressing CF-like mice [73].
–
While most research on CF inflammation has focused on epithelial cells and neutrophils, macrophages play an important role in the initiation and resolution of pulmonary inflammation. Functional abnormalities have been observed in CF macrophages from experimental models, including newborn CF pigs, and from CF patients, and found to display a constitutive proinflammatory status and hyper-responsiveness to microbial stimuli, supporting the presence of a primary defect in CF macrophages, which seems to be correlated to CFTR channel function [74][75].
–
Of note, mucus stasis per se might be responsible for the pro-inflammatory polarization of airway macrophages [76], albeit data from CF patients point to a CFTR-dependent defect in the resolution phase of inflammation, due to the inability of CF macrophages to re-polarize to the M2 immunosuppressive phenotype [77]. Notably, blockade of PI3Kγ activity promotes M1 macrophage polarization in implanted tumors, and inflammation, albeit M2 polarization has been observed in obese mice lacking PI3Kγ [78], suggesting that the cellular context and activation level of PI3Kγ might be crucial to determine the fate of macrophages.
–
Overall, these data would support the relevance of the immune response in CF disease, but whether abnormalities in immune cells, including changes to macrophage polarization, could be corrected using CFTR-directed therapies remains an open question. Whereas blockade of PI3Kγ activity by small-molecule inhibitors may represent a valid approach to down-modulate neutrophil recruitment and burst in inflamed tissues, the resulting increased susceptibility to infection might be a potential side effect. Therefore, focused therapeutic windows should be defined for the use of these molecules in CF patients.
PI3Kγ IN INFECTIVE DISEASES
Bacterial infections
Streptococcus pneumoniae is the most prevalent gram-positive bacterium causing community-acquired pneumonia, septic meningitis, and otitis media. The pathogenicity of S. pneumoniae is largely linked to its ability to produce a variety of virulence factors, among which the most relevant is pneumococcal virulence factor pneumolysin (PLY). In addition to its ability to form pores in cell membranes, PLY acts as a pathogen-associated molecular pattern by signaling via TLR4 to induce TLR4-dependent cytotoxicity in lung resident macrophages, thus further promoting the bacterial colonization of the lower respiratory tract [35][79]. Of note, TLR4 activation acts through PI3Kγ to shift macrophages towards an anti-inflammatory scenario [24]. Mechanistically, PI3Kγ and Rab8a control cytokine production by signaling through mTOR [24], which acts as a hub downstream of TLR4 to bias cytokine responses, inhibiting NFκB-dependent transcription of pro-inflammatory cytokines, like IL-6 and IL-12, while enhancing STAT3-mediated transcription of the anti-inflammatory cytokine IL-10 [80]. Consistently, either genetic deletion or pharmacologic inhibition of PI3Kγ in mice infected with S. pneumoniae causes an impaired recruitment of macrophages, associated with a reduced bacterial clearance from the lungs [81]. This, in turn, results in an impaired resolution/repair process and in progressive pneumococcal pneumonia [81]. Similar results have been observed after infection by Staphylococcus aureus, as a higher bacterial burden is present in PI3Kγ KO mice, due to the reduced recruitment of leukocytes.
–
On the contrary, PI3Kγ deficiency improves the resistance against Mycobacterium tuberculosis in the early phase of infection, by increasing T helper IL-17+ (Th17) cells number, production of IL-17, and expression of molecules associated with Th17-cells differentiation and neutrophil recruitment [82]. These findings are in accord with previous data showing increased concentrations of IL-17 in the bronco alveolar lavage fluid of PI3Kγ KO mice challenged with intranasal instillation of lipopolysaccharide (LPS) [4].
–
Moreover, a deficiency in expression of PI3Kγ, along with PI3Kδ, enhances the IL-17/G-CSF axis and induces neutrophilia [83].
–
Of note, the crosstalk between the IL-17 signaling pathway and neutrophils recruitment seems to be time-dependent [84]: while higher neutrophil counts are protective against early tuberculosis infection [85], a pathogenic role of neutrophils during the late stages of tuberculosis has been proposed [86]. Thus, whereas pharmacological inhibition of PI3Kγ may be a suitable strategy to inhibit inflammation and limit lung damage in chronic and early-stage lung diseases, it might raise concerns in acute and late-stage infections, where it could result in an impaired host defense against high bacterial burden.
–
Influenza
The role of PI3Kγ in the context of viral infections has been studied in Kaposi’s sarcoma-associated herpes virus-induced tumors, where PI3Kγ is required for the viral oncogenic signaling [87]. On the other hand, PI3Kγ is also important in the regulation of innate immune responses, as well as establishment and resolution of inflammation upon influenza infection. Influenza A (IAV) and B viruses are among the most common causes of acute respiratory diseases of viral origin, accounting for three to five million cases of severe infection and up to 650,000 deaths/year worldwide [88]. In particular, the clinical manifestation of IAV infection, a highly pathogenic strain, is characterized by an excessive inflammatory response leading to lung damage [89].
–
Response to IAV infection can be conceptually divided in three stages, which however occur simultaneously through the course of the injury [90]. First, the immune response against the influenza virus is initiated by release of type-I and type-III interferons (IFNs), mainly produced by epithelial cells, which are primarily targeted by IAV, and by dendritic cells. Second, the innate immune system (natural killer (NK) cells, macrophages, and neutrophils), is rapidly recruited to the airways by cytokines and chemokines. Finally, specificity and memory are provided by T cells. PI3Kγ plays a crucial role in all these responses, driving production of type-I and type-III IFNs, as well as recruitment of NK and CD8+ T cells, and ultimately controlling viral titers in the infected lungs.
–
PI3Kγ has a pivotal role in the recruitment and survival of macrophages and neutrophils [91][92], which, however, when excessively activated, might be harmful to the host by leading to lung damage [93][94]. Recently, PI3Kγ has been shown to be essential after IAV infection for the control of recruitment and survival of innate immune cells and for resolution of inflammation [95]. In fact, in PI3Kγ KO mice infected by IAV, the increased production of pro-inflammatory cytokines and the accumulation of activated neutrophils in the lungs contribute to lung damage and enhanced lethality. Moreover, PI3Kγ controls leukocyte survival and resolution of inflammation, as shown by the reduced number of resolving macrophages and lower IL-10 levels in PI3Kγ KO mice infected with IAV. Keeping with that, during IAV infection, this unbalance towards pro-inflammatory signals, to the detriment of pro-resolving signals, finally results in increased lung injury in PI3Kγ KO mice.
–
Recently, the contribution of PI3Kγ in regulating priming of CD8+ T cells by resident dendritic cells and NK/lymphocyte migration toward chemokine stimuli in PI3Kγ KD/KD (knockdown) [96] and PI3Kγ KO mice [45], respectively, has been shown to contribute to the enhanced susceptibility to IAV infection.
–
Consistent with these findings, PI3Kγ orchestrates the antiviral immunity and inflammatory magnitude in response to IAV by distinct mechanisms. Therefore, targeting PI3Kγ may not be useful to treat IAV infection, possibly leading to decreased control of the infection, but might be an important diagnostic marker of disease severity. Nevertheless, the contribution of the scaffold and kinase activity of PI3Kγ have not been dissected in this context, and attention should be paid to the fact that PI3Kγ KO and KD/KD mice do not necessarily have overlapping phenotypes, as previously suggested in the heart [8][97].
–
Moreover, the analysis of single-nucleotide polymorphisms (SNPs) on PIK3CG gene might be exploited for prognosis. Different genetic polymorphisms on genes encoding for host factors have been investigated to explain the heterogeneity of immune responses to influenza infection and disease outcomes [98]. SNPs on PIK3CG, or close to the gene, have been studied in genetic association studies in a number of diseases, like cardiovascular disease [99], epinephrine-induced aggregation [100] and HDL-cholesterol plasma levels [101]. Importantly, SNPs located in PIK3CG gene (rs17847825 and rs2230460) have been associated with disease protection in influenza A(H1N1)pdm09-infected patients [95], thus suggesting the possible use of PI3Kγ as a clinical prognostic factor.
CONCLUSIONS
Following the initial characterization of PI3Kγ [102][103] and the patenting of the first PI3Kγ-selective inhibitor by Novartis in 2003 for the treatment of respiratory diseases [104], drug discovery efforts in the last decade have validated the value of PI3Kγ as a promising therapeutic target, especially for inflammatory disease (for a chronological review of the patented synthetic PI3Kγ inhibitor chemotypes see [15] and [105]).
–
In particular, pharmacological targeting of PI3Kγ may be effective in the regulation of the immune system, and therefore in the control of airway diseases driven by an excessive inflammatory response. On the other hand, possible side effects can be expected upon long-term treatment or in the co-occurrence of infections, as highlighted by preclinical work [81]. Similarly, infections have been observed as a significant side effect of the dual PI3Kγδ inhibitor Duvelisib [106]. However, whether and how PI3Kγ selective inhibitors predispose to infections is still unknown, as only one compound selectivity targeting PI3Kγ, IPI-549 from Infinity Pharmaceuticals, has initiated clinical development so far, though as an anti-cancer agent [105]. Therefore, it cannot be excluded that inhibitors targeting PI3Kγ catalytic activity may have opposite effects in the lungs, and only clinical trials will define the nature and the extent of a therapeutic window for these drugs in pulmonary diseases, compared to the more advanced dual PI3Kγδ inhibitors.
–
Among these, several compounds have reached clinical trial for respiratory diseases (Table 2). For example, Duvelisib was a candidate for mild asthma, but further development in non-oncologic diseases has been stopped, as the primary end point (changes in maximum allergen-induced Forced Expiratory Volume 1 decrease) was missed in clinical trials. Significant effects were seen, however, on secondary end points, but at a dose potentially leading to serious adverse reactions [107]. Other dual γδ inhibitors that reached clinical development for respiratory diseases (like asthma and chronic obstructive pulmonary disease (COPD)) include RV1729 and RV6153, from RespiVert, and AZD8154, from AstraZeneca (Table 2). Possibly, an inhalation-based delivery of PI3Kγδ (and PI3Kγ) inhibitors could help reducing, if not overcoming, any systemic adverse effect, though impairing the response to respiratory pathogens. In line with this approach, Chiesi Farmaceutici has started a clinical study to investigate the safety, tolerability and pharmacokinetics of the inhaled CHF6523 PI3K inhibitor. As an exploratory assessment, the anti-inflammatory effect of CHF6523 on sputum and blood biomarkers in COPD subjects will be evaluated (Table 2).
–
TABLE 2. Clinical development of PI3Kγδ inhibitors for respiratory diseases. |
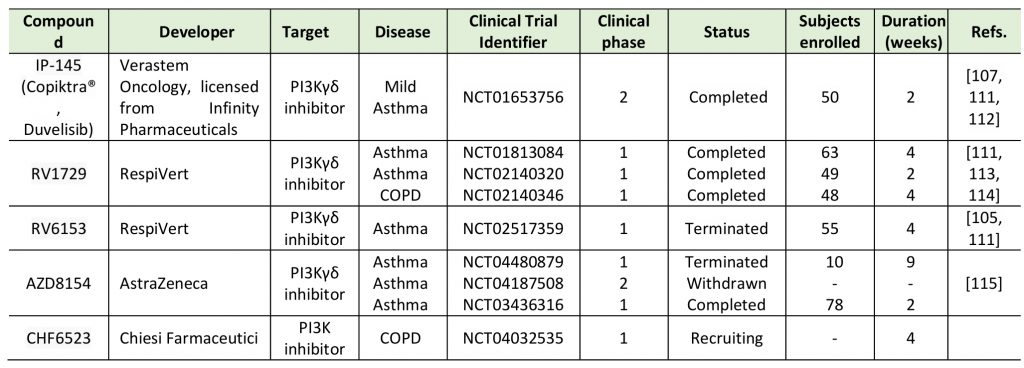 |
| [105][107][111][112][113][114][115] |
–
Moreover, considering that class IB isoforms can cooperate with class IA and IB PI3Ks in controlling, downstream signaling events, dual inhibition may be desirable to achieve a relevant therapeutic effect [15]. On the other hand, from a safety perspective, a high isoform selectivity is required, especially toward PI3Kα and β, which made development of PI3Kγ inhibitors difficult, due to the high similarity between isoform sequences. Only recently, new classes of increasingly more specific inhibitors have been generated to block PI3Kγ kinase activity [108][109][110]. However, this approach may not discriminate between the two PI3Kγ heteromeric variants, that share the same catalytic p110γ subunit combined to different regulatory subunits, which hypothetically exert distinct biological functions [27].
–
Nonetheless, PI3Kγ is a multifunctional protein, which is not only involved in the modulation of the Akt/mTOR pathway through its catalytic action, but also in the inhibition of cAMP as a scaffold protein. As cAMP elevation in lungs triggers bronchodilation and anti-inflammatory responses, better definition of the protein-protein interactions driving PI3Kγ-mediated cAMP modulation might open the way to novel therapeutic options in airway diseases.
–
Despite efforts in developing PI3Kγ inhibitors in the last decades, only one compound, the dual γδ inhibitor Duvelisib, has received approval, and its application is limited to oncological malignancies. Therefore, a more profound understanding of the biological role of PI3Kγ variants as well as of the impact of its non-catalytic functions in signal transduction is needed in order to foster new tools, and expand fields of intervention for PI3Kγ targeting.
REFERENCES
- Burke JE, Williams RL (2015). Synergy in activating class I PI3Ks. Trends Biochem Sci 40(2): 88-100. 10.1016/j.tibs.2014.12.003
- Vanhaesebroeck B (2006). Charging the batteries to heal wounds through PI3K. Nat Chem Biol 2(9): 453-455. 10.1038/nchembio0906-453
- Crackower MA, Oudit GY, Kozieradzki I, Sarao R, Sun H, Sasaki T, Hirsch E, Suzuki A, Shioi T, Irie-Sasaki J, Sah R, Cheng HY, Rybin VO, Lembo G, Fratta L, Oliveira-dos-Santos AJ, Benovic JL, Kahn CR, Izumo S, Steinberg SF, Wymann MP, Backx PH, Penninger JM (2002). Regulation of myocardial contractility and cell size by distinct PI3K-PTEN signaling pathways. Cell 110(6): 737-749. 10.1016/s0092-8674(02)00969-8
- Harris SJ, Ciuclan L, Finan PM, Wymann MP, Walker C, Westwick J, Ward SG, Thomas MJ (2012). Genetic ablation of PI3Kγ results in defective IL-17RA signalling in T lymphocytes and increased IL-17 levels. Eur J Immunol 42(12): 3394-3404. 10.1002/eji.201242463
- Li M, Sala V, De Santis MC, Cimino J, Cappello P, Pianca N, Di Bona A, Margaria JP, Martini M, Lazzarini E, Pirozzi F, Rossi L, Franco I, Bornbaum J, Heger J, Rohrbach S, Perino A, Tocchetti CG, Lima BHF, Teixeira MM, Porporato PE, Schulz R, Angelini A, Sandri M, Ameri P, Sciarretta S, Lima-Júnior RCP, Mongillo M, Zaglia T, Morello F, et al. (2018). Phosphoinositide 3-Kinase Gamma Inhibition Protects From Anthracycline Cardiotoxicity and Reduces Tumor Growth. Circulation 138(7): 696-711. 10.1161/circulationaha.117.030352
- Naga Prasad SV, Esposito G, Mao L, Koch WJ, Rockman HA (2000). Gbetagamma-dependent phosphoinositide 3-kinase activation in hearts with in vivo pressure overload hypertrophy. J Biol Chem 275(7): 4693-4698. 10.1074/jbc.275.7.4693
- Patrucco E, Notte A, Barberis L, Selvetella G, Maffei A, Brancaccio M, Marengo S, Russo G, Azzolino O, Rybalkin SD, Silengo L, Altruda F, Wetzker R, Wymann MP, Lembo G, Hirsch E (2004). PI3Kgamma modulates the cardiac response to chronic pressure overload by distinct kinase-dependent and -independent effects. Cell 118(3): 375-387. 10.1016/j.cell.2004.07.017
- Perino A, Ghigo A, Ferrero E, Morello F, Santulli G, Baillie GS, Damilano F, Dunlop AJ, Pawson C, Walser R, Levi R, Altruda F, Silengo L, Langeberg LK, Neubauer G, Heymans S, Lembo G, Wymann MP, Wetzker R, Houslay MD, Iaccarino G, Scott JD, Hirsch E (2011). Integrating cardiac PIP3 and cAMP signaling through a PKA anchoring function of p110γ. Mol Cell 42(1): 84-95. 10.1016/j.molcel.2011.01.030
- Perrino C, Schroder JN, Lima B, Villamizar N, Nienaber JJ, Milano CA, Naga Prasad SV (2007). Dynamic regulation of phosphoinositide 3-kinase-gamma activity and beta-adrenergic receptor trafficking in end-stage human heart failure. Circulation 116(22): 2571-2579. 10.1161/circulationaha.107.706515
- Yu Q, Li W, Jin R, Yu S, Xie D, Zheng X, Zhong W, Cheng X, Hu S, Li M, Zheng Q, Li G, Song Z (2019). PI3Kgamma (Phosphoinositide 3-Kinase gamma) Regulates Vascular Smooth Muscle Cell Phenotypic Modulation and Neointimal Formation Through CREB (Cyclic AMP-Response Element Binding Protein)/YAP (Yes-Associated Protein) Signaling. Arterioscler Thromb Vasc Biol 39(3): e91-e105. 10.1161/ATVBAHA.118.312212
- Jin R, Yu S, Song Z, Quillin JW, Deasis DP, Penninger JM, Nanda A, Granger DN, Li G (2010). Phosphoinositide 3-kinase-gamma expression is upregulated in brain microglia and contributes to ischemia-induced microglial activation in acute experimental stroke. Biochem Biophys Res Commun 399(3): 458-464. 10.1016/j.bbrc.2010.07.116
- Sasaki T, Irie-Sasaki J, Jones RG, Oliveira-dos-Santos AJ, Stanford WL, Bolon B, Wakeham A, Itie A, Bouchard D, Kozieradzki I, Joza N, Mak TW, Ohashi PS, Suzuki A, Penninger JM (2000). Function of PI3Kgamma in thymocyte development, T cell activation, and neutrophil migration. Science 287(5455): 1040-1046. 10.1126/science.287.5455.1040.
- Takeda AJ, Maher TJ, Zhang Y, Lanahan SM, Bucklin ML, Compton SR, Tyler PM, Comrie WA, Matsuda M, Olivier KN, Pittaluga S, McElwee JJ, Long Priel DA, Kuhns DB, Williams RL, Mustillo PJ, Wymann MP, Koneti Rao V, Lucas CL (2019). Human PI3Kgamma deficiency and its microbiota-dependent mouse model reveal immunodeficiency and tissue immunopathology. Nat Commun 10(1): 4364. 10.1038/s41467-019-12311-5
- Madeddu P, Kraenkel N, Barcelos LS, Siragusa M, Campagnolo P, Oikawa A, Caporali A, Herman A, Azzolino O, Barberis L, Perino A, Damilano F, Emanueli C, Hirsch E (2008). Phosphoinositide 3-kinase gamma gene knockout impairs postischemic neovascularization and endothelial progenitor cell functions. Arterioscler Thromb Vasc Biol 28(1): 68-76. 10.1161/atvbaha.107.145573
- Ruckle T, Schwarz MK, Rommel C (2006). PI3Kgamma inhibition: towards an ‘aspirin of the 21st century’? Nat Rev Drug Discov 5(11): 903-918. 10.1038/nrd2145
- Lionetti V, Lisi A, Patrucco E, De Giuli P, Milazzo MG, Ceci S, Wymann M, Lena A, Gremigni V, Fanelli V, Hirsch E, Ranieri VM (2006). Lack of phosphoinositide 3-kinase-gamma attenuates ventilator-induced lung injury. Crit Care Med 34(1): 134-141. 10.1097/01.ccm.0000190909.70601.2c
- Montoro DT, Haber AL, Biton M, Vinarsky V, Lin B, Birket SE, Yuan F, Chen S, Leung HM, Villoria J, Rogel N, Burgin G, Tsankov AM, Waghray A, Slyper M, Waldman J, Nguyen L, Dionne D, Rozenblatt-Rosen O, Tata PR, Mou H, Shivaraju M, Bihler H, Mense M, Tearney GJ, Rowe SM, Engelhardt JF, Regev A, Rajagopal J (2018). A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 560(7718): 319-324. 10.1038/s41586-018-0393-7
- Ciraolo E, Iezzi M, Marone R, Marengo S, Curcio C, Costa C, Azzolino O, Gonella C, Rubinetto C, Wu H, Dastrù W, Martin EL, Silengo L, Altruda F, Turco E, Lanzetti L, Musiani P, Rückle T, Rommel C, Backer JM, Forni G, Wymann MP, Hirsch E (2008). Phosphoinositide 3-kinase p110beta activity: key role in metabolism and mammary gland cancer but not development. Sci Signal 1(36): ra3. 10.1126/scisignal.1161577
- Kurig B, Shymanets A, Bohnacker T, Prajwal, Brock C, Ahmadian MR, Schaefer M, Gohla A, Harteneck C, Wymann MP, Jeanclos E, Nürnberg B (2009). Ras is an indispensable coregulator of the class IB phosphoinositide 3-kinase p87/p110gamma. Proc Natl Acad Sci U S A 106(48): 20312-20317. 10.1073/pnas.0905506106
- Shymanets A, Prajwal, Bucher K, Beer-Hammer S, Harteneck C, Nürnberg B (2013). p87 and p101 subunits are distinct regulators determining class IB phosphoinositide 3-kinase (PI3K) specificity. J Biol Chem 288(43): 31059-31068. 10.1074/jbc.M113.508234
- Schmid MC, Avraamides CJ, Dippold HC, Franco I, Foubert P, Ellies LG, Acevedo LM, Manglicmot JR, Song X, Wrasidlo W, Blair SL, Ginsberg MH, Cheresh DA, Hirsch E, Field SJ, Varner JA (2011). Receptor tyrosine kinases and TLR/IL1Rs unexpectedly activate myeloid cell PI3kγ, a single convergent point promoting tumor inflammation and progression. Cancer Cell 19(6): 715-727. 10.1016/j.ccr.2011.04.016
- Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT (2017). The PI3K Pathway in Human Disease. Cell 170(4): 605-635. 10.1016/j.cell.2017.07.029
- Luo L, Wall AA, Tong SJ, Hung Y, Xiao Z, Tarique AA, Sly PD, Fantino E, Marzolo MP, Stow JL (2018). TLR Crosstalk Activates LRP1 to Recruit Rab8a and PI3Kγ for Suppression of Inflammatory Responses. Cell Rep 24(11): 3033-3044. 10.1016/j.celrep.2018.08.028
- Luo L, Wall AA, Yeo JC, Condon ND, Norwood SJ, Schoenwaelder S, Chen KW, Jackson S, Jenkins BJ, Hartland EL, Schroder K, Collins BM, Sweet MJ, Stow JL (2014). Rab8a interacts directly with PI3Kγ to modulate TLR4-driven PI3K and mTOR signalling. Nat Commun 5: 4407. 10.1038/ncomms5407
- Wall AA, Luo L, Hung Y, Tong SJ, Condon ND, Blumenthal A, Sweet MJ, Stow JL (2017). Small GTPase Rab8a-recruited Phosphatidylinositol 3-Kinase γ Regulates Signaling and Cytokine Outputs from Endosomal Toll-like Receptors. J Biol Chem 292(11): 4411-4422. 10.1074/jbc.M116.766337
- Laffargue M, Calvez R, Finan P, Trifilieff A, Barbier M, Altruda F, Hirsch E, Wymann MP (2002). Phosphoinositide 3-kinase gamma is an essential amplifier of mast cell function. Immunity 16(3): 441-451. 10.1016/s1074-7613(02)00282-0
- Nurnberg B, Beer-Hammer S (2019). Function, Regulation and Biological Roles of PI3Kgamma Variants. Biomolecules 9(9): 427. 10.3390/biom9090427.
- Wang T, Gross C, Desai AA, Zemskov E, Wu X, Garcia AN, Jacobson JR, Yuan JX, Garcia JG, Black SM (2017). Endothelial cell signaling and ventilator-induced lung injury: molecular mechanisms, genomic analyses, and therapeutic targets. Am J Physiol Lung Cell Mol Physiol 312(4): L452-L476. 10.1152/ajplung.00231.2016
- Choi WI, Quinn DA, Park KM, Moufarrej RK, Jafari B, Syrkina O, Bonventre JV, Hales CA (2003). Systemic microvascular leak in an in vivo rat model of ventilator-induced lung injury. Am J Respir Crit Care Med 167(12): 1627-1632. 10.1164/rccm.200210-1216OC
- Fanelli V, Puntorieri V, Assenzio B, Martin EL, Elia V, Bosco M, Delsedime L, Del Sorbo L, Ferrari A, Italiano S, Ghigo A, Slutsky AS, Hirsch E, Ranieri VM (2010). Pulmonary-derived phosphoinositide 3-kinase gamma (PI3Kgamma) contributes to ventilator-induced lung injury and edema. Intensive Care Med 36(11): 1935-1945. 10.1007/s00134-010-2018-y
- Sayner SL (2011). Emerging themes of cAMP regulation of the pulmonary endothelial barrier. Am J Physiol Lung Cell Mol Physiol 300(5): L667-678. 10.1152/ajplung.00433.2010
- Shi CS, Huang TH, Lin CK, Li JM, Chen MH, Tsai ML, Chang CC (2016). VEGF Production by Ly6C+high Monocytes Contributes to Ventilator-Induced Lung Injury. PLoS One 11(10): e0165317. 10.1371/journal.pone.0165317
- Aderem A, Ulevitch RJ (2000). Toll-like receptors in the induction of the innate immune response. Nature 406(6797): 782-787. 10.1038/35021228
- Perros F, Lambrecht BN, Hammad H (2011). TLR4 signalling in pulmonary stromal cells is critical for inflammation and immunity in the airways. Respir Res 12: 125. 10.1186/1465-9921-12-125
- Villar J, Cabrera NE, Casula M, Flores C, Valladares F, Diaz-Flores L, Muros M, Slutsky AS, Kacmarek RM (2010). Mechanical ventilation modulates TLR4 and IRAK-3 in a non-infectious, ventilator-induced lung injury model. Respir Res 11: 27. 10.1186/1465-9921-11-27
- Ding X, Tong Y, Jin S, Chen Z, Li T, Billiar TR, Pitt BR, Li Q, Zhang LM (2018). Mechanical ventilation enhances extrapulmonary sepsis-induced lung injury: role of WISP1-αvβ5 integrin pathway in TLR4-mediated inflammation and injury. Crit Care 22(1): 302. 10.1186/s13054-018-2237-0
- Hu G, Malik AB, Minshall RD (2010). Toll-like receptor 4 mediates neutrophil sequestration and lung injury induced by endotoxin and hyperinflation. Crit Care Med 38(1): 194-201. 10.1097/CCM.0b013e3181bc7c17
- Li H, Su X, Yan X, Wasserloos K, Chao W, Kaynar AM, Liu ZQ, Leikauf GD, Pitt BR, Zhang LM (2010). Toll-like receptor 4-myeloid differentiation factor 88 signaling contributes to ventilator-induced lung injury in mice. Anesthesiology 113(3): 619-629. 10.1097/ALN.0b013e3181e89ab2
- Vaneker M, Joosten LA, Heunks LM, Snijdelaar DG, Halbertsma FJ, van Egmond J, Netea MG, van der Hoeven JG, Scheffer GJ (2008). Low-tidal-volume mechanical ventilation induces a toll-like receptor 4-dependent inflammatory response in healthy mice. Anesthesiology 109(3): 465-472. 10.1097/ALN.0b013e318182aef1
- Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR, Lasky JA, Lynch DA, Ryu JH, Swigris JJ, Wells AU, Ancochea J, Bouros D, Carvalho C, Costabel U, Ebina M, Hansell DM, Johkoh T, Kim DS, King TE, Kondoh Y, Myers J, Müller NL, Nicholson AG, Richeldi L, Selman M, Dudden RF, et al. (2011). An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 183(6): 788-824. 10.1164/rccm.2009-040GL
- Hutchinson J, Fogarty A, Hubbard R, McKeever T (2015). Global incidence and mortality of idiopathic pulmonary fibrosis: a systematic review. Eur Respir J 46(3): 795-806. 10.1183/09031936.00185114
- Ley B, Collard HR, King TE (2011). Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 183(4): 431-440. 10.1164/rccm.201006-0894CI
- Idiopathic Pulmonary Fibrosis Clinical Research N, Martinez FJ, de Andrade JA, Anstrom KJ, King TE, Jr., Raghu G (2014). Randomized trial of acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med 370(22): 2093-2101. 10.1056/NEJMoa1401739
- Richeldi L, Collard HR, Jones MG (2017). Idiopathic pulmonary fibrosis. Lancet 389(10082): 1941-1952. 10.1016/S0140-6736(17)30866-8
- Garcia CK (2018). Insights from human genetic studies of lung and organ fibrosis. J Clin Invest 128(1): 36-44. 10.1172/jci93556
- Sauleda J, Núñez B, Sala E, Soriano JB (2018). Idiopathic Pulmonary Fibrosis: Epidemiology, Natural History, Phenotypes. Med Sci 6(4): 110. 10.3390/medsci6040110
- Le Cras TD, Korfhagen TR, Davidson C, Schmidt S, Fenchel M, Ikegami M, Whitsett JA, Hardie WD (2010). Inhibition of PI3K by PX-866 prevents transforming growth factor-alpha-induced pulmonary fibrosis. Am J Pathol 176(2): 679-686. 10.2353/ajpath.2010.090123
- Mercer PF, Woodcock HV, Eley JD, Platé M, Sulikowski MG, Durrenberger PF, Franklin L, Nanthakumar CB, Man Y, Genovese F, McAnulty RJ, Yang S, Maher TM, Nicholson AG, Blanchard AD, Marshall RP, Lukey PT, Chambers RC (2016). Exploration of a potent PI3 kinase/mTOR inhibitor as a novel anti-fibrotic agent in IPF. Thorax 71(8): 701-711. 10.1136/thoraxjnl-2015-207429
- Campa CC, Silva RL, Margaria JP, Pirali T, Mattos MS, Kraemer LR, Reis DC, Grosa G, Copperi F, Dalmarco EM, Lima-Junior RCP, Aprile S, Sala V, Dal Bello F, Prado DS, Alves-Filho JC, Medana C, Cassali GD, Tron GC, Teixeira MM, Ciraolo E, Russo RC, Hirsch E (2018). Inhalation of the prodrug PI3K inhibitor CL27c improves lung function in asthma and fibrosis. Nat Commun 9(1): 5232. 10.1038/s41467-018-07698-6
- Conte E, Gili E, Fruciano M, Korfei M, Fagone E, Iemmolo M, Lo Furno D, Giuffrida R, Crimi N, Guenther A, Vancheri C (2013). PI3K p110γ overexpression in idiopathic pulmonary fibrosis lung tissue and fibroblast cells: in vitro effects of its inhibition. Lab Invest 93(5): 566-576. 10.1038/labinvest.2013.6
- Russo RC, Garcia CC, Barcelos LS, Rachid MA, Guabiraba R, Roffê E, Souza AL, Sousa LP, Mirolo M, Doni A, Cassali GD, Pinho V, Locati M, Teixeira MM (2011). Phosphoinositide 3-kinase γ plays a critical role in bleomycin-induced pulmonary inflammation and fibrosis in mice. J Leukoc Biol 89(2): 269-282. 10.1189/jlb.0610346
- Sontake V, Gajjala PR, Kasam RK, Madala SK (2019). New therapeutics based on emerging concepts in pulmonary fibrosis. Expert Opin Ther Targets 23(1): 69-81. 10.1080/14728222.2019.1552262
- Sokai A, Tanizawa K, Handa T, Kanatani K, Kubo T, Ikezoe K, Nakatsuka Y, Tokuda S, Oga T, Hirai T, Nagai S, Chin K, Mishima M (2017). Importance of serial changes in biomarkers in idiopathic pulmonary fibrosis. ERJ Open Res 3(3): 00019-2016. 10.1183/23120541.00019-2016
- Zank DC, Bueno M, Mora AL, Rojas M (2018). Idiopathic Pulmonary Fibrosis: Aging, Mitochondrial Dysfunction, and Cellular Bioenergetics. Front Med 5: 10. 10.3389/fmed.2018.00010
- Doukas J, Eide L, Stebbins K, Racanelli-Layton A, Dellamary L, Martin M, Dneprovskaia E, Noronha G, Soll R, Wrasidlo W, Acevedo LM, Cheresh DA (2009). Aerosolized phosphoinositide 3-kinase gamma/delta inhibitor TG100-115 [3-[2,4-diamino-6-(3-hydroxyphenyl)pteridin-7-yl]phenol] as a therapeutic candidate for asthma and chronic obstructive pulmonary disease. J Pharmacol Exp Ther 328(3): 758-765. 10.1124/jpet.108.144311
- Lim DH, Cho JY, Song DJ, Lee SY, Miller M, Broide DH (2009). PI3K gamma-deficient mice have reduced levels of allergen-induced eosinophilic inflammation and airway remodeling. Am J Physiol Lung Cell Mol Physiol 296(2): L210-219. 10.1152/ajplung.90275.2008
- Takeda M, Ito W, Tanabe M, Ueki S, Kato H, Kihara J, Tanigai T, Chiba T, Yamaguchi K, Kayaba H, Imai Y, Okuyama K, Ohno I, Sasaki T, Chihara J (2009). Allergic airway hyperresponsiveness, inflammation, and remodeling do not develop in phosphoinositide 3-kinase gamma-deficient mice. J Allergy Clin Immunol 123(4): 805-812. 10.1016/j.jaci.2008.11.047
- Saito Y, Takeda M, Nishikawa J, Konno Y, Tamaki M, Itoga M, Kobayashi Y, Moritoki Y, Ito W, Chihara J, Ueki S (2014). The effect of pharmacological PI3Kgamma inhibitor on eotaxin-induced human eosinophil functions. Pulm Pharmacol Ther 27(2): 164-169. 10.1016/j.pupt.2013.11.006
- Singh AK, Pantouris G, Borosch S, Rojanasthien S, Cho TY (2017). Structural basis for decreased induction of class IB PI3-kinases expression by MIF inhibitors. J Cell Mol Med 21(1): 142-153. 10.1111/jcmm.12949
- Deshpande DA, Penn RB (2006). Targeting G protein-coupled receptor signaling in asthma. Cell Signal 18(12): 2105-2120. 10.1016/j.cellsig.2006.04.008
- Eum SY, Maghni K, Tolloczko B, Eidelman DH, Martin JG (2005). IL-13 may mediate allergen-induced hyperresponsiveness independently of IL-5 or eotaxin by effects on airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 288(3): L576-584. 10.1152/ajplung.00380.2003
- Laporte SA, Oakley RH, Holt JA, Barak LS, Caron MG (2000). The interaction of beta-arrestin with the AP-2 adaptor is required for the clustering of beta 2-adrenergic receptor into clathrin-coated pits. J Biol Chem 275(30): 23120-23126. 10.1074/jbc.M002581200
- Sathish V, Thompson MA, Bailey JP, Pabelick CM, Prakash YS, Sieck GC (2009). Effect of proinflammatory cytokines on regulation of sarcoplasmic reticulum Ca2+ reuptake in human airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 297(1): L26-34. 10.1152/ajplung.00026.2009
- Jiang H, Abel PW, Toews ML, Deng C, Casale TB, Xie Y, Tu Y (2010). Phosphoinositide 3-kinase gamma regulates airway smooth muscle contraction by modulating calcium oscillations. J Pharmacol Exp Ther 334(3): 703-709. 10.1124/jpet.110.168518
- Saha SK, Berry MA, Parker D, Siddiqui S, Morgan A, May R, Monk P, Bradding P, Wardlaw AJ, Pavord ID, Brightling CE (2008). Increased sputum and bronchial biopsy IL-13 expression in severe asthma. J Allergy Clin Immunol 121(3): 685-691. 10.1016/j.jaci.2008.01.005
- Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY, Karp CL, Donaldson DD (1998). Interleukin-13: central mediator of allergic asthma. Science 282(5397): 2258-2261. 10.1126/science.282.5397.2258
- Walter DM, McIntire JJ, Berry G, McKenzie AN, Donaldson DD, DeKruyff RH, Umetsu DT (2001). Critical role for IL-13 in the development of allergen-induced airway hyperreactivity. J Immunol 167(8): 4668-4675. 10.4049/jimmunol.167.8.4668.
- Marwick JA, Chung KF, Adcock IM (2010). Phosphatidylinositol 3-kinase isoforms as targets in respiratory disease. Ther Adv Respir Dis 4(1): 19-34. 10.1177/1753465809352792
- Elborn JS (2016). Cystic fibrosis. Lancet 388(10059): 2519-2531. 10.1016/s0140-6736(16)00576-6
- Sly PD, Gangell CL, Chen L, Ware RS, Ranganathan S, Mott LS, Murray CP, Stick SM, Investigators AC (2013). Risk factors for bronchiectasis in children with cystic fibrosis. N Engl J Med 368(21): 1963-1970. 10.1056/NEJMoa1301725
- Stick S, Tiddens H, Aurora P, Gustafsson P, Ranganathan S, Robinson P, Rosenfeld M, Sly P, Ratjen F (2013). Early intervention studies in infants and preschool children with cystic fibrosis: are we ready? Eur Respir J 42(2): 527-538. 10.1183/09031936.00108212
- Margaroli C, Tirouvanziam R (2016). Neutrophil plasticity enables the development of pathological microenvironments: implications for cystic fibrosis airway disease. Mol Cell Pediatr 3(1): 38. 10.1186/s40348-016-0066-2
- Galluzzo M, Ciraolo E, Lucattelli M, Hoxha E, Ulrich M, Campa CC, Lungarella G, Doring G, Zhou-Suckow Z, Mall M, Hirsch E, De Rose V (2015). Genetic Deletion and Pharmacological Inhibition of PI3K γ Reduces Neutrophilic Airway Inflammation and Lung Damage in Mice with Cystic Fibrosis-Like Lung Disease. Mediators Inflamm 2015(545417. 10.1155/2015/545417
- Lévêque M, Le Trionnaire S, Del Porto P, Martin-Chouly C (2017). The impact of impaired macrophage functions in cystic fibrosis disease progression. J Cyst Fibros 16(4): 443-453. 10.1016/j.jcf.2016.10.011
- Paemka L, McCullagh BN, Abou Alaiwa MH, Stoltz DA, Dong Q, Randak CO, Gray RD, McCray PB (2017). Monocyte derived macrophages from CF pigs exhibit increased inflammatory responses at birth. J Cyst Fibros 16(4): 471-474. 10.1016/j.jcf.2017.03.007
- Saini Y, Dang H, Livraghi-Butrico A, Kelly EJ, Jones LC, O’Neal WK, Boucher RC (2014). Gene expression in whole lung and pulmonary macrophages reflects the dynamic pathology associated with airway surface dehydration. BMC Genomics 15: 726. 10.1186/1471-2164-15-726
- Tarique AA, Sly PD, Holt PG, Bosco A, Ware RS, Logan J, Bell SC, Wainwright CE, Fantino E (2017). CFTR-dependent defect in alternatively-activated macrophages in cystic fibrosis. J Cyst Fibros 16(4): 475-482. 10.1016/j.jcf.2017.03.011
- Breasson L, Sardi C, Becattini B, Zani F, Solinas G (2018). PI3Kγ ablation does not promote diabetes in. FASEB J 32(1): 319-329. 10.1096/fj.201700372RR
- Ding X, Jin S, Tong Y, Jiang X, Chen Z, Mei S, Zhang L, Billiar TR, Li Q (2017). TLR4 signaling induces TLR3 up-regulation in alveolar macrophages during acute lung injury. Sci Rep 7: 34278. 10.1038/srep34278
- Katholnig K, Linke M, Pham H, Hengstschlager M, Weichhart T (2013). Immune responses of macrophages and dendritic cells regulated by mTOR signalling. Biochem Soc Trans 41(4): 927-933. 10.1042/BST20130032
- Maus UA, Backi M, Winter C, Srivastava M, Schwarz MK, Ruckle T, Paton JC, Briles D, Mack M, Welte T, Maus R, Bohle RM, Seeger W, Rommel C, Hirsch E, Lohmeyer J, Preissner KT (2007). Importance of phosphoinositide 3-kinase gamma in the host defense against pneumococcal infection. Am J Respir Crit Care Med 175(9): 958-966. 10.1164/rccm.200610-1533OC
- Cavalcanti-Neto MP, Prado RQ, Piñeros AR, Sérgio CA, Bertolini TB, Gembre AF, Ramos SG, Bonato VL (2018). Improvement of the resistance against early Mycobacterium tuberculosis-infection in the absence of PI3Kγ enzyme is associated with increase of CD4+IL-17+ cells and neutrophils. Tuberculosis 113: 1-9. 10.1016/j.tube.2018.08.009
- Bucher K, Schmitt F, Mothes B, Blumendeller C, Schäll D, Piekorz R, Hirsch E, Nürnberg B, Beer-Hammer S (2017). Deficiency of PI3-Kinase catalytic isoforms p110γ and p110δ in mice enhances the IL-17/G-CSF axis and induces neutrophilia. Cell Commun Signal 15(1): 28. 10.1186/s12964-017-0185-y
- Leisching GR (2018). Susceptibility to Tuberculosis Is Associated With PI3K-Dependent Increased Mobilization of Neutrophils. Front Immunol 9: 1669. 10.3389/fimmu.2018.01669
- Martineau AR, Newton SM, Wilkinson KA, Kampmann B, Hall BM, Nawroly N, Packe GE, Davidson RN, Griffiths CJ, Wilkinson RJ (2007). Neutrophil-mediated innate immune resistance to mycobacteria. J Clin Invest 117(7): 1988-1994. 10.1172/jci31097
- Philips JA (2017). Neutrophils: Double agents for TB. Sci Transl Med 9(394). 10.1126/scitranslmed.aan6195
- Martin D, Galisteo R, Molinolo AA, Wetzker R, Hirsch E, Gutkind JS (2011). PI3Kγ mediates kaposi’s sarcoma-associated herpesvirus vGPCR-induced sarcomagenesis. Cancer Cell 19(6): 805-813. 10.1016/j.ccr.2011.05.005
- Iuliano AD, Roguski KM, Chang HH, Muscatello DJ, Palekar R, Tempia S, Cohen C, Gran JM, Schanzer D, Cowling BJ, Wu P, Kyncl J, Ang LW, Park M, Redlberger-Fritz M, Yu H, Espenhain L, Krishnan A, Emukule G, van Asten L, Pereira da Silva S, Aungkulanon S, Buchholz U, Widdowson MA, Bresee JS, Network GSI-aMC (2018). Estimates of global seasonal influenza-associated respiratory mortality: a modelling study. Lancet 391(10127): 1285-1300. 10.1016/s0140-6736(17)33293-2
- Brandes M, Klauschen F, Kuchen S, Germain RN (2013). A systems analysis identifies a feedforward inflammatory circuit leading to lethal influenza infection. Cell 154(1): 197-212. 10.1016/j.cell.2013.06.013
- Herold S, Becker C, Ridge KM, Budinger GR (2015). Influenza virus-induced lung injury: pathogenesis and implications for treatment. Eur Respir J 45(5): 1463-1478. 10.1183/09031936.00186214
- Hirsch E, Katanaev VL, Garlanda C, Azzolino O, Pirola L, Silengo L, Sozzani S, Mantovani A, Altruda F, Wymann MP (2000). Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science 287(5455): 1049-1053. 10.1126/science.287.5455.1049
- Yang KY, Arcaroli J, Kupfner J, Pitts TM, Park JS, Strasshiem D, Perng RP, Abraham E (2003). Involvement of phosphatidylinositol 3-kinase gamma in neutrophil apoptosis. Cell Signal 15(2): 225-233. 10.1016/s0898-6568(02)00063-3
- Akaike T, Noguchi Y, Ijiri S, Setoguchi K, Suga M, Zheng YM, Dietzschold B, Maeda H (1996). Pathogenesis of influenza virus-induced pneumonia: involvement of both nitric oxide and oxygen radicals. Proc Natl Acad Sci U S A 93(6): 2448-2453. 10.1073/pnas.93.6.2448
- Narasaraju T, Yang E, Samy RP, Ng HH, Poh WP, Liew AA, Phoon MC, van Rooijen N, Chow VT (2011). Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am J Pathol 179(1): 199-210. 10.1016/j.ajpath.2011.03.013
- Garcia CC, Tavares LP, Dias ACF, Kehdy F, Alvarado-Arnez LE, Queiroz-Junior CM, Galvão I, Lima BH, Matos AR, Gonçalves APF, Soriani FM, Moraes MO, Marques JT, Siqueira MM, Machado AMV, Sousa LP, Russo RC, Teixeira MM (2018). Phosphatidyl Inositol 3 Kinase-Gamma Balances Antiviral and Inflammatory Responses During Influenza A H1N1 Infection: From Murine Model to Genetic Association in Patients. Front Immunol 9: 975. 10.3389/fimmu.2018.00975
- Nobs SP, Schneider C, Heer AK, Huotari J, Helenius A, Kopf M (2016). PI3Kγ Is Critical for Dendritic Cell-Mediated CD8+ T Cell Priming and Viral Clearance during Influenza Virus Infection. PLoS Pathog 12(3): e1005508. 10.1371/journal.ppat.1005508
- Ghigo A, Perino A, Mehel H, Zahradnikova A, Jr., Morello F, Leroy J, Nikolaev VO, Damilano F, Cimino J, De Luca E, Richter W, Westenbroek R, Catterall WA, Zhang J, Yan C, Conti M, Gomez AM, Vandecasteele G, Hirsch E, Fischmeister R (2012). Phosphoinositide 3-kinase gamma protects against catecholamine-induced ventricular arrhythmia through protein kinase A-mediated regulation of distinct phosphodiesterases. Circulation 126(17): 2073-2083. 10.1161/CIRCULATIONAHA.112.114074
- To KK, Zhou J, Chan JF, Yuen KY (2015). Host genes and influenza pathogenesis in humans: an emerging paradigm. Curr Opin Virol 14: 7-15. 10.1016/j.coviro.2015.04.010
- Wain LV, Verwoert GC, O’Reilly PF, Shi G, Johnson T, Johnson AD, Bochud M, Rice KM, Henneman P, Smith AV, Ehret GB, Amin N, Larson MG, Mooser V, Hadley D, Dörr M, Bis JC, Aspelund T, Esko T, Janssens AC, Zhao JH, Heath S, Laan M, Fu J, Pistis G, Luan J, Arora P, Lucas G, Pirastu N, Pichler I, et al. (2011). Genome-wide association study identifies six new loci influencing pulse pressure and mean arterial pressure. Nat Genet 43(10): 1005-1011. 10.1038/ng.922
- Johnson AD, Yanek LR, Chen MH, Faraday N, Larson MG, Tofler G, Lin SJ, Kraja AT, Province MA, Yang Q, Becker DM, O’Donnell CJ, Becker LC (2010). Genome-wide meta-analyses identifies seven loci associated with platelet aggregation in response to agonists. Nat Genet 42(7): 608-613. 10.1038/ng.604
- Kächele M, Hennige AM, Machann J, Hieronimus A, Lamprinou A, Machicao F, Schick F, Fritsche A, Stefan N, Nürnberg B, Häring HU, Staiger H (2015). Variation in the Phosphoinositide 3-Kinase Gamma Gene Affects Plasma HDL-Cholesterol without Modification of Metabolic or Inflammatory Markers. PLoS One 10(12): e0144494. 10.1371/journal.pone.0144494
- Stephens L, Smrcka A, Cooke FT, Jackson TR, Sternweis PC, Hawkins PT (1994). A novel phosphoinositide 3 kinase activity in myeloid-derived cells is activated by G protein beta gamma subunits. Cell 77(1): 83-93. 10.1016/0092-8674(94)90237-2
- Stoyanov B, Volinia S, Hanck T, Rubio I, Loubtchenkov M, Malek D, Stoyanova S, Vanhaesebroeck B, Dhand R, Nurnberg B, et al. (1995). Cloning and characterization of a G protein-activated human phosphoinositide-3 kinase. Science 269(5224): 690-693. 10.1126/science.7624799
- Bruce I, F. P., Leblanc C, Mccarthy C, Whitehead L, Blair NE, Bloomfield GC, Hayler J, Kirman L, Oza MS, Shukla L (2003). 5-Phenylthiazole derivatives and their use as phosphatidylinositol 3-kinase (PI3K) inhibitors for the treatment of allergic and inflammatory diseases. WO-03072557.
- Perry MWD, Abdulai R, Mogemark M, Petersen J, Thomas MJ, Valastro B, Westin Eriksson A (2019). Evolution of PI3Kgamma and delta Inhibitors for Inflammatory and Autoimmune Diseases. J Med Chem 62(10): 4783-4814. 10.1021/acs.jmedchem.8b01298
- Horwitz SM, Koch R, Porcu P, Oki Y, Moskowitz A, Perez M, Myskowski P, Officer A, Jaffe JD, Morrow SN, Allen K, Douglas M, Stern H, Sweeney J, Kelly P, Kelly V, Aster JC, Weaver D, Foss FM, Weinstock DM (2018). Activity of the PI3K-delta, gamma inhibitor duvelisib in a phase 1 trial and preclinical models of T-cell lymphoma. Blood 131(8): 888-898. 10.1182/blood-2017-08-802470
- Schmalbach T, Fuhr R, Albayaty M, Allen K, Douglas M, Dunbar J, McLaughlin J, Alexander L, McKee C (2015). Duvelisib, a PI3K-δ,γ inhibitor, in subjects with mild asthma. Eur Respir J 46(suppl 59): PA2122. 10.1183/13993003.congress-2015.PA2122
- Bandarage UK, Aronov AM, Cao J, Come JH, Cottrell KM, Davies RJ, Giroux S, Jacobs M, Mahajan S, Messersmith D, Moody CS, Swett R, Xu J (2021). Discovery of a Novel Series of Potent and Selective Alkynylthiazole-Derived PI3Kgamma Inhibitors. ACS Med Chem Lett 12(1): 129-135. 10.1021/acsmedchemlett.0c00573
- Drew SL, Thomas-Tran R, Beatty JW, Fournier J, Lawson KV, Miles DH, Mata G, Sharif EU, Yan X, Mailyan AK, Ginn E, Chen J, Wong K, Soni D, Dhanota P, Chen PY, Shaqfeh SG, Meleza C, Pham AT, Chen A, Zhao X, Banuelos J, Jin L, Schindler U, Walters MJ, Young SW, Walker NP, Leleti MR, Powers JP, Jeffrey JL (2020). Discovery of Potent and Selective PI3Kgamma Inhibitors. J Med Chem 63(19): 11235-11257. 10.1021/acs.jmedchem.0c01203
- Miles DH, Yan X, Thomas-Tran R, Fournier J, Sharif EU, Drew SL, Mata G, Lawson KV, Ginn E, Wong K, Soni D, Dhanota P, Shaqfeh SG, Meleza C, Chen A, Pham AT, Park T, Swinarski D, Banuelos J, Schindler U, Walters MJ, Walker NP, Zhao X, Young SW, Chen J, Jin L, Leleti MR, Powers JP, Jeffrey JL (2020). Discovery of Potent and Selective 7-Azaindole Isoindolinone-Based PI3Kgamma Inhibitors. ACS Med Chem Lett 11(11): 2244-2252. 10.1021/acsmedchemlett.0c00387
- Harding SD, Faccenda E, Southan C, Pawson AJ, Maffia P, Alexander SPH, Davenport AP, Fabbro D, Levi-Schaffer F, Spedding M, Davies JA (2020). The IUPHAR Guide to Immunopharmacology: connecting immunology and pharmacology. Immunology 160(1): 10-23. 10.1111/imm.13175
- Winkler DG, Faia KL, DiNitto JP, Ali JA, White KF, Brophy EE, Pink MM, Proctor JL, Lussier J, Martin CM, Hoyt JG, Tillotson B, Murphy EL, Lim AR, Thomas BD, Macdougall JR, Ren P, Liu Y, Li LS, Jessen KA, Fritz CC, Dunbar JL, Porter JR, Rommel C, Palombella VJ, Changelian PS, Kutok JL (2013). PI3K-delta and PI3K-gamma inhibition by IPI-145 abrogates immune responses and suppresses activity in autoimmune and inflammatory disease models. Chem Biol 20(11): 1364-1374. 10.1016/j.chembiol.2013.09.017
- Mardh CK, Root J, Uddin M, Stenvall K, Malmgren A, Karabelas K, Thomas M (2017). Targets of Neutrophil Influx and Weaponry: Therapeutic Opportunities for Chronic Obstructive Airway Disease. J Immunol Res 2017: 5273201. 10.1155/2017/5273201
- Norman P (2014). Evaluation of WO2013136076: two crystalline forms of the phosphatidylinositol 3-kinase-delta inhibitor RV-1729. Expert Opin Ther Pat 24(4): 471-475. 10.1517/13543776.2014.865725
- Sadiq MW, Asimus S, Kristensson C, Hagberg A, Mäenpää J, Valastro B, Fuhr R, Koernicke T, Keen C, Brailsford W, Betts J (2019). Safety, tolerability and pharmacokinetics (PK) of AZD8154 (a selective PI3K?d inhibitor) after single ascending inhaled doses in healthy volunteers. Eur Respir J 54(suppl 63): PA4220. 10.1183/13993003.congress-2019.PA4220
- Thomas MJ, Smith A, Head DH, Milne L, Nicholls A, Pearce W, Vanhaesebroeck B, Wymann MP, Hirsch E, Trifilieff A, Walker C, Finan P, Westwick J (2005). Airway inflammation: chemokine-induced neutrophilia and the class I phosphoinositide 3-kinases. Eur J Immunol 35(4): 1283-1291. 10.1002/eji.200425634
- Puri KD, Doggett TA, Huang CY, Douangpanya J, Hayflick JS, Turner M, Penninger J, Diacovo TG (2005). The role of endothelial PI3Kgamma activity in neutrophil trafficking. Blood 106(1): 150-157. 10.1182/blood-2005-01-0023
- Yum HK, Arcaroli J, Kupfner J, Shenkar R, Penninger JM, Sasaki T, Yang KY, Park JS, Abraham E (2001). Involvement of phosphoinositide 3-kinases in neutrophil activation and the development of acute lung injury. J Immunol 167(11): 6601-6608. 10.4049/jimmunol.167.11.6601
- Miyahara T, Hamanaka K, Weber DS, Drake DA, Anghelescu M, Parker JC (2007). Phosphoinositide 3-kinase, Src, and Akt modulate acute ventilation-induced vascular permeability increases in mouse lungs. Am J Physiol Lung Cell Mol Physiol 293(1): L11-21. 10.1152/ajplung.00279.2005
- Ong E, Gao XP, Predescu D, Broman M, Malik AB (2005). Role of phosphatidylinositol 3-kinase-gamma in mediating lung neutrophil sequestration and vascular injury induced by E. coli sepsis. Am J Physiol Lung Cell Mol Physiol 289(6): L1094-1103. 10.1152/ajplung.00179.2005
–
ACKNOWLEDGMENTS
This work was supported by the Italian Cystic Fibrosis Research Foundation (FFC#25/2014, FFC#23/2015 and FFC#8/2018 to EH, FFC#4/2016 and FFC#11/2017 to AG), Compagnia di Sanpaolo (CSTO161109 to EH), Cariplo Foundation (#2015-0880 to AG and #2018-0498 to EH), Roche Foundation (Bando Roche per la Ricer-ca 2019 to AG).
COPYRIGHT
© 2021

Roles of phosphatidyl inositol 3 kinase gamma (PI3Kγ) in respiratory diseases by Sala et al. is licensed under a Creative Commons Attribution 4.0 International License.


