Reviews:
Cell Stress, Vol. 3, No. 8, pp. 240 - 266; doi: 10.15698/cst2019.08.194
Immunometabolic cross-talk in the inflamed heart

1 William Harvey Research Institute, Queen Mary University of London, Charterhouse Square, London EC1M 6BQ, United Kingdom.
2 School of Biological and Chemical Sciences, Queen Mary University of London, G.E. Fogg Building, Mile End Road, London E1 4NS, United Kingdom.
3 Centre for Inflammation and Therapeutic Innovation, Queen Mary University of London, Charterhouse Square, London EC1M 6BQ, United Kingdom.
Keywords: heart, inflammation, metabolism, innate immune response, adaptive immune responses.
Abbreviatons:
AMPK – AMP-activated protein kinase,
ATP – adenosine triphosphate,
CAD – coronary artery disease,
CK – creatine kinase,
CMR – cardiac magnetic resonance,
CRP – C-reactive protein,
DAG – diacylglycerol,
DC – dendritic cell,
DCM – dilated cardiomyopathy,
ERR – estrogen-related receptor,
ETC – electron transport chain,
FA – fatty acid,
FFA – free FA,
GC – germinal center,
HFpEF – HF with preserved ejection fraction,
HIF – hypoxia¬inducible factor,
IL – interleukin,
LDH – lactate dehydrogenase,
LV – left ventricular,
LPS – lipopolysaccharide,
MCT – monocarboxylate transporter,
MI – myocardial infarction,
MIF – migration inhibitory factor,
mTOR – mammalian target of rapamycin,
NK – natural killer,
OXPHOS – oxidative phosphorylation,
PARP – poly-(ADP-ribose)-polymerase,
PCr – phosphocreatine,
PDH – pyruvate dehydrogenase,
PFK – phosphofructokinase,
PKC – protein kinase C,
PPAR – peroxisome proliferator¬activated receptor,
ROS – reactive oxygen species,
T1D – type 1 diabetes,
T2D – type 2 diabetes,
TAG – triacylglycerol,
TCR – T cell receptor,
TNF – tumor necrosis factor,
Treg – regulatory T cell,
TTA – tetradecythioacetic acid.
Received originally: 22/01/2019 Received in revised form: 02/05/2019
Accepted: 03/05/2019
Published: 07/06/2019
Correspondence:
Federica M. Marelli-Berg, William Harvey Research Institute, Queen Mary University of London, Charterhouse Square, London EC1M 6BQ, United Kingdom; f.marelli-berg@qmul.ac.uk
Dunja Aksentijevic, William Harvey Research Institute, Queen Mary University of London, Charterhouse Square, London EC1M 6BQ, United Kingdom; d.aksentijevic@qmul.ac.uk
Conflict of interest statement: None.
Please cite this article as: Federica M. Marelli-Berg and Dunja Aksentijevic (2019). Immunometabolic cross-talk in the inflamed heart. Cell Stress 3(8): 240-266. doi: 10.15698/cst2019.08.194
Abstract
Inflammatory processes underlie many diseases associated with injury of the heart muscle, including conditions without an obvious inflammatory pathogenic component such as hypertensive and diabetic cardiomyopathy. Persistence of cardiac inflammation can cause irreversible structural and functional deficits. Some are induced by direct damage of the heart muscle by cellular and soluble mediators but also by metabolic adaptations sustained by the inflammatory microenvironment. It is well established that both cardiomyocytes and immune cells undergo metabolic reprogramming in the site of inflammation, which allow them to deal with decreased availability of nutrients and oxygen. However, like in cancer, competition for nutrients and increased production of signalling metabolites such as lactate initiate a metabolic cross-talk between immune cells and cardiomyocytes which, we propose, might tip the balance between resolution of the inflammation versus adverse cardiac remodeling. Here we review our current understanding of the metabolic reprogramming of both heart tissue and immune cells during inflammation, and we discuss potential key mechanisms by which these metabolic responses intersect and influence each other and ultimately define the prognosis of the inflammatory process in the heart.
INTRODUCTION
Metabolic reprogramming of immune cells during tissue inflammation has been intensely investigated as reviewed elsewhere [1][2][3][4][5]. Notably, three key concepts have emerged from these studies. First, the effector functions of immune cells depend on metabolic reprogramming in response to activation. For example, resting immune cells utilize energetically efficient processes such as the Kreb's cycle linked to the generation of adenosine triphosphate (ATP) via oxidative phosphorylation (OXPHOS) [2] and shift to aerobic glycolysis in response to inflammatory stimuli [6]. Second, the metabolic status of the whole body has been shown to affect functional and metabolic responses of both immune cells and organs. For example, the microbiome and metabolites produced by commensal bacteria are known to affect immune cells as well as organ homeostasis [7] and systemic metabolic diseases such as diabetes and obesity can adversely impact immunity and the function of a variety of organs [7]. Finally, the metabolic adaptation of immune cells is strongly affected by the microenvironment of the inflammatory site, in terms of nutrient availability, oxygen tension and the production of signalling metabolites by the diseased tissue itself. Cancer is paradigmatic of this effect, whereby aberrant metabolic responses by immune cells in response to the tumour microenvironment result in the induction of immune paralysis and cancer survival and expansion [8]. Similar observations have been made in the context of chronic inflammation [9].
–
While much is known about the metabolic configuration of immune cells in homeostasis and inflammation as well as the effects of systemic metabolism on immune cell function, the metabolic cross-talk between immune cells and organ parenchymal cells has been a challenging issue to address. This is mainly due to the complexity of defining the relative contribution of different cellular components to the metabolic microenvironment on effector immunity and vice versa. Unravelling the influence of the tissue microenvironment on the cross-talk between immune and parenchymal cells during inflammation leading to tissue damage and organ failure is vital to identify prognostic biomarkers and novel targets for therapeutic intervention. A vast amount of reported observations has associated metabolic syndrome – a heart-extrinsic factor – with heart functional impairment as well as systemic sub-clinical chronic inflammation, pointing to metabolic dysfunction as a key determinant of inflammatory heart disease.
–
In this review, we summarize key concepts in metabolic adaptation in the heart and immune cells in steady-state and inflammation. We highlight cardiac inflammation as a condition in which well-defined systemic and cellular components and metabolic pathways can provide a new paradigm to understand the reciprocal regulation of immune cell and tissue metabolism in inflammatory diseases.
CAVEATS AND DEFINITIONS: WHAT IS INFLAMMATORY DISEASE OF THE HEART MUSCLE?
An enormous amount of data is available on the link between metabolic syndrome, inflammation and atherosclerosis, including coronary artery disease (CAD). A number of clinical trials has examined the effects of anti-inflammatory therapy on these conditions, with mixed outcomes as their interpretation is hugely varying [10][11][12]. This topic has been extensively reviewed elsewhere [10][11][12]. While CAD can indirectly affect cardiac function and metabolism, this review focuses on the direct effects of systemic inflammation on heart muscle inflammation (myocarditis), function and metabolism. Atherosclerosis is an inflammatory disease of the vasculature which impacts both cardiac function and energetics, which is however distinct from inflammation of the heart muscle itself. Myocardium inflammation is commonly associated with an acute viral infection of the heart which normally resolves but can sometimes persist as an autoimmune condition, leading to heart muscle dysfunction and ultimately, heart failure (HF). Myocarditis is notoriously difficult to diagnose, and its viral aetiology appears to account for only a fraction of clinically diagnosed heart inflammation. A key concept recently pioneered by Heymans and colleagues is that besides direct inflammatory responses in the heart, such as those detected in viral myocarditis, genetic predisposition in association with environmental factors can initiate and sustain cardiac inflammation leading to heart failure [13]. About 40% of acute HF is diagnosed as idiopathic/inflammatory dilated cardiomyopathy (iDMC). This alternative definition is not necessarily based on histological evidence, as endomyocardial biopsy has limited sensitivity [14]. Overall, persisting cardiac inflammation appears to be a feature of a large proportion of heart conditions beyond viral acute myocarditis. Typical examples include ischemic cardiomyopathy and HF with preserved ejection fraction (HFpEF) and, more recently fulminant myocarditis as a serious adverse reaction of cancer therapies that enhance T cell responses [15]. Although treatment has dramatically improved survival post-myocardial infarction (MI), more than 20% of patients subsequently develop cardiomyopathy and HF. Ischemia-induced cardiomyocyte death activates an inflammatory response that serves to clear the injured myocardium from dead cells, and stimulates repair.
–
However, cardiomyocyte necrosis is also a powerful initiator of autoimmune inflammation of the heart leading to HF (reviewed in [16]). Interleukin-1(IL-1) α and RNA released by necrotic cardiomyocytes are key danger signals that trigger the inflammatory response following MI (reviewed in [17]). IL-1 induces a proinflammatory phenotype in leukocytes and fibroblasts, and delays myofibroblast transdifferentiation. Anti-inflammatory regulatory T cells (Tregs) exert negative regulation of the inflammatory response post-MI by modulating macrophage and fibroblast phenotype. Cardiac macrophages exhibit significant heterogeneity and phenotypic plasticity and may orchestrate the reparative response following infarction [17]. The persistence of inflammation can cause immune-mediated tissue damage [16] and progressive structural changes such as left ventricular (LV) remodeling and functional impairment [18][19][20]. Collectively, these studies reveal a complex cross-talk between cells of the immune system and the myocardial parenchyma, which likely determines the fate of the inflammatory process. Similarly, systemic inflammation, and in particular that induced by metabolic syndrome, can drive disease of the myocardium. For example, there is rising consensus that HFpEF is primarily driven by inflammation induced by ageing and related comorbidities, including obesity and diabetes [21][22]. In line, therapeutic interventions with anti-inflammatory properties have been also shown to protect from experimental HFpEF [23]. A link between systemic inflammation and cardiac has also been reported by studies correlating systemic inflammation markers (ie. cytokines) with well-known cardiac arrhythmias including atrial fibrillation and ventricular tachycardia [24]. Despite this evidence, diagnostic and prognostic biomarkers of cardiac inflammation and, importantly, mechanistic studies are missing. We propose that disturbances in the metabolic configuration of the heart muscle, with its lifetime of varying workload and energy demand, are induced by direct and indirect cross-talk with inflammatory immune cells via both nutrient and oxygen competition as well as direct signals by cytokines and metabolites. If correct, this hypothesis would justify therapeutic immune targeting in heart inflammatory disease, a much controversial approach.
–
A major obstacle to resolve controversy is the lack of clear markers of heart inflammation. Endomyocardial biopsy (EMB), the gold standard for myocarditis diagnosis, includes risks of severe complications (1.5%) and has notoriously poor diagnostic sensitivity (as low as 18% for single, 43% for five biopsies). Cardiac magnetic resonance (CMR) is a powerful tool for detection of acute cardiac inflammation but has limited diagnostic accuracy for chronic inflammatory disease. Critically, CMR cannot identify cause or type of inflammatory infiltrate, features predicting prognosis.
–
C-reactive protein (CRP) measurement can certainly indicate the presence of an inflammatory process but gives little insight on its location, pathogenesis and stage. Until a breakthrough in the pathophysiology of cardiac inflammation diagnosis is achieved, CRP remains the only, albeit not ideal pathologic cue.
–
Is it possible that metabolic parameters might reveal more to the clinician than conventional inflammation cues? Reviews of recent trials targeting inflammation in CAD have argued that immunometabolic correction with statins is superior and less prone to severe side effects attached to direct immunomodulation [12]. We have also proposed that modulation of systemic and cellular metabolism might provide an optimal strategy to reduce organ inflammatory disease [7]. To this aim, a systematic analysis of the immunometabolic cross-talk between parenchymal cells and immune cells is paramount to design effective therapeutic approaches.
–
Here, we summarize the physiological metabolism of heart muscle and provide example of its alteration in paradigmatic heart diseases with a known metabolic/inflammatory pathogenesis. In parallel, we will review key concepts in inflammatory immunometabolism and its potential impact on tissue metabolic steady state and adaptation.
METABOLIC FLEXIBILITY OF THE HEALTHY HEART
The heart is predominantly an aerobic organ and relies on the oxidation of exogenous substrates, such as free fatty acids (FFA), glucose, lactate, ketone bodies, and some amino acids, to generate ATP, the major source of energy. The process of metabolic substrate selection is dynamic and depends largely on substrate availability, O2 concentration, and myocardial workload [25][26]. The heart has an enormous ATP demand— with 2% of its total ATP reserves consumed per beat, it turns over its total ATP pool in less than one min and utilizes six kg of ATP daily [27][28][29]. Fine control of mitochondrial and cytosolic ATP-generating pathways is critical to meet the energy demands of cardiac muscle [25]. Supply must be matched to demand as failure to provide an adequate amount of ATP causes a decrease in cellular free energy leading to functional decline. Under normal physiological conditions, more than 90% of ATP is generated in mitochondria [29]. Enormous myocardial ATP demand is primarily related to energy-dependent processes driving excitation-contraction (EC) coupling [30] (Figure 1). About 70–75% of total intracellular ATP is used for force generation powering work output, with the remaining 25–30% used for basal metabolism [25].
–
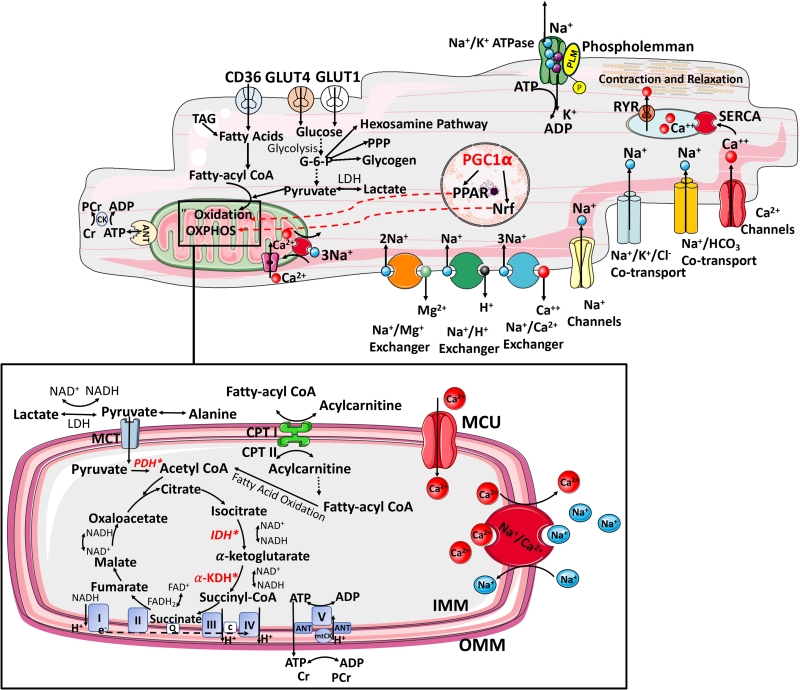 |
FIGURE 1: Interplay between ATP supply and excitation-contraction coupling in the healthy heart. The delivery of metabolic substrates, their selection and uptake are followed by OXPHOS. It involves electron shuttling from cytosolic to mitochondrial reducing equivalents, transfer of energy by electrons from reducing equivalents to ETC complexes and generation of electrochemical proton (H+) gradient within the mitochondrial intermembrane space (respiratory complexes I, II, II, III, IV). The release of H+ gradient is coupled to the synthesis of ATP from ADP + Pi by F0,F1-ATPase (complex V), contributing >95% of ATP synthesis under aerobic conditions. The final stage of myocardial ATP supply (phosphotransfer) involves delivery of ATP from mitochondria to sites of use. This involves ADP–ATP exchange across the inner mitochondrial membrane by the adenine nucleotide transporter (ANT) and propagation of local ATP/ADP disequilibria primarily by the creatine kinase (CK) [25]. TAG, triacylglycerol; PCr, phosphocreatine; ANT, adenine nucleotide transporter; GLUT, glucose transporter; CD36, fatty acid transporter; PPP, pentose phosphate pathway; LDH, lactate dehydrogenase; PDH, pyruvate dehydrogenase; CPT, carnitine palmitoyltransferase; CACT, carnitine–acylcarnitine translocase; MCU, mitochondrial calcium uniporter; α-KDH, α-ketoglutarate dehydrogenase; IDH, isocitrate dehydrogenase; mitoCK, mitochondrial creatine kinase; IMM, inner mitochondrial membrane; OMM, outer mitochondrial membrane; Q, quinone pool; c, cytochrome c; MPC, mitochondrial pyruvate carrier; e−, electrons; *Mitochondrial calcium-sensitive dehydrogenases (pyruvate dehydrogenase, isocitrate dehydrogenase and α-ketoglutarate dehydrogenase) RYR0 ryanodine receptor, SERCA- sarcoendoplasmic reticulum Ca2+ ATPase. |
–
To synthesize the ATP required to support normal function, the adult heart converts chemical energy primarily stored in FFAs (60–90%) and pyruvate (derived from glucose and lactate 10–40%) into mechanical energy for contraction [31]. There are three principal stages of myocardial ATP supply. The first stage includes metabolic substrate delivery, their selection, uptake and oxidation to generate acetyl-CoA for Kreb's cycle entry. The second stage, OXPHOS, consists of electron shuttling from cytosolic to mitochondrial reducing equivalents (primarily via the malate-aspartate shuttle), transfer of energy by electrons from reducing equivalents to O2 [via electron transport chain (ETC) complexes], and generation of an electrochemical proton gradient within the mitochondrial intermembrane space (by complexes I, III, and IV). The release of this gradient is coupled to the synthesis of ATP from ADP by F0F1-ATPase (complex V), accounting for 95% of ATP synthesis under aerobic conditions (Figure 1). The third stage is phosphotransfer referring to the delivery of ATP from mitochondria to sites of use (Figure 1) [29]. Phosphotransfer involves ADP-ATP exchange across the inner mitochondrial membrane by the adenine nucleotide transporter (ANT) and propagation of local ATP/ADP disequilibria by the creatine kinase shuttle, and to lesser extent by adenylate kinase [32] (Figure 1). However, cardiac workload varies constantly, including several-fold increase in cardiac output during exercise, thus requiring rapid and continuous matching of ATP supply to demand. This renders the heart a metabolic omnivore, giving it a high degree of substrate flexibility to rapidly switch substrate preference and utilization [31].This is also reflected in the high proportion (>30%) of cardiomyocyte volume occupied by mitochondria and mandated by the low stored ATP content, sufficient to power only ten cardiac cycles, and including phosphotransfer buffer systems, a further 20 cardiac cycles [33].
–
In order to avoid energetic inefficiency and “waste”, myocardial ATP supply is optimized via the Randle (glucose–fatty acid) cycle which regulates the opposing relationship between carbohydrate and FFA metabolism [34]. FFA enter the cytosol via metabolic transporters, such as FA translocase/CD36 (FAT/CD36), plasma membrane FA binding protein (FABPpm), and FA transport proteins (FATP1 and 6) [35] (Figure 1). In response to stimuli, such as increased insulin or activation of AMP-activated protein kinase (AMPK), FAT/CD36 translocates from intracellular vesicles to the sarcolemma to increase the uptake of FFA [36]. Upon entry into the cytosol, the non-esterified FA are esterified to fatty acyl-CoA. Depending on myocardial demand, fatty acyl-CoA is either stored in the myocardial lipid pool or enters the mitochondria for β-oxidation via the carnitine shuttle carnitine palmitoyl transferase-1 (CPT-1), the rate-limiting enzyme for mitochondrial uptake of FA [35] (Figure 1).
–
FFA oxidation triggers an increase in mitochondrial ratios of [acetyl-CoA]/[CoA] and [NADH]/[NAD+], both of which inhibit the activity of pyruvate dehydrogenase (PDH) complex. Ketone bodies, metabolic products of FFA oxidation in the liver, can also be metabolised to acetyl-CoA for entry into Kreb's cycle [37] (Figure 1).
–
Myocardial glucose uptake is facilitated by metabolic transporters, principally by the glucose transporter 1 (GLUT1, insulin-independent) and insulin-dependent GLUT4 [38]. Similar to FAT/CD36, in response to stimuli glucose transporters also ‘shuttle' between intracellular vesicles and the sarcolemma. Intracellular glucose is immediately phosphorylated by hexokinase to glucose-6-phosphate, which enters glycolysis, glycogenesis, the pentose phosphate pathway, or the hexosamine biosynthetic pathway (Figure 1). Glycolysis generates a small amount of ATP independent of O2 availability, and is regulated mainly by phosphofructokinase, which is inhibited by cytosolic citrate from the Kreb's cycle. Cytosolic citrate is also the major precursor of malonyl-CoA, which inhibits CPT-1 [39]. In normoxia, the end product of glycolysis is pyruvate, which enters mitochondria for oxidation. In hypoxia, pyruvate is reduced to lactate in the cytosol. Mitochondrial PDH is the key enzyme governing the oxidative decarboxylation of pyruvate to acetyl-CoA (Figure 1). Lactate, readily extracted from the bloodstream, can be converted to pyruvate in the cytosol and further metabolized to acetyl-CoA for ATP generation [37]. Arising as the common end product from the oxidation of a variety of substrates, acetyl-CoA enters the Kreb's cycle to produce NADH and FADH2, which donate electrons to the ETC thereby creating the proton electrochemical gradient needed to generate ATP. FFA oxidation generates more ATP compared to glucose, but at the expense of greater O2 consumption. Therefore, low O2 availability drives more metabolically efficient glucose oxidation [31].
–
By controlling intracellular concentrations of ADP and creatine, the creatine kinase (CK)-mediated phosphotransfer system stimulates both phosphotransfer and OXPHOS flux. This mechanism becomes particularly important at high workloads. Limitations in CK capacity would therefore be expected to limit myocardial contractile reserve at high workloads, assuming O2 supply is not limiting. Unlike skeletal muscle, the heart demonstrates stable time-averaged NADH/NAD+ as well as concentrations of phosphocreatine and ATP over a wide range of workloads.[40]. A study in the reperfused heart has shown that return of contractile function correlates with ATP turnover, not with [ATP] [41].
–
Regulation of cardiac metabolism
Long-term metabolic substrate utilization is governed by the activity of transcription factors that increase or suppress the expression of the key metabolic enzymes. In this context, a pivotal role is played by the peroxisome proliferatoractivated receptors (PPARs), a superfamily of nuclear receptors [42] (Figure 1). PPARα is the member of this family that is most abundantly expressed in the myocardium, where it upregulates the transcription of genes related to FA uptake and oxidation. Similarly, estrogen-related receptorα (ERRα; also known as ESRRA) and ERRγ (also known as ESRRG) target a wide set of genes related to the uptake of metabolic substrates, ATP translocation across mitochondrial membranes, and calcium handling [43] (Figure 1). PPAR activation requires coactivation with PPARγ coactivator 1α (PGC1α) or PGC1β, essential regulators of mitochondrial biogenesis [44], and the binding of PPARs to intermediates of lipid metabolism [45]. Lipid ligands inducing PPARα–PGC1α activation mainly derive from lipolysis of the intracellular triacylglycerol (TAG) pool [46].
–
ATP sensing in the heart
AMPK is a stress-activated kinase that functions as a cellular fuel gauge and a critical metabolic regulator. The activity of AMPK is primarily determined by the cellular energy state, reflected in the ratio of AMP (and ADP) to ATP. Activation of AMPK requires the phosphorylation of its α subunit at Thr172 by two upstream kinases, liver Kinase B1 (LKB1) and Ca2+/calmodulin-dependent kinase kinase (CaMKK)-β [47]. Once activated, AMPK inhibits various anabolic pathways, including protein synthesis via its action on both mTOR/p70S6K and eEF2 pathways and enhances catabolic pathways, to restore energetic balance required for cell survival [48]. Myocardial AMPK regulates genes related to mitochondrial energy metabolism, including medium chain acyl-CoA dehydrogenase (MCAD), CPT-1, cytochrome C, and uncoupling protein (UCP)-3 [49]. These transcriptional effects are mediated partly by activation of ERRα transcription factor [49]. Furthermore, AMPK regulates peroxisome proliferator activated receptor gamma co-activator (PGC)-1α, a critical modulator of cardiac gene expression and mitochondrial biogenesis. Activated AMPK increases the expression of PGC-1α in hypoxic cardiomyocytes [50]. AMPK phosphorylation of acetyl-CoA carboxylase (ACC2) inhibits its activity and the synthesis of malonyl-CoA, a potent inhibitor of carnitine palmitoyltransferase-1 (CPT-1). The decrease in malonyl-CoA levels, relieves the inhibition of CPT-1, the rate-limiting step for heart fatty acid oxidation [51].
–
AMPK also stimulates glucose transport and glycolysis. It increases GLUT4 translocation to the sarcolemma by phosphorylating Rab GTPase-activating proteins (GAP) that regulate Rab10, modulators of docking and fusion of GLUT4 vesicles with the plasma membrane [52]. Furthermore, AMPK decreases the endocytosis of GLUT4 leading to increased sarcolemma GLUT4 content and glucose uptake [53]. AMPK acts downstream to glucose transport, by indirectly increasing the activity of phosphofructokinase (PFK)-1, the rate-limiting enzyme in glycolysis. Activated AMPK directly phosphorylates and stimulates PFK-2 to synthesize fructose 2,6-bisphosphate, which in turn allosterically activates PFK-1 [54]. These increases in myocardial glucose transport and glycolysis are important components of the metabolic response to ischemia or hypoxia [55].
MYOCARDIAL METABOLIC ADAPTION TO STRESS
Myocardial inflammation has emerged as a pathophysiologic process contributing to cardiac hypertrophy, fibrosis and dysfunction in context of heart disease [18][56][57]. Accumulating evidence also suggests that myocardial inflammation is also implicated in the development of diabetic cardiomyopathy [58][59] Several pathological insults can trigger myocardial inflammation which initially represents an adaptive mechanism against stress [18][56][57] but becomes maladaptive if stress stimulus persists. Various pathological stressors directly induce the secretion of cytokines, chemokines [interleukin (IL8), monocyte chemoattractant protein-1 (MCP-1)] and adhesion molecules (vascular adhesion molecule 1, intracellular adhesion molecule-1 (ICAM-1)] in cardiomyocytes, fibroblasts and endothelial cells that promote myocardial recruitment of monocytes and lymphocytes [18][56][57][58]. However, clear distinction exists between chronic myocardial inflammation and the myocardial inflammation associated with viral myocarditis. Systemic inflammation is subclinical and contributes to the development of cardiac abnormalities in the long term. In contrast, cardiac metabolism can be affected by local inflammatory processes – such as in myocarditis and MI leading to adverse remodeling and HF in rapid manner. Here we summarize two paradigmatic examples of cardiac metabolic adaptation in common conditions of systemic inflammation.
–
Metabolism of the failing heart
HF imposes an enormous, worldwide clinical and economic burden, with its increasing prevalence due to progressive ageing of the general population. First identified in the early 20th century, and now a well-established energy starvation hypothesis, it is proposed that maladaptive metabolic remodelling precedes, initiates and maintains adverse contractile dysfunction in hypertrophy and HF [29][30]. Advances in analytical technologies have improved our insights into the “engine out of fuel” metabolic HF phenomenon and helped to classify metabolic alterations leading to myocardial energy starvation. HF is accompanied by derangements of all three fundamental steps of energy metabolism: substrate uptake and utilization, OXPHOS, and energetics [29]. Using in vivio 31P nuclear magnetic resonance (NMR), Neubauer and colleagues [29] found that the myocardial phosphocreatine-to-ATP ratio (PCr:ATP) can be used as a reliable prognostic indicator of dilated cardiomyopathy (DCM) where 44% of DCM patients with a PCr:ATP of <1.6 died of cardiovascular causes vs. 5% with a PCr:ATP of >1.6. There have also been numerous preclinical studies as well as clinical data inferring mitochondrial respiratory impairment (complex activities and/or altered expression of the ETC complexes, ATP synthase and adenine nucleotide translocase) in hypertrophy and HF [60][61][62].
–
The size and number of mitochondria are altered in the failing myocardium. There is evidence of mitochondrial misalignment, disorganized cristae, reduced density, membrane disruption and aggregation [63]. Reactive oxygen species (ROS) production from dysfunctional mitochondrial electron transport chain ETC/ATP synthesis intensifies oxidative damage of proteins, lipids and DNA, in a vicious amplifying cycle of mitochondrial dysfunction and ROS production, leading to cardiomyocyte loss [63]. Many studies have explored the relationship between decompensated cardiac remodelling and cardiac substrate utilization. Whereas the question of whether FFA utilization is decreased [64][65][66][67] or unchanged [68][69] during compensated hypertrophy remains subject to debate, the majority of studies reported that the development of overt cardiac dysfunction is accompanied by reduced FFA oxidation [70][71] (Figure 2A). This metabolic shift was confirmed in multiple animal models of HF, including ischaemic [72] and pacing-induced [73] HF, and was supported by clinical studies reporting a reduced FFA oxidative capacity in HF [74][75].
–
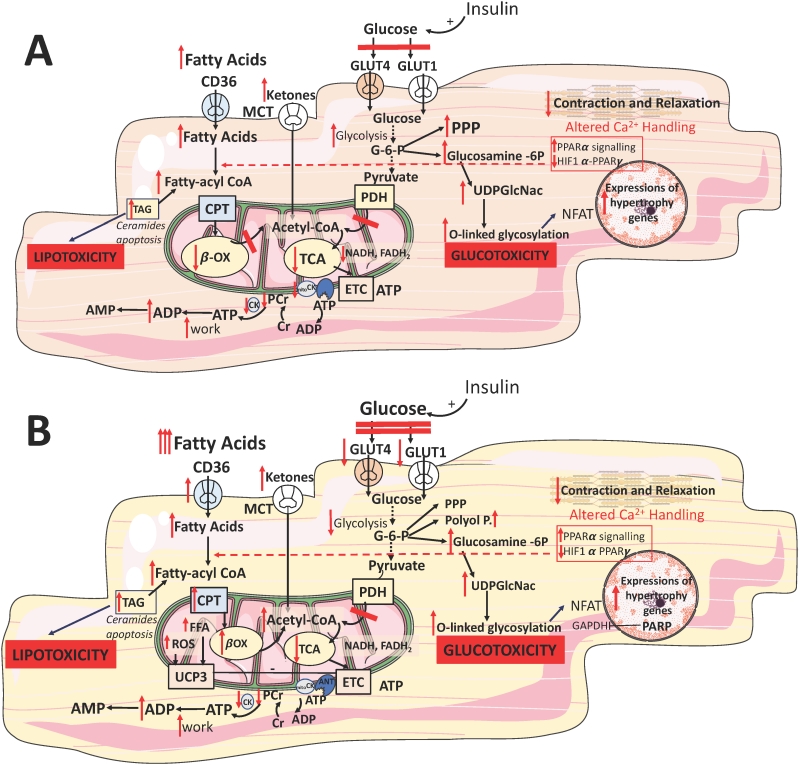 | FIGURE 2: Myocardial metabolic adaptation to stress: failing heart (A) and diabetic heart (B). Abbreviations: TAG - triacylglycerol, CPT - carnitine palmitoyl transferase, G6P- glucose 6 phosphate, ETC - electron transport chain, CK - creatine kinase, CD36- fatty acid transporter, Glucosamine 6P - glucosamine 6-phosphate, PPP - pentose phosphate pathway, GLUT - glucose transporter, MCT - monocarboxylate transporter, PDH - pyruvate dehydrogenase, TCA - Kreb's cycle, βOX - beta oxidation, PCr - phosphocreatine, Cr - creatine, NFAT - Nuclear factor of activated T-cells, PPAR - Peroxisome proliferator-activated receptor, ROS - reactive oxygen species, polyol p-polyol pathway. Hypertrophy genes: Class II HDACs, GATA, NFAT, MEF |
–
Reduced FFA oxidation might be explained at least in part by suppression of PPARα signaling [76][77] and possibly activation of the hypoxiainducible factor 1α (HIF1α)–PPARγ signalling axis [78], which impairs mitochondrial FFA transport and downregulates the expression of enzymes for FFA oxidation [79] (Figure 2A). However, a metabolic switch away from FFA oxidation is not accompanied by a decrease in myocardial FFA uptake (Figure 2A). Paradoxically, FFA plasma levels are increased in the advanced stages of HF, potentially due to sympathetic activation and consequent increased delivery of FFA to cardiac myocytes [80]. The mismatch between FFA uptake and oxidation leads to intracellular accumulation of lipids [78][81], which are partly stored as TAGs (Figure 2A), but are also chanelled into non-catabolic pathways, generating toxic lipids, such as ceramide and diacylglycerol (DAG; Figure 2A). This lipotoxicity leads to mitochondrial dysfunction and apoptosis and might contribute to the progression of HF [82][83]. Furthermore, intracellular accumulation of FFAs contributes to the development of insulin resistance by inducing posttranslational modifications of several components of the insulin signalling cascade [84][85]. In patients with decompensated HF, mechanical unloading by LV assist device implantation reduced lipid levels and restored myocardial insulin signaling [86]. Thus, collectively there is enough evidence to indicate that oversupply of substrates contributes to the progression of cardiac dysfunction, and insulin resistance has been proposed as the beneficial myocardial mechanism protecting it from the detrimental effects of fuel overload [87].
–
HF is characterized by a perturbed glucose metabolism: an increase in glucose uptake and glycolytic rates is not accompanied by a concomitant increase in glucose oxidation [71][88][89] (Figure 2A). This mismatch between increased glycolytic activity has been observed in the experimental models of cardiac pressure overload [65][69] and a study in patients with end stage HF [90]. It could be caused by multiple factors: increased activity of the ratelimiting glycolytic enzyme ATPdependent 6phosphofructokinase (PFK1) [91], unchanged or decreased pyruvate oxidation in mitochondria [92], impaired glucose and lactate oxidation related to defective mitochondrial oxidative metabolism [93][94] and myocardial insulin resistance [93][95] (Figure 2A). Therefore, although the relative contribution of glucose oxidation to ATP production increases in HF, the absolute substrate flux through glucose oxidative pathways actually decreases. Furthermore, in the late stages of HF cardiac glucose uptake is impaired as a consequence of decreased insulin sensitivity of the myocardium, thereby further reducing the availability of glucose for ATP synthesis [96].
–
Although a shift towards glucose oxidation in end stage HF could be beneficial given its higher metabolic efficiency than FFA oxidation, it is unlikely to compensate fully for the decrease in FFA utilization. An additional compensatory mechanism in cardiac hypertrophy is anaplerosis — the use of alternative oxidative pathways to generate Krebs cycle intermediates independent of acetyl-CoA [89]. Carboxylation of pyruvate to malate is a major anaplerotic reaction, which might partly account for the mismatch between glycolytic flux and glucose oxidation in mitochondria [89][97]. However, anaplerotic reactions are energetically less efficient than pyruvate oxidation because they bypass oxidative decarboxylation and other ATP-producing stages of the Kreb's cycle [70].
–
Two reports have shown an upregulation of enzymes and metabolic intermediates involved in ketone body oxidation in animal models [98] and patients with end stage HF [74], indicating that ketone bodies might become a more relevant energy source in this setting (Figure 2A). Ketone bodies are not readily available from food, but produced in the liver by incomplete oxidation of FFA released from adipose tissue in response to fasting. Because the ATP production:oxygen consumption (P:O) ratio of the ketone body β-hydroxybutyrate (2.50) is higher than that of FFA palmitate (2.33), βhydroxybutyrate was proposed as the ‘super fuel' [99] and that stimulating ketone body over FFA oxidation might be an adaptation to increase myocardial metabolic efficiency. However, this hypothesis is the subject to the same caveat affecting the general myocardial energy starvation model — it assumes that an increase in ATP production efficiency would ameliorate cardiac dysfunction [42]. Consequently, the question of whether an increased utilization of ketone bodies is an adaptive or maladaptive response remains unanswered.
–
ATP produced by OXPHOS needs to be transmitted to cellular regions of high ATP demand. Inside mitochondria, the phosphate bond of ATP is transferred to creatine by the mitochondrial creatine kinase (mitoCK) to form phosphocreatine (PCr), which rapidly diffuses to the cytosol, where the phosphoryl group is transferred back to ADP by the cytosolic CK (Figure 2A) [32]. Thus, the CR-mediated shuttle operates as ATP buffer, preventing an increase in cytosolic ADP by promptly regenerating ATP. These optimized control mechanisms maintain stable levels of high energy phosphates even during haemodynamic challenges to the heart [29]. Many studies using 31P-MRS have reported reduced PCr:ATP ratios in patients with HF [100], predicting LV dysfunction and poor outcomes [100]. Strong evidence from the literature indicates that HF is associated with severe impairment of the CK system (Figure 2A), which affects the ATP delivery to the contractile apparatus and ion pumps. However, the results from the mouse models of creatine/CK deficiency challenge the idea that PCr is essential to sustain cardiac function in response to pathological stressors [101]. Thus, the causative role of the CK-mediated phosphotransfer defect in the pathogenesis of HF is still subject to debate.
–
A number of intracellular signalling pathways have been implicated in the aetiology and regulation of cardiac hypertrophy and HF (reviewed in [102]) including the AMPK-mediated pathway. In chronic pressure overload-induced cardiac hypertrophy, the AMPK activity was shown to increase [103], suggesting that AMPK activation could be necessary compensatory consequence of pressure-overload. This increase in AMPK activity has been suggested to be a major contributor to enhanced glucose uptake in hypertrophy [103], which may be a beneficial metabolic adaptation for an energy-starved heart. However, in contrast, increased AMPK activity in the hypertrophic heart may also contribute to the loss of metabolic flexibility and progression to failure by accelerating FFA oxidation at the expense of glucose oxidation. This would uncouple glycolysis from glucose oxidation, resulting in increased proton production. Increase in proton production is detrimental to the metabolically stressed heart. It leads to accelerated Ca2+ overload due to increased Na+/H+ and Na+/Ca2+ exchange activities. Subsequently, coupled with alterations in Ca2+ handling, larger amounts of ATP are required for the maintenance of intracellular ion gradients thus rendering hypertrophied heart more inefficient by depriving it of valuable ATP needed for the restoration of mechanical function. This may explain why the hypertrophied hearts are more susceptible to ischaemia–reperfusion injury [104][105]. However, the most recent evidence suggests that AMPK activation counteracts cardiac hypertrophy by reducing O-GlcNAcylation of proteins such as troponin T [48].
–
Inflammation and metabolic insufficiency in heart failure – the missing link
In most patients with chronic HF, circulating cytokines are elevated and their levels correlate with the severity of HF and prognosis [106][107]. Experimental models of HF suggest that pro-inflammatory mediators play an important role in the development and progression of HF [63][108]. However, many trials of anti-inflammatory therapy for patients with HF have shown neutral or negative effects on outcomes [108].
–
Impact of cytokines on cardiac function and remodelling in failing heart has been well documented (reviewed in [57][109]), however, there are very few studies examining the impact on cardiac metabolism. The link between HF and inflammation was first recognized in by Levine et al. [110] who reported elevated levels of tumour necrosis factor (TNF) in HF patients with a reduced ejection fraction (EF). Sustained increases in TNF-α have been related to ischaemic myocardial injury, cardiac hypertrophy, and chronic HF. Spontaneously hypertensive rats show increased myocardial TNF-α production, which contributes to remodelling, decreased cardiac function, and faster progression to HF [111]. Likewise, the failing human heart produces large amounts of TNF-α [112], while it has been proposed that persistent intra-cardiac expression of TNF-α contributes to the development of cardiac allograft hypertrophy [113]. Ubiquitous inducible factor named nuclear factor-κB (NF-κB) controls activation of NF-κB itself is involved in various cardiovascular diseases, such as cardiac hypertrophy and HF [113]. Increased TNF-α levels reduce PGC-1α and pyruvate dehydrogenase kinase (PDK4) expression in human cardiac AC16 cells in vitro as well as in heart of TNF1.6 mice, a murine model of cardiac-specific TNF-α overexpression and cytokine-induced cardiomyopathy [112][114]. A recent in vitro study has shown that the p65 subunit of NF-κB directly represses PGC-1α activity in cultured cardiac cells, thereby leading to a reduction in PDK4 expression and the subsequent increase in glucose oxidation observed during the proinflammatory states such as chronic ischaemia, cardiac hypertrophy, and HF [115].
–
Metabolic remodeling in diabetic cardiomyopathy
Diabetes is a risk factor for cardiovascular mortality and cardiac remodeling with specific changes to myocardial metabolism, energetics, structure, and function. Diabetic cardiomyopathy is a distinct cardiomyopathy, independent of ischaemia or hypertension, describing the direct effects of systemic diabetes-linked metabolic alterations on myocardial function [116]. Metabolically, diabetes is characterized by rapid defective (type 1 diabetes, T1D) or gradual impairment (type 2 diabetes, T2D) of insulin secretion, leading to increased extracellular glucose and greater reliance on fatty acid oxidation. In both T1D and T2D, failure of insulin to suppress hormone sensitive lipase in adipose tissue and very low-density lipoprotein secretion in the liver increases circulating FFAs [35]. This, in turn, activates PPARα, a transcription factor that upregulates FFA metabolism while decreasing GLUT4, resulting in systemic hyperglycaemia [117]. Early in T2D, the primary problem, the lack of response to insulin in peripheral organs, is over-compensated by increased insulin secretion, resulting in hyperinsulinaemia [118]. Hyperinsulinaemia may be prolonged and may cause a gradual loss of pancreatic function, resulting in hypoinsulinaemia and hyperglycaemia [37]. There is a long-standing idea that insulin resistance and systemic adiposity increase the risk of cardiovascular (CV) events, however a new school of thought is emerging that defines myocardial insulin resistance as a defense against glucotoxicity and oxidative stress [87][119].
–
The systemic gradual impairment of insulin production and signalling in diabetes is associated with increased myocardial FFA uptake whilst mitochondrial FFA uptake and oxidation is reduced (Figure 2B). This leads to cytosolic accumulation of TAG, DAG and ceramide (Figure 2B) [120]. Such intermediates are pro-apoptotic as they compromise ATP production via the activation of several stress kinases, including protein kinase C (PKC) [121]. PKC inhibits the metabolic action of insulin by phosphorylating the serine/threonine residues on the insulin receptor and/or its substrates [122], disrupting insulin signalling, and inhibiting insulin-stimulated translocation of GLUT4 (Figure 2B). PKC activation triggers apoptosis and leads to lower basal expression of HIF1α and vascular endothelial growth factor [121]. Pharmacological PKC inhibition was shown to ameliorate FFA-mediated inhibition of basal and insulin-stimulated glucose oxidation. It normalized diastolic function in the STZ-treated T1D heart without altering the circulating metabolites [123].
–
In several clinical studies, proton (1H)-MRS has revealed that diabetic patients have between 1.5- and 2.3-fold higher myocardial TAG levels compared to non-diabetic controls, the levels predicting concentric LV remodeling and subclinical, asymptomatic contractile dysfunction [124][125][126]. Increased availability of plasma FFA increases the flux through myocardial FFA oxidation via activation of the PPARα transcription factor [120][127]. This leads to the upregulation of enzymes involved in FFA oxidation, including acyl-CoA dehydrogenases (Figure 2B). This metabolic shift is the principal driver of the energetic inefficiency of the diabetic heart. Specifically, unlike glucose oxidation, FFA oxidation requires 11% more O2 per carbon. Furthermore, FFA induce expression of mitochondrial uncoupling protein (UCP)-3 through PPAR-α, driving the dissipation of the mitochondrial proton gradient (Figure 2B).
–
This deteriorates efficiency of ATP production as more O2 is required for ATP synthesis, a process defined as mitochondrial uncoupling [128]. A similar concept emerged for UCP2 and UCP3 in HF [98]. In patients undergoing coronary bypass surgery, upregulation of cardiac UCP-3 correlated positively with plasma concentrations of FFA [129] (Figure 2B). The db/db mice have increased myocardial UCP-3 that increased mitochondrial inefficiency following ischaemia [130].
–
In mice, elevation of UCP-3 expression is mediated via increased FFA stimulation of nuclear transcription factor, PPARα [120] (Figure 2B). Activation of the metabolic-sensing ‘master switch' AMPK by metformin was shown to promote both cellular uptake of glucose and β-oxidation of FFAs, attenuating remodeling and HF in diabetes [35]. AMPK also promotes autophagy, providing important nutrients from the breakdown of macromolecules and organelles [131]. In a diabetic mouse HF model, autophagy is impaired and metformin treatment enhances autophagic activity leading to the preservation of cardiac function through an AMPK-dependent mechanism [47]. Nevertheless, animal experiments involving pharmacological activation of PPAR in diabetic hearts remain inconclusive [35], possibly due to the agent specificity for the various PPAR isoforms. Apart for tetradecythioacetic acid (TTA), a PPARα agonist with potent antioxidant properties [132], all other agonists demonstrated reduction in circulating FFA and increased glucose oxidation. Thus, overall cardiac effects were inconsistent: studies employing a PPARγ agonist rosiglitazone and TTA demonstrated improved ischaemic tolerance [131], whereas others using BM17.0744 or 2-(2-(4-phenoxy-2-propylphenoxy)ethyl)indole-5-acetic acid (PPARα and PPARγ agonists, respectively) showed no difference [134][135].
–
Acetoacetate and β-hydroxybutyrate (β-OHB) are ketone bodies generated by the liver from non-esterified FAs in response to hypoinsulinaemia and hypoglycaemia. Ketone bodies are oxidized by most body tissues to form acetyl-CoA. Due to its association with life-threatening acidosis in diabetic patients, ketosis has always been a feared status. However, increased ketone metabolism in the diabetic heart has been recently reported [136]. Furthermore, a recent study showed that ketosis may potentially be protective in T2D [128]. In patients with T2D, ketone bodies are more efficient fuel sources than glucose since insulin is not required for their utilization. Therefore, ketogenesis in acute hyperglycaemic crisis may be lifesaving, because it supplies the myocardium with a sufficient amount of ATP [128]. Given that exogenous d-β-hydroxybutyrate, consumed as a ketone ester drink, was metabolized by exercising skeletal muscle to increase endurance performance in athletes and healthy rats [137][138], it may be that increased ketone metabolism in the diabetic heart is compensating for defects in mitochondrial energy transduction associated with acute insulin deficiency [139].
–
Inflammatory signalling promotes metabolic derangement in diabetic cardiomyopathy
Development of myocardial inflammation in diabetes may involve several molecular mechanisms. In principal, these mechanisms converge towards the activation of the NF-κB pathway which is highly active in the diabetic heart and vasculature contributing to damage by promoting the upregulation of cytokines (IL-1β, IL-6, IL-18, TNF-α, TGF-β1), chemokines and adhesion molecules [140][141][142]. Cardiomyocyte-specific overexpression of IκB-α protein, which suppresses the canonical NF-κB signalling pathway, was observed to prevent streptozotocin-induced diabetic cardiomyopathy through the inhibition of the renin-angiotensin system [143]. Other work also showed that pharmacological inhibition of NF-κB mitigates cardiac oxidative stress induced by T2D and reduces mitochondrial abnormalities [144]. Chronic myocardial inflammation may contribute to cardiac dysfunction by inducing metabolic perturbations that can impair energetics, particularly in response to metabolic stress. Infusion of IL-6 was found to impair cardiac glucose metabolism through a SOCS3-dependent inhibition of IRS-1 [58]. In contrast, genetic disruption of IL-6 gene reduced inflammation and reversed glucose metabolism defects induced by high fat diet, which was paralleled by SOCS3 inhibition and IRS-1 reactivation [145]. Furthermore, cardiac expression of PGC-1α, a master regulator of mitochondrial function and biogenesis [142], was found to be inhibited by a chronic inflammatory process, through a mechanism dependent on NF-κB activation. Exposure of AC16 cells to TNF-α reduced the levels of PGC-1α through the activation of NF-κB [114]. This may be due to p65 binding and sequestering PGC-1α, thereby leading to its inhibition and downregulation [115]. PGC-1α inhibition in response to NF-κB activation was found to cause increased glucose utilization through the downregulation of PDK4 [115].
–
Hyperglycaemia in T2D also induces cellular damage via four major pathways: activation of the PKC pathway via DAG, increased hexosamine pathway flux, increased advanced glycation end products, and increased polyol pathway flux [146][147]. All pathways increased ROS production and activated nuclear poly-(ADP-ribose)-polymerase (PARP), which cleaves NAD+ into nicotinamide and ADP-ribose [146] (Figure 2B). Overactivation of PARP in hyperglycaemia forces the cell to synthesize NAD+ via the salvage pathway which consumes ATP [148]. The process leads to the ribosylation and inactivation of glyceraldehyde-3-phosphate dehydrogenase (GAPDH), which in turn increases glycolytic intermediates and activates the proinflammatory transcription factor NF-κB [146] (Figure 2B). Multiple studies link diabetes to inflammation and it is associated with increased levels of CRP and IL-6 [149]. Other proposed mechanisms for increased inflammation in diabetic cardiomyopathy include oxidative stress via a Ras-related C3 botulinum toxin substrate 1 (RAC1)-mediated activation of NADPH oxidase and endoplasmic reticulum (ER) stress [150].
–
Many interventions have demonstrated beneficial effects in diabetic cardiomyopathy, attributable in part to reduced cardiac inflammation. These interventions include angiotensin 1 (AT-1) receptor antagonism, activation of the kallikrein–kinin system, inhibition of p38 mitogen-activated protein kinase (MAPK) signalling, gene deletion of kinin receptor b1, inhibition of interleukin converting enzyme, atorvastatin treatment, anti-TNF-α treatment, inactivation of GSK-3β, and cannabidiol treatment ( reviewed in [151]).
METABOLIC ADAPTION OF IMMUNE CELLS IN INFLAMMATORY MICROENVIRONMENTS
Aerobic glycolysis, i.e. glucose metabolism to lactate in the presence of abundant oxygen, is the preferential pathway adopted by activated and effector immune cells to meet the demands for ATP required for proliferation and the synthesis of inflammatory mediators. As discussed above, although less efficient than oxidative phosphorylation, aerobic glycolysis offers the advantage of a quick release of ATP and a carbon source feeding a range of biosynthetic pathways. Proliferation and differentiation of antigen-activated T lymphocytes is accompanied by metabolic reprogramming towards aerobic glycolysis, anabolic growth and biomass accumulation [152]. Similarly, activated pro-inflammatory myeloid cells mainly utilize glycolysis with minimal oxidative phosphorylation [153][154][155][156][157][158][159]. In these cells, interruption of the Kreb's cycle allows the generation of molecules that are important for pro-inflammatory functions [153][159]. For example, pro-inflammatory macrophages generate high levels of the metabolite succinate, which promotes HIF1α activity and IL-1 production [153]. Increased levels of citrate promote the generation of the antimicrobial metabolite itaconic acid [160][161]. Dendritic cell (DC) activation leads to a metabolic switch from fatty acid-oxidation and OXPHOS to glycolysis [159], crucial for their antigen-presenting functions [162][163]. Neutrophils also rely on glycolysis for their effector functions [154][155][164], including the formation of neutrophil extracellular traps, which are activated via an mTORC1 signaling pathway [154][155][164][165][166]. Finally, B lymphocytes and natural killer (NK) cells also increase rates of glycolysis in response to various activation stimuli [167][168][169][170]. A key molecular mediator of activation-induced glycolysis in most leukocytes is the mammalian target of rapamycin (mTOR) [4][171]. For example, mTOR complex 1 (mTORC1) activity is essential for induction and maintenance of glycolysis in T cells [4][172] as well as for cytokine-induced glycolysis in NK cells [167]. Key transcription factors involved in the metabolic reprogramming of immune cells towards glycolysis include HIF1α and c-Myc [172][173][174]. HIF1α and c-Myc directly induce transcription of genes encoding glycolytic enzymes and glucose transporters.
–
The development of inflammation leads to dramatic changes in tissue microenvironment, with oxygen and nutrient availability becoming a constraining factor for immune cell function. Albeit this event has not yet been investigated in the context of cardiac inflammation, it is well known that cancerous cells in solid tumours deplete glucose from the microenvironment, thus dampening the ability of adaptive immune cells to utilize glycolysis for their effector functions and promoting the development of an immune-suppressive milieu [8][175]. Similarly, bacterial infection can lead to local nutrient deprivation due to elevated levels of oxygen and glucose consumption by the invading bacteria [176]. Virus-infected human hepatocytes, fibroblasts and epithelial cells can be reprogrammed to upregulate glycolysis to allow viral replication [177][178][179]. In parallel to nutrient depletion, inflammation-associated hypoxia arises as the result of multiple factors, including the accumulation of metabolically active leukocytes using aerobic glycolysis, the generation of interstitial oedema leading to an increase in the inter-capillary distance and inflammation-associated fever, which increases oxygen consumption. All of these factors contribute to the development of a hypoxic environment at the inflammatory site, which can in turn modulate the responses of infiltrating immune cells as well as parenchymal cells. Immune cells are able to adapt to oxygen- and nutrient-depleted environment by engaging the HIF1α and AMPK pathways, respectively.
–
Immune cell adaptation to hypoxia
HIF1α is a basic helix-loop-helix transcription factor that is strongly induced by hypoxia. In hypoxic conditions, HIF1α binds the constitutively expressed HIF1α (also known as Arnt, the aryl hydrocarbon receptor nuclear transporter), which prevents its degradation and allows its translocation to the nucleus. Here, it binds to hypoxia response elements located in the promoters of a number of genes [180][181], including glycolytic enzymes and glucose transporters, VEGF, and the chemokine receptor CXCR4.
–
In addition to hypoxia, HIF1α-mediated transcription can be induced by circumstances associated with pathological stress including cancer, inflammatory mediators, and bacterial infection. HIF1α is expressed by most immune cell populations including macrophages, neutrophils, DCs, as well in T and B lymphocytes and innate lymphoid cells [182]. By regulating immune cell metabolic adaptation to hypoxia, HIF1α plays a key role in modulating their effector functions [183][184][185]. In physiologically hypoxic environments (i.e. lymph nodes), HIF1α contributes to innate and adaptive immune cell homeostasis, whereas in pathological hypoxia, HIF1α signalling can promote immune cell dysfunction and tissue damage [186].
–
HIF1α has been implicated in the regulation T-cell development, proliferation, survival, and cytokine production (e.g., IFN-γ), and lack of HIF1α expression has been associated with overproduction of proinflammatory cytokines [187][188][189][190][191]. HIF1α protein stabilization and nuclear translocation can occur in multiple contexts in T cells. While hypoxic exposure of T cells results in modest HIF1α stabilization [192], T cell receptor (TCR) stimulation results in robust HIF1α protein expression, which is further enhanced in hypoxic conditions [192][193]. TCR-induced HIF1α expression relies upon the PI3 kinase/mTOR pathway [193]. In addition, the pro-inflammatory cytokine IL-6 can induce HIF1α expression in T cells via the STAT3 transcription factor in vitro [194]. Of note, HIF1α is not required for the initiation of aerobic glycolysis in activated T cells; instead, c-Myc is uniquely mediating this metabolic shift [172]. However, HIF1α is instrumental in promoting sustained glycolytic responses in Th17 cell differentiation [174]. Th17 cells are induced by IL-6 and TGF-β in a HIF1α-dependent manner [195][196], while the key transcription factor that drives Th17 differentiation is the retinoic acid-related orphan receptor γt (RORγt) [197]. It has been suggested that the mTOR-HIF1α axis controls Th17 differentiation through transcriptional activation of RORγt and sustained glycolysis [198].
–
In addition to its role in the development of Th17 responses, HIF1α can also promote the development of anti-inflammatory Tregs thus controlling the homeostatic balance between these T cell subsets with opposite biological effects [174][190][191][194][199]. Ambient hypoxia (1% oxygen) was shown to induce selective, robust induction of FoxP3, a key transcriptional regulator for Tregs, likely via HIF1α regulation of the FoxP3 promoter. The same study showed that Treg-intrinsic HIF1α is required for optimal Treg function in vitro and in vivo [200].
–
Hypoxia and HIF1α stabilization play an important role in B cell development and function. Lack of HIF1α expression in lymphoid tissues of chimeric mice causes abnormal B cell development and autoimmunity [201], due to the impairment of hypoxia-induced cell cycle arrest in B cells [202]. More recently, HIF1α was shown to control the expression of the TASK-2 potassium channels in B cells, which are required for B cell proliferation, survival or cytokine production [203]. HIF1α induces glycolytic metabolism in germinal center (GC) B cells, thus regulating the GC reaction and antibody production [204].
–
Macrophages are key effectors of innate immunity and – based on their phenotypic and functional features – have been classified into M1 (classic) and M2 (regulatory) macrophages. Classically activated macrophages (M1) play a key role in the innate defense against bacterial infections via the production of large amounts of nitric oxide (NO) by inducible nitric oxide synthase (iNOS) [205]. Alternatively activated (M2) macrophage differentiation is induced by IL-4 and IL-13. These exert anti-inflammatory and pro-angiogenic functions, thus promoting wound healing, tissue repair and regeneration [206][207]. Macrophage-specific loss of HIF1α expression decreases ATP production, which in turn adversely affects survival, invasion, motility, aggregation and bactericidal activity of murine and human macrophages [208][209][210]. In addition, mice with myeloid cell-specific deletion of HIF-1α are resistant to lipopolysaccharide (LPS)-induced death [211], suggesting a key role for HIF-1α in classical macrophage polarization [212]. Indeed, LPS-mediated activation and pro-inflammatory (M1) polarization in macrophages perturbs the Krebs cycle, leading to the accumulation of the intermediates fumarate and succinate. These in turn increase HIF1α expression and stabilization, which activates a glycolytic reprogramming promoting the acquisition of a pro-inflammatory phenotype [153][213][214][215]. In contrast, alternatively activated (M2) macrophages engage oxidative phosphorylation to meet energy demands [212].
–
HIF1α stabilization is known to regulate several DC functions, such as survival, differentiation, migration, maturation and antigen presentation. In addition, HIF1α regulates interferon-γ (IFN-γ), IL-22 and IL-10 production in human and murine DCs [216][217][218][219]. As in other immune cells HIF1α is involved in metabolic reprogramming of DCs following activation. For example, LPS-induced DC activation under normoxia induces HIF1α expression to greater levels than those induced by hypoxia [220]. HIF1α-induced glycolysis is required for DC maturation [220].
–
The HIF signaling pathway also increases survival, β2 integrin expression, generation of antimicrobial peptides and glycolytic responses in murine and human neutrophils [208][209][221][222][223].
–
Immune cell adaptation to nutrient depletion
Immune cells also have a degree of metabolic plasticity in response to limiting glucose availability. For instance, when glucose levels are low, effector T cells have the ability to adapt and increase glutamine uptake and glutaminolysis to support cellular metabolism [224]. However, in conditions of severe cellular ATP depletion, the nutrient-sensor AMPK becomes activated and induces a catabolic program to preserve survival of immune cells. AMPK is an evolutionarily conserved serine/threonine kinase integrator of energy-sensing signals in the immune system function, which specializes in modulating the cellular responses to an energy challenge [225]. Once activated by stimuli like nutrient deprivation, hypoxia, or excessive ROS production, AMPK activates ATP-producing catabolic pathways while inhibiting ATP-consuming anabolic pathways to restore energy homeostasis. Indeed, AMPK activation is induced by a high cellular AMP / ATP ratio and by upstream kinases [226][227]. In tumor cells, cardiomyocytes and macrophages/monocytes, AMPK activation boosts the glycolysis rate by increasing glucose uptake and through by phosphorylation and activation of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 2/3, which produces fructose-2,6-bisphosphate, an allosteric activator of glycolytic enzyme phosphofructokinase-1 [228][229].
–
In parallel, FA synthesis is reduced via phosphorylation of acetyl-CoA carboxylase (ACC) [230][231], which catalyzes the rate-limiting step in FA synthesis by converting acetyl-coA to malonyl-coA, and (2) sterol regulatory element-binding protein 1c (SREBP1c), a transcription factor that increases the expression of lipogenic enzymes, including ACC1 and FA synthase [232][233]. In addition to reducing lipid anabolism, AMPK activates lipid catabolism and mitochondrial biogenesis [229]. Specifically, AMPK increases FA uptake by increasing translocation of the FA transporter CD36 to the plasma membrane [234]. Once inside cells, FAs are transported into the mitochondria for β-oxidation by carnitine palmitoyltransferase-1 (CPT-1). AMPK boosts CPT-1 activity and activates FAO by inducing the inhibitory phosphorylation of ACC2, which is localized to the outer membrane of the mitochondria near CPT-1 where it inhibits production of malonyl-CoA, a potent allosteric inhibitor of CPT-1[233].
–
AMPK also inhibits glycogen synthesis through inhibitory phosphorylation of glycogen synthase (GS). Furthermore, AMPK also activates glycogen breakdown by phosphorylating and activating glycogen phosphorylase (GP). By indirectly inhibiting mTORC1 through the phosphorylation of TSC2 and raptor, AMPK also inhibits protein synthesis and cap-dependent translation during both initiation and elongation steps [235][236][237]. Autophagy is a lysosome-dependent self-digestive process activated during nutrient deficiency to preserve cellular integrity. Autophagy is directly initiated by AMPK during severe energy challenges through activation of the Unc-51-like kinase (ULK)-1, a mammalian homolog of ATG1 which removes damaged mitochondria thus maintaining mitochondrial integrity during nutrient starvation [238][239]. In addition, AMPK indirectly promotes autophagy by inhibiting mTORC1, which phosphorylates ULK1 to prevent interaction with AMPK. Further, AMPK phosphorylates and activates FOXO transcription factors, which in turn upregulate expression of several autophagy inducers, such as Bnip3, LC3 and ATG12 [240].
–
Although the AMPK and HIF pathways share fundamental functions in the maintenance of cellular metabolic homeostasis, they use different strategies to carry out these functions. Specifically, while HIF factors activate anabolic processes to generate energy, AMPK signalling promotes catabolic mechanisms. In addition, they can control each other's function in a context-dependent manner. For example, HIF1α activates aerobic glycolysis, an effect which is counteracted by AMPK activation [241] possibly via inhibition of protein synthesis [242]. HIF1α and AMPK also exert antagonistic effects in inflammatory microenvironments, e.g. in cancer. HIF-1α signalling has an essential role in the activation and function of immune cells [208][243]. In contrast, AMPK activation inhibits inflammatory responses by activating the survival factors SIRT1 and FoxO [5][244]. In addition, the anti-inflammatory cytokines TGFβ and IL10 have been shown to activate AMPK in macrophages via an as yet unidentified mechanism [245].
–
Substantial evidence supports an indirect, reciprocal regulation between the AMPK and HIF-1α pathways via context-dependent and tissue-selective mediators. For example, AMPK signalling regulates HIF1α nuclear translocation [246], a key step of the hypoxia response. In addition, AMPK is a potent activator of SIRT1, a member of NAD+-dependent sirtuin family, which can either inhibit or activate the signalling of HIF factors. In turn, SIRT1 can directly and indirectly inhibit HIF1α transcriptional activity in cultured cells [247].
–
Macrophage migration inhibitory factor (MIF) is an evolutionarily conserved, multifunctional cytokine, which has been implicated in the pathogenesis of many cancers and inflammatory diseases [248][249]. MIF is a secreted pro-inflammatory cytokine expressed not only in immune cells but also in many non-immune cells, such as endothelial and epithelial cells. It is known that this pleiotropic factor transmits signals through a cell surface receptor composed of CD74/CD44 proteins, e.g. activating the ERK1/2, PI3K/Akt, and NF-κB pathways. Interestingly, several studies have demonstrated that the MIF gene is a hypoxia-inducible, HIF1α-dependent gene [250][251][252]. MIF has been shown to activate AMPK signalling eliciting anti-inflammatory effect such as protection from rat heart ischemia reperfusion injury [253], delay of cellular senescence [250][254] and prevention of liver fibrosis in mice [255]. However, the anti-inflammatory effects of the HIF1α-MIF-AMPK pathway can promote tumorigenesis, known to be induced by MIF [248].
METABOLIC CROSS TALK BETWEEN IMMUNE CELLS AND CARDIOMYOCYTES DURING INFLAMMATION
Although the immune system has evolved to protect against external stressors including infection, its powerful effector mechanism has the potential to injure and impair function of healthy tissues. Organs with continuous function critical for survival such as eye, brain and the heart are readily impaired by the immune inflammatory responses. Thus, as discussed in section “CAVEATS AND DEFINITIONS: WHAT IS INFLAMMATORY DISEASE OF THE HEART MUSCLE?”, the role of inflammation in many cardiac diseases is now widely accepted [58]. There are very few T cells found in healthy cardiac muscle thus maintaining the relative state of immune quiescence which discourages T cell recruitment, activation and presence of resident memory T cells [15]. However, there are significant numbers of resident myocardial macrophages and DCs. The extensive myocardial microvascular network which ensures sufficient oxygen and metabolite supply for continuous cardiac work, provides ample opportunity for circulating T cells to migrate into heart. While direct damage to the myocardium by infiltrating effector immune cells has been widely studied [14], the role of metabolic cross-talk between inflammatory cells and the cardiac parenchyma itself is less well understood.
–
As discussed above, both cardiomyocytes and immune cells undergo metabolic adaptation in response to the inflammatory stimuli (cardiac and systemic). As a consequence, these cells can influence each other via the production of soluble mediators and metabolites that can function as signaling molecules (Figure 3).
–
Tumor necrosis factor α (TNFα)
Chronic cardiomyocyte exposure to TNFα directly inhibits and downregulates expression of PPAR-γ coactivator-1α (PGC1α) via NFkB (p65) activation [115]. A close relationship exists among PGC-1 function, insulin sensitivity, and T2D with its expression downregulated in T2D subjects [256][257][258]. A common polymorphism of the PGC1α gene (Gly482Ser), expressing reduced PGC1α activity, increases a risk of T2D [259]. PGC1α is a master regulator of myocardial FFA and glucose oxidative metabolism. It controls expression of transcription factors regulating FFA oxidation, glucose uptake and metabolism, mitochondrial function and biogenesis [259]. PGC1α has been reported to activate the expression of insulin-sensitive GLUT4 [260][261]. These observations would suggest that reduced levels and compromised activity of PGC1α are driving the metabolic remodelling in cardiac pathologies. The rescue of PGC1α expression by reducing proinflammatory cytokine load could both prevent and ameliorate metabolic remodelling in HF and diabetes: improve mitochondrial function and organisation, reduce insulin resistance by enhancing glucose uptake (GLUT4) and metabolism (PDK4), reducing myocardial steatosis by promoting FFA β-oxidation (due to relationship between myocardial triglyceride accumulation and insulin resistance) [258][262]. Thus, by altering the nature of immune response we could prevent the origin of pathologic metabolic remodelling (myocardial glucose-FFA oxidation switch), reduced mitochondrial number and activity, as well as rescue metabolic flexibility of hypertrophic heart. However, whether T-cell mediated systemic inflammation significantly contributes to the energetic deficit of HF and diabetes and how the reduction of inflammation by reduction in pro-inflammatory T-cell migration/activation impacts cardiac metabolic (in)flexibility in HF and diabetes needs demonstrating.
–
Interleukin-6 (IL-6)
The pro-inflammatory cytokine IL-6 is known for its effect on systemic metabolism [263][264]. For example, IL-6 promotes FFA mobilization [265], suggesting that IL-6 contributes to shifting metabolic adaptation toward FAO rather than glycolysis. The increase of IL-6 plasma levels during exercise has been proposed to promote energy supply via mobilization of FFA. IL-6 can signal in cardiomyocytes independently of the canonical STAT1/3 pathway, via Erk1/2 and PI3K activation [266]. IL-6R receptors are expressed primarily in myocardial interstitial cells such as fibroblasts and monocytes [267], rather than cardiomyocytes, but are upregulated by cardiomyocytes in response to pro-inflammatory signals as well as mechanical stretch [267][268]. Overall, IL-6 plays an important protective role in myocardial remodelling in a model of Angiotensin II-dependent hypertension [269] and other IL-6 family cytokines such as leukaemia inhibitory factor (LIF) and cardiotrophin-1 (CT-1) can also promote cardiomyocyte survival and hypertrophy via LIFRβ signalling [270][271]. However, chronic exposure to IL-6 has been shown to impair myocardial glucose metabolism (via SOC3 inhibition and IRS activation) in diabetic cardiomyopathy in mice [58].
–
AMPK-regulating inflammatory cytokines
The traditional paradigm of AMPK as an energy stress-activated kinase has been expanded to include a diverse array of cytokines as AMPK-activators during inflammatory processes [54]. Short-term coronary infusions of pro-inflammatory cytokines such as IL-6, appear to reduce AMPK α subunit content and activation [145]. Fat diet feeding in mice was shown to elevate plasma levels of IL-6, which may contribute to the downregulation of myocardial AMPK seen in this model [145]. MIF is a master-regulator of inflammatory cytokines, and is highly expressed in cardiomyocytes. Hypoxia increases cardiac MIF expression, via a HIF1α-dependent mechanism [272], and ischemia triggers MIF secretion. Endogenous cardiac MIF has an important autocrine-paracrine action to modulate AMPK activation during ischemia and hypoxia in the heart [253]. Extracellular MIF activates AMPK via its cell surface receptor CD74 with subsequent activation of the signal transducer CD44 [253]. MIF knockout mice have impaired heart AMPK activation and are more susceptible to ischemic injury [253].
–
Lactate
Highly glycolytic, activated immune cells produce and secrete high amounts of the glycolysis end product lactate in inflamed tissue, including rheumatoid arthritis and cancer [273]. Extracellular lactate can directly signal to immune cells themselves and tissue parenchymal cells via lactate receptors as well as by affecting metabolic pathways when enriched in the cytosol following uptake by lactate transporters.
–
In immune cells lactate mainly signals via the surface-expressed G-protein-coupled receptor GPR81 [274]. GPR81-mediated signals are strong inhibitors of immune effector functions. For example, extracellular lactate reduces the LPS-induced IL-1β production by murine macrophages and human peripheral blood mononuclear cells (PBMCs) in vitro [274]. In vivo, high lactate concentrations in the tumour microenvironment contribute to the immune-suppressed microenvironment by inhibiting cytokine production by- and migration of- monocytes and macrophages [275][276]. Administration of sodium lactate has been shown to reduce the inflammatory response in hepatitis [274] and to prevent the development of inflammation in a colitis model [277]. The anti-inflammatory effects of lactate induced by GPR81 are mediated by inhibition of NF-κB and inflammasome activation [274].
–
Lactate uptake by immune cells via monocarboxylate transporters (MCTs) can directly affect their cellular metabolism and function independently of GPR8. Expression and modulation of MCTs can control immune cell function in T cells and macrophages [9][278] by inhibiting the glycolytic pathway [279][280]. A key enzyme in lactate metabolism is lactate dehydrogenase (LDH), which has a higher affinity for pyruvate compared with lactate. Thus, it converts pyruvate into lactate and NAD+. LDH has been implicated in IFNγ-production by T cells [281] and anti-tumour activity of macrophages [282]. Lactate also represents a source of energy for heart muscle by boosting mitochondrial energy metabolism. Lactate amounts to about 10 % of energy production in the healthy heart [283], and this fraction can substantially increase during exercise or different pathophysiological conditions.
–
In cardiomyocytes lactate production in the cytosol is balanced by oxidation in the mitochondria. Intracellular lactate is shuttled from the cytosol by MCT1 on the mitochondrial membrane [284][285]. Under normal physiological conditions, low levels of ROS upregulate MCT1 expression by transcriptional mechanisms, thus promoting the transport of lactate into the mitochondria for oxidative metabolism [286]. Not only physical activity but also different pathological conditions could change the hearts' preference from FAs to lactate as energy source [287][288]. In a rat model of congestive heart failure, e.g., a significant increase in the expression of the lactate transporter MCT 1 was observed [289]. Other studies on rats could demonstrate that an increase in blood lactate level has positive effects on heart function during a hemorrhagic or septic shock [290][291].
–
However, excess of extracellular and cytosolic lactate as a result of protracted inflammation has been linked to cardiomyocyte apoptosis in several experimental models of cardiovascular diseases, including myocardial infarction [292], ischemia/reperfusion injury [293], dilated cardiomyopathy [294] and end-stage heart failure [295]. Cardiac MCTs can function as acid loaders or extruders, depending on the transport direction [296], hence the concentration of lactate in the cytosol is directly proportional to the amount of extracellular lactate.
–
Mechanistically, under pathological conditions, high intracellular lactate concentration promotes the excessive generation of ROS. High levels of ROS can cause oxidative stress and mitochondrial damage, which lead to the activation of mitochondrial-dependent apoptosis [297], a highly regulated program of cell death that can be activated in cardiomyocytes by multiple stressors including cytokines [298], oxidative stress [299] and DNA damage [300]. A significant association has been identified between the lactate signaling cascade and cardiovascular diseases, such as myocardial infarction [284], atrial fibrillation [285] and heart failure [286].
BREAKING THE VICIOUS CIRCLE: CAN TARGETING METABOLISM REDUCE CARDIAC INFLAMMATION?
As previously discussed, although targeting inflammatory mediators and mechanism in heart disease is a rapidly developing and promising strategy, the lack of diagnostic knowledge of disease-specific mechanisms and stage of inflammation remains a major challenge. On the other hand, several drugs that are currently used in clinic for patients with diabetes, dyslipidemia and metabolic dysfunctions have been shown to impact immune cell function. Understanding the cross-talk between immune cells and myocardium might provide the basis for the repositioning of ‘old' drugs as immunomodulators. Metformin (dimethylbiguanide) widely prescribed for T2D activates AMPK [301], and reduces redox shuttle enzyme mitochondrial glycerophosphate dehydrogenase [302]. Beyond its effects on glucose metabolism, metformin decreases inflammatory markers in plasma, including soluble intercellular adhesion molecule, vascular cell adhesion molecule-1, macrophage migration inhibitory factor, and CRP [303][304]. The anti-inflammatory effects of metformin are likely related to its ability to inhibit mTOR through AMPK activation [305].
–
Statins, inhibitors of the enzyme 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase (which catalyzes the formation of mevalonate, the rate-limiting step for cholesterol synthesis), are the most efficient and widely used agents in treating cardiovascular diseases. Originally designed to target elevated lipids, the “traditional” cause of atherosclerosis, statins might also confer cardiovascular benefit by modulating inflammation [306][307][308]. These effects are independent of the HMG-CoA reductase inhibition [309][310], while rely upon isoprenoid (and downstream prenylated proteins) biosynthesis from mevalonate [311][312]. Through this pathway, statins can deviate T-cell differentiation towards the generation of Tregs instead of pro-inflammatory Th17 cells via a mechanism dependent on protein geranylgeranylation [313][314].
–
Targeting the PPARs nuclear receptors agonists has also been effective in controlling inflammatory responses in metabolic diseases. In humans, activation of PPARα using fenofibrate or bezafibrate has been shown to decrease plasma levels of several acute phase response proteins that are increased during inflammatory conditions and ameliorates endotoxemia [315]. PPARγ its pharmacological agonists promotes the anti-inflammatory differentiation of macrophages [316][317] and the function of Tregs in adipose tissue (AT) [318]. Anti-inflammatory effects of PPAR agonists have been reported in a number of model diseases, including inflammatory bowel disease, central nervous system inflammation, LPS-induced cardiac and pulmonary inflammation [319], although the beneficial effect of these drugs in diabetic heart disease remains unclear [35], as previously discussed.
CONCLUDING REMARKS
This overview of the metabolic plasticity of immune cells and cardiac tissue during inflammation highlights the complexity of immunometabolic events which can determine either the resolution of the inflammatory process or ultimately lead to loss of organ function.
–
Overall, and perhaps in an over-simplified fashion, the metabolic adaptation of immune cells to the inflammatory microenvironment occurs in synchrony with the evolution of the inflammatory response. Thus, when immune cells localize in the inflammatory site, the hypoxia response mediated by HIF promotes a pro-inflammatory effector function sustained by glycolysis. With the progression of inflammation and increased nutrient consumption relative to supply, the metabolic adaptation mediated by AMPK and lactate favours the development of an anti-inflammatory environment, thus possibly contributing to the resolution of inflammation. The tissue also adapts metabolically and functionally to the changing environment dictated by inflammation in order to cope with damaging events. If this model (Figure 3) is correct, it is plausible that abnormal metabolic reprogramming by either immune cells or cardiomyocytes might underlie the progression of inflammation and irreversible tissue damage. As a consequence, pharmacological modulation of abnormal metabolic adaptation may provide an effective approach to treat inflammation-associated heart disease.
–
 | FIGURE 3: Immunometabolic cross-talk in the inflamed heart. During inflammation, inflammatory infiltrates and heart parenchymal cells (including cardiomyocytes, compete with oxygen and nutrients. This leads to the activation of the HIF and AMPK pathways and metabolic reprogramming of both cellular components (1). The production of cytokines (2) and signalling metabolites, such as lactate (3) can lead to further metabolic reprogramming and eventually, to cellular dysfunction (4). MO, macrophages, T, T-lymphocytes; B, B lymphocytes; DC, dendritic cells; NK, NK cells; GN, granulocytes; EOS, eosinophils; CMC, cardiomyocytes; SC, stromal cells; EC, endothelial cells. |
REFERENCES
- MacIver NJ, Michalek RD, Rathmell JC (2013). Metabolic regulation of T lymphocytes. Annu Rev Immunol 31(259-283. doi: 10.1146/annurev-immunol-032712-095956
- Pearce EL, Pearce EJ (2013). Metabolic pathways in immune cell activation and quiescence. Immunity 38(4): 633-643. doi: 10.1016/j.immuni.2013.04.005
- Wang R, Green DR (2012). Metabolic checkpoints in activated T cells. Nat Immunol 13(10): 907-915. doi: 10.1038/ni.2386
- Pollizzi KN, Powell JD (2014). Integrating canonical and metabolic signalling programmes in the regulation of T cell responses. Nat Rev Immunol 14(7): 435-446. doi: 10.1038/nri3701
- O'Neill LA, Hardie DG (2013). Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature 493(7432): 346-355. doi: 10.1038/nature11862
- MacIver NJ, Michalek RD, Rathmell JC (2013). Metabolic regulation of T lymphocytes. Annu Rev Immunol 31: 259-283. doi: 10.1146/annurev-immunol-032712-095956
- Norata GD, Caligiuri G, Chavakis T, Matarese G, Netea MG, Nicoletti A, O'Neill LA, Marelli-Berg FM (2015). The Cellular and Molecular Basis of Translational Immunometabolism. Immunity 43(3): 421-434. doi: 10.1016/j.immuni.2015.08.023
- Chang CH, Qiu J, O'Sullivan D, Buck MD, Noguchi T, Curtis JD, Chen Q, Gindin M, Gubin MM, van der Windt GJ, Tonc E, Schreiber RD, Pearce EJ, Pearce EL (2015). Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 162(6): 1229-1241. doi: 10.1016/j.cell.2015.08.016
- Haas R, Smith J, Rocher-Ros V, Nadkarni S, Montero-Melendez T, D'Acquisto F, Bland EJ, Bombardieri M, Pitzalis C, Perretti M, Marelli-Berg FM, Mauro C (2015). Lactate Regulates Metabolic and Pro-inflammatory Circuits in Control of T Cell Migration and Effector Functions. PLoS Biol 13(7): e1002202. doi: 10.1371/journal.pbio.1002202
- Zhao TX, Mallat Z (2019). Targeting the Immune System in Atherosclerosis: JACC State-of-the-Art Review. J Am Coll Cardiol 73(13): 1691-1706. doi: 10.1016/j.jacc.2018.12.083
- Koenig W (2017). Inflammation Revisited: Atherosclerosis in The Post-CANTOS Era. Eur Cardiol 12(2): 89-91. doi: 10.15420/ecr.2017:18:1
- Reklou A, Doumas M, Imprialos K, Stavropoulos K, Patoulias D, Athyros VG (2018). Reduction of Vascular Inflammation, LDL-C, or Both for the Protection from Cardiovascular Events? Open Cardiovasc Med J 12: 29-40. doi: 10.2174/1874192401812010029
- Bondue A, Arbustini E, Bianco A, Ciccarelli M, Dawson D, De Rosa M, Hamdani N, Hilfiker-Kleiner D, Meder B, Leite-Moreira AF, Thum T, Tocchetti CG, Varricchi G, Van der Velden J, Walsh R, Heymans S (2018). Complex roads from genotype to phenotype in dilated cardiomyopathy: scientific update from the Working Group of Myocardial Function of the European Society of Cardiology. Cardiovasc Res114(10): 1287-1303. doi: 10.1093/cvr/cvy122
- Stephenson E, Savvatis K, Mohiddin SA, Marelli-Berg FM (2017). T-cell immunity in myocardial inflammation: pathogenic role and therapeutic manipulation. Br J Pharmacol 174(22): 3914-3925. doi: 10.1111/bph.13613
- Grabie N, Lichtman AH, Padera R (2019). T cell checkpoint regulators in the heart. Cardiovasc Res 115(5): 869-877. doi: 10.1093/cvr/cvz025
- Ferrini A, Stevens MM, Sattler S, Rosenthal N (2019). Toward Regeneration of the Heart: Bioengineering Strategies for Immunomodulation. Front Cardiovasc Med 6: 26. doi: 10.3389/fcvm.2019.00026
- Frangogiannis NG (2015). Inflammation in cardiac injury, repair and regeneration. Curr Opin Cardiol 30(3): 240-245. doi: 10.1097/HCO.0000000000000158
- Prabhu SD, Frangogiannis NG (2016). The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis. Circ Res 119(1): 91-112. doi: 10.1161/CIRCRESAHA.116.303577
- Sattler S, Fairchild P, Watt FM, Rosenthal N, Harding SE (2017). The adaptive immune response to cardiac injury-the true roadblock to effective regenerative therapies? NPJ Regen Med 2: 19. doi: 10.1038/s41536-017-0022-3
- Hansson EM, Lindsay ME, Chien KR (2009). Regeneration next: toward heart stem cell therapeutics. Cell Stem Cell 5(4): 364-377. doi: 10.1016/j.stem.2009.09.004
- Paulus WJ, Tschope C (2013). A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol 62(4): 263-271. doi: 10.1016/j.jacc.2013.02.092
- Franssen C, Chen S, Unger A, Korkmaz HI, De Keulenaer GW, Tschope C, Leite-Moreira AF, Musters R, Niessen HW, Linke WA, Paulus WJ, Hamdani N (2016). Myocardial Microvascular Inflammatory Endothelial Activation in Heart Failure With Preserved Ejection Fraction. JACC Heart Fail 4(4): 312-324. doi: 10.1016/j.jchf.2015.10.007
- Eisenberg T, Abdellatif M, Schroeder S, Primessnig U, Stekovic S, Pendl T, Harger A, Schipke J, Zimmermann A, Schmidt A, Tong M, Ruckenstuhl C, Dammbrueck C, Gross AS, Herbst V, Magnes C, Trausinger G, Narath S, Meinitzer A, Hu Z, Kirsch A, Eller K, Carmona-Gutierrez D, Buttner S, Pietrocola F, Knittelfelder O, Schrepfer E, Rockenfeller P, Simonini C, Rahn A, et al. (2016). Cardioprotection and lifespan extension by the natural polyamine spermidine. Nature Med 22(12): 1428-1438. doi: 10.1038/nm.4222
- Yalta T, Yalta K (2018). Systemic Inflammation and Arrhythmogenesis: A Review of Mechanistic and Clinical Perspectives. Angiology 69(4): 288-296. doi: 10.1177/0003319717709380
- Aksentijevic D, O'Brien BA, Eykyn TR, Shattock MJ (2018). Is there a causal link between intracellular Na elevation and metabolic remodelling in cardiac hypertrophy? Biochem Soc Trans 46(4): 817-827. doi: 10.1042/BST20170508
- Taegtmeyer H, Golfman L, Sharma S, Razeghi P, van Arsdall M (2004). Linking gene expression to function: metabolic flexibility in the normal and diseased heart. Ann N Y Acad Sci 1015(202-213. doi: 10.1196/annals.1302.017
- Balaban RS (2002). Cardiac energy metabolism homeostasis: role of cytosolic calcium. J Mol Cell Cardiol 34(10): 1259-1271. doi: 10.1016/s0022-2828(02)92082-1
- Mootha VK, Arai AE, Balaban RS (1997). Maximum oxidative phosphorylation capacity of the mammalian heart. Am J Physiol 272(2 Pt 2): H769-775. doi: 10.1152/ajpheart.1997.272.2.H769
- Neubauer S (2007). The failing heart–an engine out of fuel. N Engl J Med 356(11): 1140-1151. doi: 10.1056/NEJMra063052
- Peterzan MA, Lygate CA, Neubauer S, Rider OJ (2017). Metabolic remodeling in hypertrophied and failing myocardium: a review. Am J Physiol Heart Circ Physiol 313(3): H597-H616. doi: 10.1152/ajpheart.00731.2016
- Stanley WC, Recchia FA, Lopaschuk GD (2005). Myocardial substrate metabolism in the normal and failing heart. Physiol Rev 85(3): 1093-1129. doi: 10.1152/physrev.00006.2004
- Aksentijevic D, Lygate CA, Makinen K, Zervou S, Sebag-Montefiore L, Medway D, Barnes H, Schneider JE, Neubauer S (2010). High-energy phosphotransfer in the failing mouse heart: role of adenylate kinase and glycolytic enzymes. Eur J Heart Fail 12(12): 1282-1289. doi: 10.1093/eurjhf/hfq174
- Ingwall J (2002). ATP and the Heart. New York: Springer Science & Business Media.
- Hue L, Taegtmeyer H (2009). The Randle cycle revisited: a new head for an old hat. Am J Physiol Endocrinol Metab 297(3): E578-591. doi: 10.1152/ajpendo.00093.2009
- Chong CR, Clarke K, Levelt E (2017). Metabolic Remodeling in Diabetic Cardiomyopathy. Cardiovasc Res. doi: 10.1093/cvr/cvx018
- Heather LC, Cole MA, Atherton HJ, Coumans WA, Evans RD, Tyler DJ, Glatz JF, Luiken JJ, Clarke K (2010). Adenosine monophosphate-activated protein kinase activation, substrate transporter translocation, and metabolism in the contracting hyperthyroid rat heart. Endocrinology 151(1): 422-431. doi: 10.1210/en.2009-0593
- Heather LC, Clarke K (2011). Metabolism, hypoxia and the diabetic heart. J Mol Cell Cardiol 50(4): 598-605. doi: 10.1016/j.yjmcc.2011.01.007
- Aksentijevic D, Bhandari S, Seymour AM (2009). Insulin resistance and altered glucose transporter 4 expression in experimental uremia. Kidney Int 75(7): 711-718. doi: 10.1038/ki.2008.691
- Aksentijevic D, McAndrew DJ, Karlstadt A, Zervou S, Sebag-Montefiore L, Cross R, Douglas G, Regitz-Zagrosek V, Lopaschuk GD, Neubauer S, Lygate CA (2014). Cardiac dysfunction and peri-weaning mortality in malonyl-coenzyme A decarboxylase (MCD) knockout mice as a consequence of restricting substrate plasticity. J Mol Cell Cardiol 75: 76-87. doi: 10.1016/j.yjmcc.2014.07.008
- Balaban RS, Kantor HL, Katz LA, Briggs RW (1986). Relation between work and phosphate metabolite in the in vivo paced mammalian heart. Science 232(4754): 1121-1123. doi: 10.1126/science.3704638
- Carvajal K, Zarrinpashneh E, Szarszoi O, Joubert F, Athea Y, Mateo P, Gillet B, Vaulont S, Viollet B, Bigard X, Bertrand L, Ventura-Clapier R, Hoerter JA (2007). Dual cardiac contractile effects of the alpha2-AMPK deletion in low-flow ischemia and reperfusion. Am J Physiol Heart Circ Physiol 292(6): H3136-3147. doi: 10.1152/ajpheart.00683.2006
- Bertero E, Maack C (2018). Metabolic remodelling in heart failure. Nat Rev Cardiol 15(8): 457-470. doi: 10.1038/s41569-018-0044-6
- Dufour CR, Wilson BJ, Huss JM, Kelly DP, Alaynick WA, Downes M, Evans RM, Blanchette M, Giguere V (2007). Genome-wide orchestration of cardiac functions by the orphan nuclear receptors ERRalpha and gamma. Cell Metab 5(5): 345-356. doi: 10.1016/j.cmet.2007.03.007
- Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP (2000). Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest 106(7): 847-856. doi: 10.1172/JCI10268
- Rowe GC, Jiang A, Arany Z (2010). PGC-1 coactivators in cardiac development and disease. Circ Res 107(7): 825-838. doi: 10.1161/CIRCRESAHA.110.223818
- Haemmerle G, Moustafa T, Woelkart G, Buttner S, Schmidt A, van de Weijer T, Hesselink M, Jaeger D, Kienesberger PC, Zierler K, Schreiber R, Eichmann T, Kolb D, Kotzbeck P, Schweiger M, Kumari M, Eder S, Schoiswohl G, Wongsiriroj N, Pollak NM, Radner FP, Preiss-Landl K, Kolbe T, Rulicke T, Pieske B, Trauner M, Lass A, Zimmermann R, Hoefler G, Cinti S, et al. (2011). ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-alpha and PGC-1. Nat Med 17(9): 1076-1085. doi: 10.1038/nm.2439
- Xie Z, He C, Zou MH (2011). AMP-activated protein kinase modulates cardiac autophagy in diabetic cardiomyopathy. Autophagy 7(10): 1254-1255. doi: 10.4161/auto.7.10.16740
- Gelinas R, Mailleux F, Dontaine J, Bultot L, Demeulder B, Ginion A, Daskalopoulos EP, Esfahani H, Dubois-Deruy E, Lauzier B, Gauthier C, Olson AK, Bouchard B, Des Rosiers C, Viollet B, Sakamoto K, Balligand JL, Vanoverschelde JL, Beauloye C, Horman S, Bertrand L (2018). AMPK activation counteracts cardiac hypertrophy by reducing O-GlcNAcylation. Nat Commun 9(1): 374. doi: 10.1038/s41467-017-02795-4
- Hu X, Xu X, Lu Z, Zhang P, Fassett J, Zhang Y, Xin Y, Hall JL, Viollet B, Bache RJ, Huang Y, Chen Y (2011). AMP activated protein kinase-alpha2 regulates expression of estrogen-related receptor-alpha, a metabolic transcription factor related to heart failure development. Hypertension 58(4): 696-703. doi: 10.1161/HYPERTENSIONAHA.111.174128
- Zhu L, Wang Q, Zhang L, Fang Z, Zhao F, Lv Z, Gu Z, Zhang J, Wang J, Zen K, Xiang Y, Wang D, Zhang CY (2010). Hypoxia induces PGC-1alpha expression and mitochondrial biogenesis in the myocardium of TOF patients. Cell Res 20(6): 676-687. doi: 10.1038/cr.2010.46
- Kudo N, Barr AJ, Barr RL, Desai S, Lopaschuk GD (1995). High rates of fatty acid oxidation during reperfusion of ischemic hearts are associated with a decrease in malonyl-CoA levels due to an increase in 5'-AMP-activated protein kinase inhibition of acetyl-CoA carboxylase. J Biol Chem 270(29): 17513-17520. doi: 10.1074/jbc.270.29.17513
- Chavez JA, Roach WG, Keller SR, Lane WS, Lienhard GE (2008). Inhibition of GLUT4 translocation by Tbc1d1, a Rab GTPase-activating protein abundant in skeletal muscle, is partially relieved by AMP-activated protein kinase activation. J Biol Chem 283(14): 9187-9195. doi: 10.1074/jbc.M708934200
- Yang J, Holman GD (2005). Insulin and contraction stimulate exocytosis, but increased AMP-activated protein kinase activity resulting from oxidative metabolism stress slows endocytosis of GLUT4 in cardiomyocytes. J Biol Chem 280(6): 4070-4078. doi: 10.1074/jbc.M410213200
- Zaha VG, Young LH (2012). AMP-activated protein kinase regulation and biological actions in the heart. Circ Res 111(6): 800-814. doi: 10.1161/CIRCRESAHA.111.255505
- Tian R, Abel ED (2001). Responses of GLUT4-deficient hearts to ischemia underscore the importance of glycolysis. Circulation 103(24): 2961-2966. doi: 10.1161/01.cir.103.24.2961
- Frieler RA, Mortensen RM (2015). Immune cell and other noncardiomyocyte regulation of cardiac hypertrophy and remodeling. Circulation 131(11): 1019-1030. doi: 10.1161/CIRCULATIONAHA.114.008788
- Mann DL (2015). Innate immunity and the failing heart: the cytokine hypothesis revisited. Circ Res 116(7): 1254-1268. doi: 10.1161/CIRCRESAHA.116.302317
- Frati G, Schirone L, Chimenti I, Yee D, Biondi-Zoccai G, Volpe M, Sciarretta S (2017). An overview of the inflammatory signalling mechanisms in the myocardium underlying the development of diabetic cardiomyopathy. Cardiovasc Res 113(4): 378-388. doi: 10.1093/cvr/cvx011
- Boudina S, Abel ED (2007). Diabetic cardiomyopathy revisited. Circulation 115(25): 3213-3223. doi: 10.1161/CIRCULATIONAHA.106.679597
- Garnier A, Fortin D, Delomenie C, Momken I, Veksler V, Ventura-Clapier R (2003). Depressed mitochondrial transcription factors and oxidative capacity in rat failing cardiac and skeletal muscles. J Physiol 551(Pt 2): 491-501. doi: 10.1113/jphysiol.2003.045104
- Lemieux H, Semsroth S, Antretter H, Hofer D, Gnaiger E (2011). Mitochondrial respiratory control and early defects of oxidative phosphorylation in the failing human heart. Int J Biochem Cell Biol 43(12): 1729-1738. doi: 10.1016/j.biocel.2011.08.008
- Sharov VG, Todor AV, Silverman N, Goldstein S, Sabbah HN (2000). Abnormal mitochondrial respiration in failed human myocardium. J Mol Cell Cardiol 32(12): 2361-2367. doi: 10.1006/jmcc.2000.1266
- Nishida K, Otsu K (2017). Inflammation and metabolic cardiomyopathy. Cardiovasc Res 113(4): 389-398. doi: 10.1093/cvr/cvx012
- Aksentijevic A, Brandt KR, Tsakanikos E, Thorpe MJA (2019). It takes me back: The mnemonic time-travel effect. Cognition 182: 242-250. doi: 10.1016/j.cognition.2018.10.007
- Allard MF, Schonekess BO, Henning SL, English DR, Lopaschuk GD (1994). Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am J Physiol 267(2 Pt 2): H742-750. doi: 10.1152/ajpheart.1994.267.2.H742
- Christe ME, Rodgers RL (1994). Altered glucose and fatty acid oxidation in hearts of the spontaneously hypertensive rat. J Mol Cell Cardiol 26(10): 1371-1375. doi: 10.1006/jmcc.1994.1155
- Doenst T, Pytel G, Schrepper A, Amorim P, Farber G, Shingu Y, Mohr FW, Schwarzer M (2010). Decreased rates of substrate oxidation ex vivo predict the onset of heart failure and contractile dysfunction in rats with pressure overload. Cardiovasc Res 86(3): 461-470. doi: 10.1093/cvr/cvp414
- Degens H, de Brouwer KF, Gilde AJ, Lindhout M, Willemsen PH, Janssen BJ, van der Vusse GJ, van Bilsen M (2006). Cardiac fatty acid metabolism is preserved in the compensated hypertrophic rat heart. Basic Res Cardiol 101(1): 17-26. doi: 10.1007/s00395-005-0549-0
- Kato T, Niizuma S, Inuzuka Y, Kawashima T, Okuda J, Tamaki Y, Iwanaga Y, Narazaki M, Matsuda T, Soga T, Kita T, Kimura T, Shioi T (2010). Analysis of metabolic remodeling in compensated left ventricular hypertrophy and heart failure. Circ Heart Fail 3(3): 420-430. doi: 10.1161/CIRCHEARTFAILURE.109.888479
- Doenst T, Nguyen TD, Abel ED (2013). Cardiac metabolism in heart failure: implications beyond ATP production. Circ Res 113(6): 709-724. doi: 10.1161/CIRCRESAHA.113.300376
- Kolwicz SC, Jr., Purohit S, Tian R (2013). Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ Res 113(5): 603-616. doi: 10.1161/CIRCRESAHA.113.302095
- Heather LC, Cole MA, Lygate CA, Evans RD, Stuckey DJ, Murray AJ, Neubauer S, Clarke K (2006). Fatty acid transporter levels and palmitate oxidation rate correlate with ejection fraction in the infarcted rat heart. Cardiovasc Res 72(3): 430-437. doi: 10.1016/j.cardiores.2006.08.020
- Osorio JC, Stanley WC, Linke A, Castellari M, Diep QN, Panchal AR, Hintze TH, Lopaschuk GD, Recchia FA (2002). Impaired myocardial fatty acid oxidation and reduced protein expression of retinoid X receptor-alpha in pacing-induced heart failure. Circulation 106(5): 606-612. PMID: 12147544
- Bedi KC, Jr., Snyder NW, Brandimarto J, Aziz M, Mesaros C, Worth AJ, Wang LL, Javaheri A, Blair IA, Margulies KB, Rame JE (2016). Evidence for Intramyocardial Disruption of Lipid Metabolism and Increased Myocardial Ketone Utilization in Advanced Human Heart Failure. Circulation 133(8): 706-716. doi: 10.1161/CIRCULATIONAHA.115.017545
- Davila-Roman VG, Vedala G, Herrero P, de las Fuentes L, Rogers JG, Kelly DP, Gropler RJ (2002). Altered myocardial fatty acid and glucose metabolism in idiopathic dilated cardiomyopathy. J Am Coll Cardiol 40(2): 271-277. doi: 10.1016/s0735-1097(02)01967-8
- Barger PM, Brandt JM, Leone TC, Weinheimer CJ, Kelly DP (2000). Deactivation of peroxisome proliferator-activated receptor-alpha during cardiac hypertrophic growth. J Clin Invest 105(12): 1723-1730. doi: 10.1172/JCI9056
- Lahey R, Wang X, Carley AN, Lewandowski ED (2014). Dietary fat supply to failing hearts determines dynamic lipid signaling for nuclear receptor activation and oxidation of stored triglyceride. Circulation 130(20): 1790-1799. doi: 10.1161/CIRCULATIONAHA.114.011687
- Krishnan J, Suter M, Windak R, Krebs T, Felley A, Montessuit C, Tokarska-Schlattner M, Aasum E, Bogdanova A, Perriard E, Perriard JC, Larsen T, Pedrazzini T, Krek W (2009). Activation of a HIF1alpha-PPARgamma axis underlies the integration of glycolytic and lipid anabolic pathways in pathologic cardiac hypertrophy. Cell Metab 9(6): 512-524. doi: 10.1016/j.cmet.2009.05.005
- Sack MN, Rader TA, Park S, Bastin J, McCune SA, Kelly DP (1996). Fatty acid oxidation enzyme gene expression is downregulated in the failing heart. Circulation 94(11): 2837-2842. doi: 10.1161/01.cir.94.11.2837
- Opie LH, Knuuti J (2009). The adrenergic-fatty acid load in heart failure. J Am Coll Cardiol 54(18): 1637-1646. doi: 10.1016/j.jacc.2009.07.024
- Sharma S, Adrogue JV, Golfman L, Uray I, Lemm J, Youker K, Noon GP, Frazier OH, Taegtmeyer H (2004). Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J 18(14): 1692-1700. doi: 10.1096/fj.04-2263com
- Goldberg IJ, Trent CM, Schulze PC (2012). Lipid metabolism and toxicity in the heart. Cell Metab 15(6): 805-812. doi: 10.1016/j.cmet.2012.04.006
- Wende AR, Abel ED (2010). Lipotoxicity in the heart. Biochim Biophys acta 1801(3): 311-319. doi: 10.1016/j.bbalip.2009.09.023
- Kim JK, Fillmore JJ, Sunshine MJ, Albrecht B, Higashimori T, Kim DW, Liu ZX, Soos TJ, Cline GW, O'Brien WR, Littman DR, Shulman GI (2004). PKC-theta knockout mice are protected from fat-induced insulin resistance. J Clin Invest 114(6): 823-827. doi: 10.1172/JCI22230
- Yuan M, Konstantopoulos N, Lee J, Hansen L, Li ZW, Karin M, Shoelson SE (2001). Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science 293(5535): 1673-1677. doi: 10.1126/science.1061620
- Chokshi A, Drosatos K, Cheema FH, Ji R, Khawaja T, Yu S, Kato T, Khan R, Takayama H, Knoll R, Milting H, Chung CS, Jorde U, Naka Y, Mancini DM, Goldberg IJ, Schulze PC (2012). Ventricular assist device implantation corrects myocardial lipotoxicity, reverses insulin resistance, and normalizes cardiac metabolism in patients with advanced heart failure. Circulation 125(23): 2844-2853. doi: 10.1161/CIRCULATIONAHA.111.060889
- Taegtmeyer H, Beauloye C, Harmancey R, Hue L (2013). Insulin resistance protects the heart from fuel overload in dysregulated metabolic states. Am J Physiol Heart Circ Physiol 305(12): H1693-1697. doi: 10.1152/ajpheart.00854.2012
- Pound KM, Sorokina N, Ballal K, Berkich DA, Fasano M, Lanoue KF, Taegtmeyer H, O'Donnell JM, Lewandowski ED (2009). Substrate-enzyme competition attenuates upregulated anaplerotic flux through malic enzyme in hypertrophied rat heart and restores triacylglyceride content: attenuating upregulated anaplerosis in hypertrophy. Circ Res 104(6): 805-812. doi: 10.1161/CIRCRESAHA.108.189951
- Sorokina N, O'Donnell JM, McKinney RD, Pound KM, Woldegiorgis G, LaNoue KF, Ballal K, Taegtmeyer H, Buttrick PM, Lewandowski ED (2007). Recruitment of compensatory pathways to sustain oxidative flux with reduced carnitine palmitoyltransferase I activity characterizes inefficiency in energy metabolism in hypertrophied hearts. Circulation 115(15): 2033-2041. doi: 10.1161/CIRCULATIONAHA.106.668665
- Diakos NA, Navankasattusas S, Abel ED, Rutter J, McCreath L, Ferrin P, McKellar SH, Miller DV, Park SY, Richardson RS, Deberardinis R, Cox JE, Kfoury AG, Selzman CH, Stehlik J, Fang JC, Li DY, Drakos SG (2016). Evidence of Glycolysis Up-Regulation and Pyruvate Mitochondrial Oxidation Mismatch During Mechanical Unloading of the Failing Human Heart: Implications for Cardiac Reloading and Conditioning. JACC Basic Transl Sci 1(6): 432-444. doi: 10.1016/j.jacbts.2016.06.009
- Nascimben L, Ingwall JS, Lorell BH, Pinz I, Schultz V, Tornheim K, Tian R (2004). Mechanisms for increased glycolysis in the hypertrophied rat heart. Hypertension 44(5): 662-667. doi: 10.1161/01.HYP.0000144292.69599.0c
- Atherton HJ, Dodd MS, Heather LC, Schroeder MA, Griffin JL, Radda GK, Clarke K, Tyler DJ (2011). Role of pyruvate dehydrogenase inhibition in the development of hypertrophy in the hyperthyroid rat heart: a combined magnetic resonance imaging and hyperpolarized magnetic resonance spectroscopy study. Circulation 123(22): 2552-2561. doi: 10.1161/CIRCULATIONAHA.110.011387
- Mori J, Basu R, McLean BA, Das SK, Zhang L, Patel VB, Wagg CS, Kassiri Z, Lopaschuk GD, Oudit GY (2012). Agonist-induced hypertrophy and diastolic dysfunction are associated with selective reduction in glucose oxidation: a metabolic contribution to heart failure with normal ejection fraction. Circ Heart Fail 5(4): 493-503. doi: 10.1161/CIRCHEARTFAILURE.112.966705
- Zhabyeyev P, Gandhi M, Mori J, Basu R, Kassiri Z, Clanachan A, Lopaschuk GD, Oudit GY (2013). Pressure-overload-induced heart failure induces a selective reduction in glucose oxidation at physiological afterload. Cardiovasc Res 97(4): 676-685. doi: 10.1093/cvr/cvs424
- Zhang L, Jaswal JS, Ussher JR, Sankaralingam S, Wagg C, Zaugg M, Lopaschuk GD (2013). Cardiac insulin-resistance and decreased mitochondrial energy production precede the development of systolic heart failure after pressure-overload hypertrophy. Circ Heart Fail 6(5): 1039-1048. doi: 10.1161/CIRCHEARTFAILURE.112.000228
- Swan JW, Anker SD, Walton C, Godsland IF, Clark AL, Leyva F, Stevenson JC, Coats AJ (1997). Insulin resistance in chronic heart failure: relation to severity and etiology of heart failure. J Am Coll Cardiol 30(2): 527-532. doi: 10.1016/s0735-1097(97)00185-x
- Lydell CP, Chan A, Wambolt RB, Sambandam N, Parsons H, Bondy GP, Rodrigues B, Popov KM, Harris RA, Brownsey RW, Allard MF (2002). Pyruvate dehydrogenase and the regulation of glucose oxidation in hypertrophied rat hearts. Cardiovasc Res 53(4): 841-851. doi: 10.1016/s0008-6363(01)00560-0
- Aubert G, Martin OJ, Horton JL, Lai L, Vega RB, Leone TC, Koves T, Gardell SJ, Kruger M, Hoppel CL, Lewandowski ED, Crawford PA, Muoio DM, Kelly DP (2016). The Failing Heart Relies on Ketone Bodies as a Fuel. Circulation 133(8): 698-705. doi: 10.1161/CIRCULATIONAHA.115.017355
- Cahill GF, Jr. (2006). Fuel metabolism in starvation. Annu Rev Nutr 26: 1-22. doi: 10.1146/annurev.nutr.26.061505.111258
- Neubauer S, Horn M, Cramer M, Harre K, Newell JB, Peters W, Pabst T, Ertl G, Hahn D, Ingwall JS, Kochsiek K (1997). Myocardial phosphocreatine-to-ATP ratio is a predictor of mortality in patients with dilated cardiomyopathy. Circulation 96(7): 2190-2196. doi: 10.1161/01.cir.96.7.2190
- Lygate CA, Aksentijevic D, Dawson D, ten Hove M, Phillips D, de Bono JP, Medway DJ, Sebag-Montefiore L, Hunyor I, Channon KM, Clarke K, Zervou S, Watkins H, Balaban RS, Neubauer S (2013). Living without creatine: unchanged exercise capacity and response to chronic myocardial infarction in creatine-deficient mice. Circ Res 112(6): 945-955. doi: 10.1161/CIRCRESAHA.112.300725
- Frey N, Olson EN (2003). Cardiac hypertrophy: the good, the bad, and the ugly. Annu Rev Physiol 65: 45-79. doi: 10.1146/annurev.physiol.65.092101.142243
- Tian R, Musi N, D'Agostino J, Hirshman MF, Goodyear LJ (2001). Increased adenosine monophosphate-activated protein kinase activity in rat hearts with pressure-overload hypertrophy. Circulation 104(14): 1664-1669. doi: 10.1161/hc4001.097183
- Allard MF, Flint JD, English JC, Henning SL, Salamanca MC, Kamimura CT, English DR (1994). Calcium overload during reperfusion is accelerated in isolated hypertrophied rat hearts. J Mol Cell Cardiol 26(12): 1551-1563. doi: 10.1006/jmcc.1994.1175
- Schonekess BO, Allard MF, Lopaschuk GD (1996). Recovery of glycolysis and oxidative metabolism during postischemic reperfusion of hypertrophied rat hearts. Am J Physiol 271(2 Pt 2): H798-805. doi: 10.1152/ajpheart.1996.271.2.H798
- Deswal A, Petersen NJ, Feldman AM, Young JB, White BG, Mann DL (2001). Cytokines and cytokine receptors in advanced heart failure: an analysis of the cytokine database from the Vesnarinone trial (VEST). Circulation 103(16): 2055-2059. doi: 10.1016/s1062-1458(01)00394-4
- Seta Y, Shan K, Bozkurt B, Oral H, Mann DL (1996). Basic mechanisms in heart failure: the cytokine hypothesis. J Card Fail 2(3): 243-249. doi: 10.1016/s1071-9164(96)80047-9
- Briasoulis A, Androulakis E, Christophides T, Tousoulis D (2016). The role of inflammation and cell death in the pathogenesis, progression and treatment of heart failure. Heart Fail Rev 21(2): 169-176. doi: 10.1007/s10741-016-9533-z
- Dick SA, Epelman S (2016). Chronic Heart Failure and Inflammation: What Do We Really Know? Circ Res 119(1): 159-176. doi: 10.1161/CIRCRESAHA.116.308030
- Levine B, Kalman J, Mayer L, Fillit HM, Packer M (1990). Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med 323(4): 236-241. doi: 10.1056/NEJM199007263230405
- Bergman MR, Kao RH, McCune SA, Holycross BJ (1999). Myocardial tumor necrosis factor-alpha secretion in hypertensive and heart failure-prone rats. Am J Physiol 277(2): H543-550. doi: 10.1152/ajpheart.1999.277.2.H543
- Kubota T, McTiernan CF, Frye CS, Slawson SE, Lemster BH, Koretsky AP, Demetris AJ, Feldman AM (1997). Dilated cardiomyopathy in transgenic mice with cardiac-specific overexpression of tumor necrosis factor-alpha. Circ Res 81(4): 627-635. doi: 10.1161/01.res.81.4.627
- Gupta S, Young D, Maitra RK, Gupta A, Popovic ZB, Yong SL, Mahajan A, Wang Q, Sen S (2008). Prevention of cardiac hypertrophy and heart failure by silencing of NF-kappaB. J Mol Biol 375(3): 637-649. doi: 10.1016/j.jmb.2007.10.006
- Palomer X, Alvarez-Guardia D, Rodriguez-Calvo R, Coll T, Laguna JC, Davidson MM, Chan TO, Feldman AM, Vazquez-Carrera M (2009). TNF-alpha reduces PGC-1alpha expression through NF-kappaB and p38 MAPK leading to increased glucose oxidation in a human cardiac cell model. Cardiovasc Res 81(4): 703-712. doi: 10.1093/cvr/cvn327
- Alvarez-Guardia D, Palomer X, Coll T, Davidson MM, Chan TO, Feldman AM, Laguna JC, Vazquez-Carrera M (2010). The p65 subunit of NF-kappaB binds to PGC-1alpha, linking inflammation and metabolic disturbances in cardiac cells. Cardiovasc Res 87(3): 449-458. doi: 10.1093/cvr/cvq080
- Maack C, Lehrke M, Backs J, Heinzel FR, Hulot JS, Marx N, Paulus WJ, Rossignol P, Taegtmeyer H, Bauersachs J, Bayes-Genis A, Brutsaert D, Bugger H, Clarke K, Cosentino F, De Keulenaer G, Dei Cas A, Gonzalez A, Huelsmann M, Iaccarino G, Lunde IG, Lyon AR, Pollesello P, Rena G, Riksen NP, Rosano G, Staels B, van Laake LW, Wanner C, Farmakis D, et al. (2018). Heart failure and diabetes: metabolic alterations and therapeutic interventions: a state-of-the-art review from the Translational Research Committee of the Heart Failure Association-European Society of Cardiology. Eur Heart J. doi: 10.1093/eurheartj/ehy596
- Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A, Han X, Gross RW, Kozak R, Lopaschuk GD, Kelly DP (2002). The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J Clin Invest 109(1): 121-130. doi: 10.1172/JCI14080
- Kaufman BA, Li C, Soleimanpour SA (2015). Mitochondrial regulation of beta-cell function: maintaining the momentum for insulin release. Mol Aspects Med 42(91-104. doi: 10.1016/j.mam.2015.01.004
- Sakai K, Matsumoto K, Nishikawa T, Suefuji M, Nakamaru K, Hirashima Y, Kawashima J, Shirotani T, Ichinose K, Brownlee M, Araki E (2003). Mitochondrial reactive oxygen species reduce insulin secretion by pancreatic beta-cells. Biochem Biophys Res Commun 300(1): 216-222. doi: 10.1016/s0006-291x(02)02832-2
- Murray AJ, Panagia M, Hauton D, Gibbons GF, Clarke K (2005). Plasma free fatty acids and peroxisome proliferator-activated receptor alpha in the control of myocardial uncoupling protein levels. Diabetes 54(12): 3496-3502. doi: 10.2337/diabetes.54.12.3496
- Geraldes P, King GL (2010). Activation of protein kinase C isoforms and its impact on diabetic complications. Circ Res 106(8): 1319-1331. doi: 10.1161/CIRCRESAHA.110.217117
- Jiang ZY, Lin YW, Clemont A, Feener EP, Hein KD, Igarashi M, Yamauchi T, White MF, King GL (1999). Characterization of selective resistance to insulin signaling in the vasculature of obese Zucker (fa/fa) rats. J Clin Invest 104(4): 447-457. doi: 10.1172/JCI5971
- Arikawa E, Ma RC, Isshiki K, Luptak I, He Z, Yasuda Y, Maeno Y, Patti ME, Weir GC, Harris RA, Zammit VA, Tian R, King GL (2007). Effects of insulin replacements, inhibitors of angiotensin, and PKCbeta's actions to normalize cardiac gene expression and fuel metabolism in diabetic rats. Diabetes 56(5): 1410-1420. doi: 10.2337/db06-0655
- Levelt E, Rodgers CT, Clarke WT, Mahmod M, Ariga R, Francis JM, Liu A, Wijesurendra RS, Dass S, Sabharwal N, Robson MD, Holloway CJ, Rider OJ, Clarke K, Karamitsos TD, Neubauer S (2016). Cardiac energetics, oxygenation, and perfusion during increased workload in patients with type 2 diabetes mellitus. Eur Heart J 37(46): 3461-3469. doi: 10.1093/eurheartj/ehv442
- McGavock JM, Lingvay I, Zib I, Tillery T, Salas N, Unger R, Levine BD, Raskin P, Victor RG, Szczepaniak LS (2007). Cardiac steatosis in diabetes mellitus: a 1H-magnetic resonance spectroscopy study. Circulation 116(10): 1170-1175. doi: 10.1161/CIRCULATIONAHA.106.645614
- Rijzewijk LJ, van der Meer RW, Smit JW, Diamant M, Bax JJ, Hammer S, Romijn JA, de Roos A, Lamb HJ (2008). Myocardial steatosis is an independent predictor of diastolic dysfunction in type 2 diabetes mellitus. J Am Coll Cardiol 52(22): 1793-1799. doi: 10.1016/j.jacc.2008.07.062
- Cole MA, Murray AJ, Cochlin LE, Heather LC, McAleese S, Knight NS, Sutton E, Jamil AA, Parassol N, Clarke K (2011). A high fat diet increases mitochondrial fatty acid oxidation and uncoupling to decrease efficiency in rat heart. Basic Res Cardiol 106(3): 447-457. doi: 10.1007/s00395-011-0156-1
- Kruljac I, Cacic M, Cacic P, Ostojic V, Stefanovic M, Sikic A, Vrkljan M (2017). Diabetic ketosis during hyperglycemic crisis is associated with decreased all-cause mortality in patients with type 2 diabetes mellitus. Endocrine 55(1): 139-143. doi: 10.1007/s12020-016-1082-7
- Murray AJ, Anderson RE, Watson GC, Radda GK, Clarke K (2004). Uncoupling proteins in human heart. Lancet 364(9447): 1786-1788. doi: 10.1016/S0140-6736(04)17402-3
- Banke NH, Lewandowski ED (2015). Impaired cytosolic NADH shuttling and elevated UCP3 contribute to inefficient citric acid cycle flux support of postischemic cardiac work in diabetic hearts. J Mol Cell Cardiol 79: 13-20. doi: 10.1016/j.yjmcc.2014.10.015
- Gustafsson AB, Gottlieb RA (2008). Recycle or die: the role of autophagy in cardioprotection. J Mol Cell Cardiol 44(4): 654-661. doi: 10.1016/j.yjmcc.2008.01.010
- Khalid AM, Hafstad AD, Larsen TS, Severson DL, Boardman N, Hagve M, Berge RK, Aasum E (2011). Cardioprotective effect of the PPAR ligand tetradecylthioacetic acid in type 2 diabetic mice. Am J Physiol Heart Circ Physiol 300(6): H2116-2122. doi: 10.1152/ajpheart.00357.2010
- Sidell RJ, Cole MA, Draper NJ, Desrois M, Buckingham RE, Clarke K (2002). Thiazolidinedione treatment normalizes insulin resistance and ischemic injury in the zucker Fatty rat heart. Diabetes 51(4): 1110-1117. doi: 10.2337/diabetes.51.4.1110
- Aasum E, Belke DD, Severson DL, Riemersma RA, Cooper M, Andreassen M, Larsen TS (2002). Cardiac function and metabolism in Type 2 diabetic mice after treatment with BM 17.0744, a novel PPAR-alpha activator. Am J Physiol Heart Circ Physiol 283(3): H949-957. doi: 10.1152/ajpheart.00226.2001
- Carley AN, Semeniuk LM, Shimoni Y, Aasum E, Larsen TS, Berger JP, Severson DL (2004). Treatment of type 2 diabetic db/db mice with a novel PPARgamma agonist improves cardiac metabolism but not contractile function. Am J Physiol Endocrinol Metab 286(3): E449-455. doi: 10.1152/ajpendo.00329.2003
- Mizuno Y, Harada E, Nakagawa H, Morikawa Y, Shono M, Kugimiya F, Yoshimura M, Yasue H (2017). The diabetic heart utilizes ketone bodies as an energy source. Metabolism 77: 65-72. doi: 10.1016/j.metabol.2017.08.005
- Cox PJ, Kirk T, Ashmore T, Willerton K, Evans R, Smith A, Murray AJ, Stubbs B, West J, McLure SW, King MT, Dodd MS, Holloway C, Neubauer S, Drawer S, Veech RL, Griffin JL, Clarke K (2016). Nutritional Ketosis Alters Fuel Preference and Thereby Endurance Performance in Athletes. Cell Metab 24(2): 256-268. doi: 10.1016/j.cmet.2016.07.010
- Murray AJ, Knight NS, Cole MA, Cochlin LE, Carter E, Tchabanenko K, Pichulik T, Gulston MK, Atherton HJ, Schroeder MA, Deacon RM, Kashiwaya Y, King MT, Pawlosky R, Rawlins JN, Tyler DJ, Griffin JL, Robertson J, Veech RL, Clarke K (2016). Novel ketone diet enhances physical and cognitive performance. FASEB J 30(12): 4021-4032. doi: 10.1096/fj.201600773R
- Sato K, Kashiwaya Y, Keon CA, Tsuchiya N, King MT, Radda GK, Chance B, Clarke K, Veech RL (1995). Insulin, ketone bodies, and mitochondrial energy transduction. FASEB J 9(8): 651-658. doi: 10.1096/fasebj.9.8.7768357
- Baker RG, Hayden MS, Ghosh S (2011). NF-kappaB, inflammation, and metabolic disease. Cell Metab 13(1): 11-22. doi: 10.1016/j.cmet.2010.12.008
- Gordon JW, Shaw JA, Kirshenbaum LA (2011). Multiple facets of NF-kappaB in the heart: to be or not to NF-kappaB. Circ Res 108(9): 1122-1132. doi: 10.1161/CIRCRESAHA.110.226928
- Shah MS, Brownlee M (2016). Molecular and Cellular Mechanisms of Cardiovascular Disorders in Diabetes. Circ Res 118(11): 1808-1829. doi: 10.1161/CIRCRESAHA.116.306923
- Thomas CM, Yong QC, Rosa RM, Seqqat R, Gopal S, Casarini DE, Jones WK, Gupta S, Baker KM, Kumar R (2014). Cardiac-specific suppression of NF-kappaB signaling prevents diabetic cardiomyopathy via inhibition of the renin-angiotensin system. Am J Physiol Heart Circ Physiol 307(7): H1036-1045. doi: 10.1152/ajpheart.00340.2014
- Mariappan N, Elks CM, Sriramula S, Guggilam A, Liu Z, Borkhsenious O, Francis J (2010). NF-kappaB-induced oxidative stress contributes to mitochondrial and cardiac dysfunction in type II diabetes. Cardiovasc Res 85(3): 473-483. doi: 10.1093/cvr/cvp305
- Ko HJ, Zhang Z, Jung DY, Jun JY, Ma Z, Jones KE, Chan SY, Kim JK (2009). Nutrient stress activates inflammation and reduces glucose metabolism by suppressing AMP-activated protein kinase in the heart. Diabetes 58(11): 2536-2546. doi: 10.2337/db08-1361
- Du X, Matsumura T, Edelstein D, Rossetti L, Zsengeller Z, Szabo C, Brownlee M (2003). Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J Clin Invest 112(7): 1049-1057. doi: 10.1172/JCI18127
- Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, Brownlee M (2000). Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 404(6779): 787-790. doi: 10.1038/35008121
- Szabo C (2006). Poly(ADP-ribose) polymerase activation by reactive nitrogen species–relevance for the pathogenesis of inflammation. Nitric Oxide 14(2): 169-179. doi: 10.1016/j.niox.2005.06.008
- Pradhan AD, Manson JE, Rifai N, Buring JE, Ridker PM (2001). C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA 286(3): 327-334. doi: 10.1001/jama.286.3.327
- Li J, Zhu H, Shen E, Wan L, Arnold JM, Peng T (2010). Deficiency of rac1 blocks NADPH oxidase activation, inhibits endoplasmic reticulum stress, and reduces myocardial remodeling in a mouse model of type 1 diabetes. Diabetes 59(8): 2033-2042. doi: 10.2337/db09-1800
- Bugger H, Abel ED (2014). Molecular mechanisms of diabetic cardiomyopathy. Diabetologia 57(4): 660-671. doi: 10.1007/s00125-014-3171-6
- Buck MD, O'Sullivan D, Pearce EL (2015). T cell metabolism drives immunity. J Exp Med 212(9): 1345-1360. doi: 10.1084/jem.20151159
- Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, Zheng L, Gardet A, Tong Z, Jany SS, Corr SC, Haneklaus M, Caffrey BE, Pierce K, Walmsley S, Beasley FC, Cummins E, Nizet V, Whyte M, Taylor CT, Lin H, Masters SL, Gottlieb E, Kelly VP, Clish C, Auron PE, et al. (2013). Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 496(7444): 238-242. doi: 10.1038/nature11986
- Borregaard N, Herlin T (1982). Energy metabolism of human neutrophils during phagocytosis. J Clin Invest 70(3): 550-557. doi: 10.1172/jci110647
- Fossati G, Moulding DA, Spiller DG, Moots RJ, White MR, Edwards SW (2003). The mitochondrial network of human neutrophils: role in chemotaxis, phagocytosis, respiratory burst activation, and commitment to apoptosis. J Immunol 170(4): 1964-1972. doi: 10.4049/jimmunol.170.4.1964
- Maianski NA, Geissler J, Srinivasula SM, Alnemri ES, Roos D, Kuijpers TW (2004). Functional characterization of mitochondria in neutrophils: a role restricted to apoptosis. Cell Death Differ 11(2): 143-153. doi: 10.1038/sj.cdd.4401320
- Newsholme P, Curi R, Gordon S, Newsholme EA (1986). Metabolism of glucose, glutamine, long-chain fatty acids and ketone bodies by murine macrophages. Biochem J 239(1): 121-125. doi: 10.1042/bj2390121
- Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, Cross JR, Jung E, Thompson CB, Jones RG, Pearce EJ (2010). Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 115(23): 4742-4749. doi: 10.1182/blood-2009-10-249540
- Pearce EJ, Everts B (2015). Dendritic cell metabolism. Nat Rev Immunol 15(1): 18-29. doi: 10.1038/nri3771
- Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, Chmielewski K, Stewart KM, Ashall J, Everts B, Pearce EJ, Driggers EM, Artyomov MN (2015). Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 42(3): 419-430. doi: 10.1016/j.immuni.2015.02.005
- Michelucci A, Cordes T, Ghelfi J, Pailot A, Reiling N, Goldmann O, Binz T, Wegner A, Tallam A, Rausell A, Buttini M, Linster CL, Medina E, Balling R, Hiller K (2013). Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc Natl Acad Sci U S A 110(19): 7820-7825. doi: 10.1073/pnas.1218599110
- Amiel E, Everts B, Fritz D, Beauchamp S, Ge B, Pearce EL, Pearce EJ (2014). Mechanistic target of rapamycin inhibition extends cellular lifespan in dendritic cells by preserving mitochondrial function. J Immunol 193(6): 2821-2830. doi: 10.4049/jimmunol.1302498
- Amiel E, Everts B, Freitas TC, King IL, Curtis JD, Pearce EL, Pearce EJ (2012). Inhibition of mechanistic target of rapamycin promotes dendritic cell activation and enhances therapeutic autologous vaccination in mice. J Immunol 189(5): 2151-2158. doi: 10.4049/jimmunol.1103741
- Rodriguez-Espinosa O, Rojas-Espinosa O, Moreno-Altamirano MM, Lopez-Villegas EO, Sanchez-Garcia FJ (2015). Metabolic requirements for neutrophil extracellular traps formation. Immunology 145(2): 213-224. doi: 10.1111/imm.12437
- Azevedo EP, Rochael NC, Guimaraes-Costa AB, de Souza-Vieira TS, Ganilho J, Saraiva EM, Palhano FL, Foguel D (2015). A Metabolic Shift toward Pentose Phosphate Pathway Is Necessary for Amyloid Fibril- and Phorbol 12-Myristate 13-Acetate-induced Neutrophil Extracellular Trap (NET) Formation. J Biol Chem 290(36): 22174-22183. doi: 10.1074/jbc.M115.640094
- McInturff AM, Cody MJ, Elliott EA, Glenn JW, Rowley JW, Rondina MT, Yost CC (2012). Mammalian target of rapamycin regulates neutrophil extracellular trap formation via induction of hypoxia-inducible factor 1 alpha. Blood 120(15): 3118-3125. doi: 10.1182/blood-2012-01-405993
- Donnelly RP, Loftus RM, Keating SE, Liou KT, Biron CA, Gardiner CM, Finlay DK (2014). mTORC1-dependent metabolic reprogramming is a prerequisite for NK cell effector function. J Immunol 193(9): 4477-4484. doi: 10.4049/jimmunol.1401558
- Doughty CA, Bleiman BF, Wagner DJ, Dufort FJ, Mataraza JM, Roberts MF, Chiles TC (2006). Antigen receptor-mediated changes in glucose metabolism in B lymphocytes: role of phosphatidylinositol 3-kinase signaling in the glycolytic control of growth. Blood 107(11): 4458-4465. doi: 10.1182/blood-2005-12-4788
- Dufort FJ, Bleiman BF, Gumina MR, Blair D, Wagner DJ, Roberts MF, Abu-Amer Y, Chiles TC (2007). Cutting edge: IL-4-mediated protection of primary B lymphocytes from apoptosis via Stat6-dependent regulation of glycolytic metabolism. J Immunol 179(8): 4953-4957. doi: 10.4049/jimmunol.179.8.4953
- Caro-Maldonado A, Wang R, Nichols AG, Kuraoka M, Milasta S, Sun LD, Gavin AL, Abel ED, Kelsoe G, Green DR, Rathmell JC (2014). Metabolic reprogramming is required for antibody production that is suppressed in anergic but exaggerated in chronically BAFF-exposed B cells. J Immunol 192(8): 3626-3636. doi: 10.4049/jimmunol.1302062
- Donnelly RP, Finlay DK (2015). Glucose, glycolysis and lymphocyte responses. Mol Immunol 68(2 Pt C): 513-519. doi: 10.1016/j.molimm.2015.07.034
- Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger J, Green DR (2011). The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35(6): 871-882. doi: 10.1016/j.immuni.2011.09.021
- Oestreich KJ, Read KA, Gilbertson SE, Hough KP, McDonald PW, Krishnamoorthy V, Weinmann AS (2014). Bcl-6 directly represses the gene program of the glycolysis pathway. Nat Immunol 15(10): 957-964. doi: 10.1038/ni.2985
- Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, Chi H (2011). HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med 208(7): 1367-1376. doi: 10.1084/jem.20110278
- Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, Tsui YC, Cui G, Micevic G, Perales JC, Kleinstein SH, Abel ED, Insogna KL, Feske S, Locasale JW, Bosenberg MW, Rathmell JC, Kaech SM (2015). Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 162(6): 1217-1228. doi: 10.1016/j.cell.2015.08.012
- Vitko NP, Spahich NA, Richardson AR (2015). Glycolytic dependency of high-level nitric oxide resistance and virulence in Staphylococcus aureus. MBio 6(2). doi: 10.1128/mBio.00045-15
- Ripoli M, D'Aprile A, Quarato G, Sarasin-Filipowicz M, Gouttenoire J, Scrima R, Cela O, Boffoli D, Heim MH, Moradpour D, Capitanio N, Piccoli C (2010). Hepatitis C virus-linked mitochondrial dysfunction promotes hypoxia-inducible factor 1 alpha-mediated glycolytic adaptation. J Virol 84(1): 647-660. doi: 10.1128/JVI.00769-09
- Yu Y, Maguire TG, Alwine JC (2011). Human cytomegalovirus activates glucose transporter 4 expression to increase glucose uptake during infection. J Virol 85(4): 1573-1580. doi: 10.1128/JVI.01967-10
- Thai M, Graham NA, Braas D, Nehil M, Komisopoulou E, Kurdistani SK, McCormick F, Graeber TG, Christofk HR (2014). Adenovirus E4ORF1-induced MYC activation promotes host cell anabolic glucose metabolism and virus replication. Cell Metab 19(4): 694-701. doi: 10.1016/j.cmet.2014.03.009
- Schodel J, Oikonomopoulos S, Ragoussis J, Pugh CW, Ratcliffe PJ, Mole DR (2011). High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood 117(23): e207-217. doi: 10.1182/blood-2010-10-314427
- Wenger RH, Stiehl DP, Camenisch G (2005). Integration of oxygen signaling at the consensus HRE. Sci STKE 2005(306): re12. doi: 10.1126/stke.3062005re12
- McNamee EN, Korns Johnson D, Homann D, Clambey ET (2013). Hypoxia and hypoxia-inducible factors as regulators of T cell development, differentiation, and function. Immunol Res 55(1-3): 58-70. doi: 10.1007/s12026-012-8349-8
- Caldwell CC, Tschoep J, Lentsch AB (2007). Lymphocyte function during hepatic ischemia/reperfusion injury. J Leukoc Biol 82(3): 457-464. doi: 10.1189/jlb.0107062
- Huang Y, Rabb H, Womer KL (2007). Ischemia-reperfusion and immediate T cell responses. Cell Immunol 248(1): 4-11. doi: 10.1016/j.cellimm.2007.03.009
- Rausch ME, Weisberg S, Vardhana P, Tortoriello DV (2008). Obesity in C57BL/6J mice is characterized by adipose tissue hypoxia and cytotoxic T-cell infiltration. Int J Obes 32(3): 451-463. doi: 10.1038/sj.ijo.0803744
- Semenza GL (2011). Oxygen sensing, homeostasis, and disease. N Engl J Med 365(6): 537-547. doi: 10.1056/NEJMra1011165
- Biju MP, Neumann AK, Bensinger SJ, Johnson RS, Turka LA, Haase VH (2004). Vhlh gene deletion induces Hif-1-mediated cell death in thymocytes. Mol Cell Biol 24(20): 9038-9047. doi: 10.1128/MCB.24.20.9038-9047.2004
- Neumann AK, Yang J, Biju MP, Joseph SK, Johnson RS, Haase VH, Freedman BD, Turka LA (2005). Hypoxia inducible factor 1 alpha regulates T cell receptor signal transduction. Proc Natl Acad Sci U S A 102(47): 17071-17076. doi: 10.1073/pnas.0506070102
- Caldwell CC, Kojima H, Lukashev D, Armstrong J, Farber M, Apasov SG, Sitkovsky MV (2001). Differential effects of physiologically relevant hypoxic conditions on T lymphocyte development and effector functions. J Immunol 167(11): 6140-6149. doi: 10.4049/jimmunol.167.11.6140
- Lukashev D, Klebanov B, Kojima H, Grinberg A, Ohta A, Berenfeld L, Wenger RH, Sitkovsky M (2006). Cutting edge: hypoxia-inducible factor 1alpha and its activation-inducible short isoform I.1 negatively regulate functions of CD4+ and CD8+ T lymphocytes. J Immunol 177(8): 4962-4965. doi: 10.4049/jimmunol.177.8.4962
- Thiel M, Caldwell CC, Kreth S, Kuboki S, Chen P, Smith P, Ohta A, Lentsch AB, Lukashev D, Sitkovsky MV (2007). Targeted deletion of HIF-1alpha gene in T cells prevents their inhibition in hypoxic inflamed tissues and improves septic mice survival. PLoS One 2(9): e853. doi: 10.1371/journal.pone.0000853
- Gaber T, Haupl T, Sandig G, Tykwinska K, Fangradt M, Tschirschmann M, Hahne M, Dziurla R, Erekul K, Lautenbach M, Kolar P, Burmester GR, Buttgereit F (2009). Adaptation of human CD4+ T cells to pathophysiological hypoxia: a transcriptome analysis. J Rheumatol 36(12): 2655-2669. doi: 10.3899/jrheum.090255
- Nakamura H, Makino Y, Okamoto K, Poellinger L, Ohnuma K, Morimoto C, Tanaka H (2005). TCR engagement increases hypoxia-inducible factor-1 alpha protein synthesis via rapamycin-sensitive pathway under hypoxic conditions in human peripheral T cells. J Immunol 174(12): 7592-7599. doi: 10.4049/jimmunol.174.12.7592
- Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, Bordman Z, Fu J, Kim Y, Yen HR, Luo W, Zeller K, Shimoda L, Topalian SL, Semenza GL, Dang CV, Pardoll DM, Pan F (2011). Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell 146(5): 772-784. doi: 10.1016/j.cell.2011.07.033
- Tabarkiewicz J, Pogoda K, Karczmarczyk A, Pozarowski P, Giannopoulos K (2015). The Role of IL-17 and Th17 Lymphocytes in Autoimmune Diseases. Arch Immunol Ther Exp (Warsz) 63(6): 435-449. doi: 10.1007/s00005-015-0344-z
- Yamagata T, Skepner J, Yang J (2015). Targeting Th17 Effector Cytokines for the Treatment of Autoimmune Diseases. Arch Immunol Ther Exp 63(6): 405-414. doi: 10.1007/s00005-015-0362-x
- Ivanov, II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR (2006). The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126(6): 1121-1133. doi: 10.1016/j.cell.2006.07.035
- Gnanaprakasam JNR, Sherman JW, Wang R (2017). MYC and HIF in shaping immune response and immune metabolism. Cytokine Growth Factor Rev 35(63-70. doi: 10.1016/j.cytogfr.2017.03.004
- Ben-Shoshan J, Maysel-Auslender S, Mor A, Keren G, George J (2008). Hypoxia controls CD4+CD25+ regulatory T-cell homeostasis via hypoxia-inducible factor-1alpha. Eur J Immunol 38(9): 2412-2418. doi: 10.1002/eji.200838318
- Clambey ET, McNamee EN, Westrich JA, Glover LE, Campbell EL, Jedlicka P, de Zoeten EF, Cambier JC, Stenmark KR, Colgan SP, Eltzschig HK (2012). Hypoxia-inducible factor-1 alpha-dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc Natl Acad Sci U S A 109(41): E2784-2793. doi: 10.1073/pnas.1202366109
- Kojima H, Gu H, Nomura S, Caldwell CC, Kobata T, Carmeliet P, Semenza GL, Sitkovsky MV (2002). Abnormal B lymphocyte development and autoimmunity in hypoxia-inducible factor 1alpha -deficient chimeric mice. Proc Natl Acad Sci U S A 99(4): 2170-2174. doi: 10.1073/pnas.052706699
- Goda N, Ryan HE, Khadivi B, McNulty W, Rickert RC, Johnson RS (2003). Hypoxia-inducible factor 1alpha is essential for cell cycle arrest during hypoxia. Mol Cell Biol 23(1): 359-369. doi: 10.1046/j.1356-9597.2001.00512.x
- Shin DH, Lin H, Zheng H, Kim KS, Kim JY, Chun YS, Park JW, Nam JH, Kim WK, Zhang YH, Kim SJ (2014). HIF-1alpha-mediated upregulation of TASK-2 K(+) channels augments Ca(2)(+) signaling in mouse B cells under hypoxia. J Immunol 193(10): 4924-4933. doi: 10.4049/jimmunol.1301829
- Cho SH, Raybuck AL, Stengel K, Wei M, Beck TC, Volanakis E, Thomas JW, Hiebert S, Haase VH, Boothby MR (2016). Germinal centre hypoxia and regulation of antibody qualities by a hypoxia response system. Nature 537(7619): 234-238. doi: 10.1038/nature19334
- Hu CJ, Wang LY, Chodosh LA, Keith B, Simon MC (2003). Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol Cell Biol 23(24): 9361-9374. doi: 10.1128/mcb.23.24.9361-9374.2003
- Labonte AC, Tosello-Trampont AC, Hahn YS (2014). The role of macrophage polarization in infectious and inflammatory diseases. Mol Cells 37(4): 275-285. doi: 10.14348/molcells.2014.2374
- Sheikh SZ, Plevy SE (2010). The role of the macrophage in sentinel responses in intestinal immunity. Curr Opin Gastroenterol 26(6): 578-582. doi: 10.1097/MOG.0b013e32833d4b71
- Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, Firestein GS, Gerber HP, Ferrara N, Johnson RS (2003). HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell 112(5): 645-657. doi: 10.1016/s0092-8674(03)00154-5
- Lin N, Simon MC (2016). Hypoxia-inducible factors: key regulators of myeloid cells during inflammation. J Clin Invest 126(10): 3661-3671. doi: 10.1172/JCI84426
- Mills E, O'Neill LA (2014). Succinate: a metabolic signal in inflammation. Trends Cell Biol 24(5): 313-320. doi: 10.1016/j.tcb.2013.11.008
- Peyssonnaux C, Cejudo-Martin P, Doedens A, Zinkernagel AS, Johnson RS, Nizet V (2007). Cutting edge: Essential role of hypoxia inducible factor-1alpha in development of lipopolysaccharide-induced sepsis. J Immunol 178(12): 7516-7519. doi: 10.4049/jimmunol.178.12.7516
- Galvan-Pena S, O'Neill LA (2014). Metabolic reprograming in macrophage polarization. Front Immunol 5: 420. doi: 10.3389/fimmu.2014.00420
- Peyssonnaux C, Datta V, Cramer T, Doedens A, Theodorakis EA, Gallo RL, Hurtado-Ziola N, Nizet V, Johnson RS (2005). HIF-1alpha expression regulates the bactericidal capacity of phagocytes. J Clin Invest 115(7): 1806-1815. doi: 10.1172/JCI23865
- Blouin CC, Page EL, Soucy GM, Richard DE (2004). Hypoxic gene activation by lipopolysaccharide in macrophages: implication of hypoxia-inducible factor 1alpha. Blood 103(3): 1124-1130. doi: 10.1182/blood-2003-07-2427
- O'Neill LA, Pearce EJ (2016). Immunometabolism governs dendritic cell and macrophage function. J Exp Med 213(1): 15-23. doi: 10.1084/jem.20151570
- Naldini A, Morena E, Pucci A, Miglietta D, Riboldi E, Sozzani S, Carraro F (2012). Hypoxia affects dendritic cell survival: role of the hypoxia-inducible factor-1alpha and lipopolysaccharide. J Cell Physiol 227(2): 587-595. doi: 10.1002/jcp.22761
- Wobben R, Husecken Y, Lodewick C, Gibbert K, Fandrey J, Winning S (2013). Role of hypoxia inducible factor-1alpha for interferon synthesis in mouse dendritic cells. Biol Chem 394(4): 495-505. doi: 10.1515/hsz-2012-0320
- Kohler T, Reizis B, Johnson RS, Weighardt H, Forster I (2012). Influence of hypoxia-inducible factor 1alpha on dendritic cell differentiation and migration. Eur J Immunol 42(5): 1226-1236. doi: 10.1002/eji.201142053
- Hammami A, Charpentier T, Smans M, Stager S (2015). IRF-5-Mediated Inflammation Limits CD8+ T Cell Expansion by Inducing HIF-1alpha and Impairing Dendritic Cell Functions during Leishmania Infection. PLoS Pathog 11(6): e1004938. doi: 10.1371/journal.ppat.1004938
- Jantsch J, Chakravortty D, Turza N, Prechtel AT, Buchholz B, Gerlach RG, Volke M, Glasner J, Warnecke C, Wiesener MS, Eckardt KU, Steinkasserer A, Hensel M, Willam C (2008). Hypoxia and hypoxia-inducible factor-1 alpha modulate lipopolysaccharide-induced dendritic cell activation and function. J Immunol 180(7): 4697-4705. doi: 10.4049/jimmunol.180.7.4697
- Harris AJ, Thompson AR, Whyte MK, Walmsley SR (2014). HIF-mediated innate immune responses: cell signaling and therapeutic implications. Hypoxia 2: 47-58. doi: 10.2147/HP.S50269
- Walmsley SR, Cadwallader KA, Chilvers ER (2005). The role of HIF-1alpha in myeloid cell inflammation. Trends Immunol 26(8): 434-439. doi: 10.1016/j.it.2005.06.007
- Kong T, Eltzschig HK, Karhausen J, Colgan SP, Shelley CS (2004). Leukocyte adhesion during hypoxia is mediated by HIF-1-dependent induction of beta2 integrin gene expression. Proc Natl Acad Sci U S A 101(28): 10440-10445. doi: 10.1073/pnas.0401339101
- Wang L, Yang X, Li D, Liang Z, Chen Y, Ma G, Wang Y, Li Y, Liang Y, Niu H (2018). The elevated glutaminolysis of bladder cancer and T cells in a simulated tumor microenvironment contributes to the up-regulation of PD-L1 expression by interferon-gamma. Onco Targets Ther 11: 7229-7243. doi: 10.2147/OTT.S180505
- Kahn BB, Alquier T, Carling D, Hardie DG (2005). AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab 1(1): 15-25. doi: 10.1016/j.cmet.2004.12.003
- Hardie DG (2004). The AMP-activated protein kinase pathway–new players upstream and downstream. J Cell Sci 117(Pt 23): 5479-5487. doi: 10.1242/jcs.01540
- Hawley SA, Davison M, Woods A, Davies SP, Beri RK, Carling D, Hardie DG (1996). Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J Biol Chem 271(44): 27879-27887. doi: 10.1074/jbc.271.44.27879
- Marsin AS, Bertrand L, Rider MH, Deprez J, Beauloye C, Vincent MF, Van den Berghe G, Carling D, Hue L (2000). Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Current biology 10(20): 1247-1255. doi: 10.1016/s0960-9822(00)00742-9
- Hardie DG (2011). AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev 25(18): 1895-1908. doi: 10.1101/gad.17420111
- Yeh LA, Lee KH, Kim KH (1980). Regulation of rat liver acetyl-CoA carboxylase. Regulation of phosphorylation and inactivation of acetyl-CoA carboxylase by the adenylate energy charge. J Biol Chem 255(6): 2308-2314. PMID: 6102090
- Davies SP, Sim AT, Hardie DG (1990). Location and function of three sites phosphorylated on rat acetyl-CoA carboxylase by the AMP-activated protein kinase. Eur J Biochem 187(1): 183-190. doi: 10.1111/j.1432-1033.1990.tb15293.x
- Li Y, Xu S, Mihaylova MM, Zheng B, Hou X, Jiang B, Park O, Luo Z, Lefai E, Shyy JY, Gao B, Wierzbicki M, Verbeuren TJ, Shaw RJ, Cohen RA, Zang M (2011). AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab 13(4): 376-388. doi: 10.1016/j.cmet.2011.03.009
- Hardie DG, Pan DA (2002). Regulation of fatty acid synthesis and oxidation by the AMP-activated protein kinase. Biochem Soc Trans 30(Pt 6): 1064-1070. doi: 10.1042/bst0301064
- Habets DD, Coumans WA, El Hasnaoui M, Zarrinpashneh E, Bertrand L, Viollet B, Kiens B, Jensen TE, Richter EA, Bonen A, Glatz JF, Luiken JJ (2009). Crucial role for LKB1 to AMPKalpha2 axis in the regulation of CD36-mediated long-chain fatty acid uptake into cardiomyocytes. Biochim Biophys Acta 1791(3): 212-219. doi: 10.1016/j.bbalip.2008.12.009
- Inoki K, Zhu T, Guan KL (2003). TSC2 mediates cellular energy response to control cell growth and survival. Cell 115(5): 577-590. doi: 10.1016/s0092-8674(03)00929-2
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ (2008). AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 30(2): 214-226. doi: 10.1016/j.molcel.2008.03.003
- Horman S, Browne G, Krause U, Patel J, Vertommen D, Bertrand L, Lavoinne A, Hue L, Proud C, Rider M (2002). Activation of AMP-activated protein kinase leads to the phosphorylation of elongation factor 2 and an inhibition of protein synthesis. Curr Biol 12(16): 1419-1423. doi: 10.1016/s0960-9822(02)01077-1
- Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, Asara JM, Fitzpatrick J, Dillin A, Viollet B, Kundu M, Hansen M, Shaw RJ (2011). Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331(6016): 456-461. doi: 10.1126/science.1196371
- Kim J, Kundu M, Viollet B, Guan KL (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13(2): 132-141. doi: 10.1038/ncb2152
- Mihaylova MM, Shaw RJ (2011). The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol 13(9): 1016-1023. doi: 10.1038/ncb2329
- Faubert B, Boily G, Izreig S, Griss T, Samborska B, Dong Z, Dupuy F, Chambers C, Fuerth BJ, Viollet B, Mamer OA, Avizonis D, DeBerardinis RJ, Siegel PM, Jones RG (2013). AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab 17(1): 113-124. doi: 10.1016/j.cmet.2012.12.001
- Hoppe S, Bierhoff H, Cado I, Weber A, Tiebe M, Grummt I, Voit R (2009). AMP-activated protein kinase adapts rRNA synthesis to cellular energy supply. Proc Natl Acad Sci U S A 106(42): 17781-17786. doi: 10.1073/pnas.0909873106
- Dehne N, Brune B (2009). HIF-1 in the inflammatory microenvironment. Exp Cell Res 315(11): 1791-1797. doi: 10.1016/j.yexcr.2009.03.019
- Salminen A, Hyttinen JM, Kaarniranta K (2011). AMP-activated protein kinase inhibits NF-kappaB signaling and inflammation: impact on healthspan and lifespan. J Mol Med 89(7): 667-676. doi: 10.1007/s00109-011-0748-0
- Sag D, Carling D, Stout RD, Suttles J (2008). Adenosine 5'-monophosphate-activated protein kinase promotes macrophage polarization to an anti-inflammatory functional phenotype. J Immunol 181(12): 8633-8641. doi: 10.4049/jimmunol.181.12.8633
- Chen S, Yin C, Lao T, Liang D, He D, Wang C, Sang N (2015). AMPK-HDAC5 pathway facilitates nuclear accumulation of HIF-1alpha and functional activation of HIF-1 by deacetylating Hsp70 in the cytosol. Cell Cycle 14(15): 2520-2536. doi: 10.1080/15384101.2015.1055426
- Lim JH, Lee YM, Chun YS, Chen J, Kim JE, Park JW (2010). Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol Cell 38(6): 864-878. doi: 10.1016/j.molcel.2010.05.023
- Bucala R, Donnelly SC (2007). Macrophage migration inhibitory factor: a probable link between inflammation and cancer. Immunity 26(3): 281-285. doi: 10.1016/j.immuni.2007.03.005
- Rendon BE, Willer SS, Zundel W, Mitchell RA (2009). Mechanisms of macrophage migration inhibitory factor (MIF)-dependent tumor microenvironmental adaptation. Exp Mol Pathol 86(3): 180-185. doi: 10.1016/j.yexmp.2009.01.001
- Welford SM, Bedogni B, Gradin K, Poellinger L, Broome Powell M, Giaccia AJ (2006). HIF1alpha delays premature senescence through the activation of MIF. Genes Dev 20(24): 3366-3371. doi: 10.1101/gad.1471106
- Winner M, Koong AC, Rendon BE, Zundel W, Mitchell RA (2007). Amplification of tumor hypoxic responses by macrophage migration inhibitory factor-dependent hypoxia-inducible factor stabilization. Cancer Res 67(1): 186-193. doi: 10.1158/0008-5472.CAN-06-3292
- Fu H, Luo F, Yang L, Wu W, Liu X (2010). Hypoxia stimulates the expression of macrophage migration inhibitory factor in human vascular smooth muscle cells via HIF-1alpha dependent pathway. BMC Cell Biol 11: 66. doi: 10.1186/1471-2121-11-66
- Miller EJ, Li J, Leng L, McDonald C, Atsumi T, Bucala R, Young LH (2008). Macrophage migration inhibitory factor stimulates AMP-activated protein kinase in the ischaemic heart. Nature 451(7178): 578-582. doi: 10.1038/nature06504
- Xia W, Zhang F, Xie C, Jiang M, Hou M (2015). Macrophage migration inhibitory factor confers resistance to senescence through CD74-dependent AMPK-FOXO3a signaling in mesenchymal stem cells. Stem Cell Res Ther 6: 82. doi: 10.1186/s13287-015-0076-3
- Heinrichs D, Knauel M, Offermanns C, Berres ML, Nellen A, Leng L, Schmitz P, Bucala R, Trautwein C, Weber C, Bernhagen J, Wasmuth HE (2011). Macrophage migration inhibitory factor (MIF) exerts antifibrotic effects in experimental liver fibrosis via CD74. Proc Natl Acad Sci U S A 108(42): 17444-17449. doi: 10.1073/pnas.1107023108
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, Groop LC (2003). PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34(3): 267-273. doi: 10.1038/ng1180
- Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, Miyazaki Y, Kohane I, Costello M, Saccone R, Landaker EJ, Goldfine AB, Mun E, DeFronzo R, Finlayson J, Kahn CR, Mandarino LJ (2003). Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc Natl Acad Sci U S A 100(14): 8466-8471. doi: 10.1073/pnas.1032913100
- Petersen KF, Befroy D, Dufour S, Dziura J, Ariyan C, Rothman DL, DiPietro L, Cline GW, Shulman GI (2003). Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science 300(5622): 1140-1142. doi: 10.1126/science.1082889
- Liang H, Ward WF (2006). PGC-1alpha: a key regulator of energy metabolism. Adv Physiol Educ 30(4): 145-151. doi: 10.1152/advan.00052.2006
- Baar K, Wende AR, Jones TE, Marison M, Nolte LA, Chen M, Kelly DP, Holloszy JO (2002). Adaptations of skeletal muscle to exercise: rapid increase in the transcriptional coactivator PGC-1. FASEB J 16(14): 1879-1886. doi: 10.1096/fj.02-0367com
- Michael LF, Wu Z, Cheatham RB, Puigserver P, Adelmant G, Lehman JJ, Kelly DP, Spiegelman BM (2001). Restoration of insulin-sensitive glucose transporter (GLUT4) gene expression in muscle cells by the transcriptional coactivator PGC-1. Proc Natl Acad Sci U S A 98(7): 3820-3825. doi: 10.1073/pnas.061035098
- Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI (2004). Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med 350(7): 664-671. doi: 10.1056/NEJMoa031314
- Gorski J, Namiot Z, Giedrojc J (1978). Effect of exercise on metabolism of glycogen and triglycerides in the respiratory muscles. Pflugers Arch 377(3): 251-254. doi: 10.1007/bf00584280
- Hebert JA, Kerkhoff L, Bell L, Lopez-s A (1975). Effect of exercise on lipid metabolism of rats fed high carbohydrate diets. J Nutr 105(6): 718-725. doi: 10.1093/jn/105.6.718
- Wueest S, Item F, Boyle CN, Jirkof P, Cesarovic N, Ellingsgaard H, Boni-Schnetzler M, Timper K, Arras M, Donath MY, Lutz TA, Schoenle EJ, Konrad D (2014). Interleukin-6 contributes to early fasting-induced free fatty acid mobilization in mice. Am J Physiol Regul Integr Comp Physiol 306(11): R861-867. doi: 10.1152/ajpregu.00533.2013
- Fahmi A, Smart N, Punn A, Jabr R, Marber M, Heads R (2013). p42/p44-MAPK and PI3K are sufficient for IL-6 family cytokines/gp130 to signal to hypertrophy and survival in cardiomyocytes in the absence of JAK/STAT activation. Cell Signal 25(4): 898-909. doi: 10.1016/j.cellsig.2012.12.008
- Briest W, Rassler B, Deten A, Leicht M, Morwinski R, Neichel D, Wallukat G, Ziegelhoffer T, Zimmer HG (2003). Norepinephrine-induced interleukin-6 increase in rat hearts: differential signal transduction in myocytes and non-myocytes. Pflugers Arch 446(4): 437-446. doi: 10.1007/s00424-003-1043-x
- Saito S, Aikawa R, Shiojima I, Nagai R, Yazaki Y, Komuro I (1999). Endothelin-1 induces expression of fetal genes through the interleukin-6 family of cytokines in cardiac myocytes. FEBS Lett 456(1): 103-107. doi: 10.1016/s0014-5793(99)00936-9
- Coles B, Fielding CA, Rose-John S, Scheller J, Jones SA, O'Donnell VB (2007). Classic interleukin-6 receptor signaling and interleukin-6 trans-signaling differentially control angiotensin II-dependent hypertension, cardiac signal transducer and activator of transcription-3 activation, and vascular hypertrophy in vivo. Am J Pathol 171(1): 315-325. doi: 10.2353/ajpath.2007.061078
- Wollert KC, Taga T, Saito M, Narazaki M, Kishimoto T, Glembotski CC, Vernallis AB, Heath JK, Pennica D, Wood WI, Chien KR (1996). Cardiotrophin-1 activates a distinct form of cardiac muscle cell hypertrophy. Assembly of sarcomeric units in series VIA gp130/leukemia inhibitory factor receptor-dependent pathways. J Biol Chem 271(16): 9535-9545. doi: 10.1074/jbc.271.16.9535
- Sheng Z, Pennica D, Wood WI, Chien KR (1996). Cardiotrophin-1 displays early expression in the murine heart tube and promotes cardiac myocyte survival. Development 122(2): 419-428. PMID: 8625793
- Geiger PC, Hancock C, Wright DC, Han DH, Holloszy JO (2007). IL-6 increases muscle insulin sensitivity only at superphysiological levels. Am J Physiol Endocrinol Metab 292(6): E1842-1846. doi: 10.1152/ajpendo.00701.2006
- Palsson-McDermott EM, O'Neill LA (2013). The Warburg effect then and now: from cancer to inflammatory diseases. Bioessays 35(11): 965-973. doi: 10.1002/bies.201300084
- Hoque R, Farooq A, Ghani A, Gorelick F, Mehal WZ (2014). Lactate reduces liver and pancreatic injury in Toll-like receptor- and inflammasome-mediated inflammation via GPR81-mediated suppression of innate immunity. Gastroenterology 146(7): 1763-1774. doi: 10.1053/j.gastro.2014.03.014
- Goetze K, Walenta S, Ksiazkiewicz M, Kunz-Schughart LA, Mueller-Klieser W (2011). Lactate enhances motility of tumor cells and inhibits monocyte migration and cytokine release. Int J Oncol 39(2): 453-463. doi: 10.3892/ijo.2011.1055
- Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, Cyrus N, Brokowski CE, Eisenbarth SC, Phillips GM, Cline GW, Phillips AJ, Medzhitov R (2014). Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513(7519): 559-563. doi: 10.1038/nature13490
- Iraporda C, Romanin DE, Bengoa AA, Errea AJ, Cayet D, Foligne B, Sirard JC, Garrote GL, Abraham AG, Rumbo M (2016). Local Treatment with Lactate Prevents Intestinal Inflammation in the TNBS-Induced Colitis Model. Front Immunol 7: 651. doi: 10.3389/fimmu.2016.00651
- Tan Z, Xie N, Banerjee S, Cui H, Fu M, Thannickal VJ, Liu G (2015). The monocarboxylate transporter 4 is required for glycolytic reprogramming and inflammatory response in macrophages. J Biol Chem 290(1): 46-55. doi: 10.1074/jbc.M114.603589
- Dietl K, Renner K, Dettmer K, Timischl B, Eberhart K, Dorn C, Hellerbrand C, Kastenberger M, Kunz-Schughart LA, Oefner PJ, Andreesen R, Gottfried E, Kreutz MP (2010). Lactic acid and acidification inhibit TNF secretion and glycolysis of human monocytes. J Immunol 184(3): 1200-1209. doi: 10.4049/jimmunol.0902584
- Errea A, Cayet D, Marchetti P, Tang C, Kluza J, Offermanns S, Sirard JC, Rumbo M (2016). Lactate Inhibits the Pro-Inflammatory Response and Metabolic Reprogramming in Murine Macrophages in a GPR81-Independent Manner. PLoS One 11(11): e0163694. doi: 10.1371/journal.pone.0163694
- Peng M, Yin N, Chhangawala S, Xu K, Leslie CS, Li MO (2016). Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science 354(6311): 481-484. doi: 10.1126/science.aaf6284
- Seth P, Csizmadia E, Hedblom A, Vuerich M, Xie H, Li M, Longhi MS, Wegiel B (2017). Deletion of Lactate Dehydrogenase-A in Myeloid Cells Triggers Antitumor Immunity. Cancer Res 77(13): 3632-3643. doi: 10.1158/0008-5472.CAN-16-2938
- van Hall G (2010). Lactate kinetics in human tissues at rest and during exercise. Acta Physiol 199(4): 499-508. doi: 10.1111/j.1748-1716.2010.02122.x
- Halestrap AP, Wang X, Poole RC, Jackson VN, Price NT (1997). Lactate transport in heart in relation to myocardial ischemia. Am J Cardiol 80(3A): 17A-25A. doi: 10.1016/s0002-9149(97)00454-2
- Xu J, Xu X, Si L, Xue L, Zhang S, Qin J, Wu Y, Shao Y, Chen Y, Wang X (2013). Intracellular lactate signaling cascade in atrial remodeling of mitral valvular patients with atrial fibrillation. J Cardiothorac Surg 8: 34. doi: 10.1186/1749-8090-8-34
- Evans RK, Schwartz DD, Gladden LB (2003). Effect of myocardial volume overload and heart failure on lactate transport into isolated cardiac myocytes. J Appl Physiol 94(3): 1169-1176. doi: 10.1152/japplphysiol.00778.2002
- Levy B, Mansart A, Montemont C, Gibot S, Mallie JP, Regnault V, Lecompte T, Lacolley P (2007). Myocardial lactate deprivation is associated with decreased cardiovascular performance, decreased myocardial energetics, and early death in endotoxic shock. Intensive Care Med 33(3): 495-502. doi: 10.1007/s00134-006-0523-9
- Salem JE, Stanley WC, Cabrera ME (2004). Computational studies of the effects of myocardial blood flow reductions on cardiac metabolism. Biomed Eng Online 3(1): 15. doi: 10.1186/1475-925X-3-15
- Tessier JP, Thurner B, Jungling E, Luckhoff A, Fischer Y (2003). Impairment of glucose metabolism in hearts from rats treated with endotoxin. Cardiovasc Res 60(1): 119-130. doi: 10.1016/s0008-6363(03)00320-1
- Johannsson E, Lunde PK, Heddle C, Sjaastad I, Thomas MJ, Bergersen L, Halestrap AP, Blackstad TW, Ottersen OP, Sejersted OM (2001). Upregulation of the cardiac monocarboxylate transporter MCT1 in a rat model of congestive heart failure. Circulation 104(6): 729-734. doi: 10.1161/hc3201.092286
- Kline JA, Thornton LR, Lopaschuk GD, Barbee RW, Watts JA (2000). Lactate improves cardiac efficiency after hemorrhagic shock. Shock 14(2): 215-221. doi: 10.1097/00024382-200014020-00023
- Saraste A, Pulkki K, Kallajoki M, Henriksen K, Parvinen M, Voipio-Pulkki LM (1997). Apoptosis in human acute myocardial infarction. Circulation 95(2): 320-323. PMID: 9008443
- Gao HK, Yin Z, Zhou N, Feng XY, Gao F, Wang HC (2008). Glycogen synthase kinase 3 inhibition protects the heart from acute ischemia-reperfusion injury via inhibition of inflammation and apoptosis. J Cardiovasc Pharmacol 52(3): 286-292. doi: 10.1097/FJC.0b013e318186a84d
- Aharinejad S, Andrukhova O, Lucas T, Zuckermann A, Wieselthaler G, Wolner E, Grimm M (2008). Programmed cell death in idiopathic dilated cardiomyopathy is mediated by suppression of the apoptosis inhibitor Apollon. Ann Thorac Surg 86(1): 109-114; discussion 114. doi: 10.1016/j.athoracsur.2008.03.057
- Narula J, Haider N, Virmani R, DiSalvo TG, Kolodgie FD, Hajjar RJ, Schmidt U, Semigran MJ, Dec GW, Khaw BA (1996). Apoptosis in myocytes in end-stage heart failure. N Engl J Med 335(16): 1182-1189. doi: 10.1056/NEJM199610173351603
- Vaughan-Jones RD, Spitzer KW, Swietach P (2009). Intracellular pH regulation in heart. J Mol Cell Cardiol 46(3): 318-331. doi: 10.1016/j.yjmcc.2008.10.024
- Crow MT, Mani K, Nam YJ, Kitsis RN (2004). The mitochondrial death pathway and cardiac myocyte apoptosis. Circ Res 95(10): 957-970. doi: 10.1161/01.RES.0000148632.35500.d9
- Bryant D, Becker L, Richardson J, Shelton J, Franco F, Peshock R, Thompson M, Giroir B (1998). Cardiac failure in transgenic mice with myocardial expression of tumor necrosis factor-alpha. Circulation 97(14): 1375-1381. doi: 10.1161/01.cir.97.14.1375
- Sayen MR, Gustafsson AB, Sussman MA, Molkentin JD, Gottlieb RA (2003). Calcineurin transgenic mice have mitochondrial dysfunction and elevated superoxide production. Am J Physiol Cell Physiol 284(2): C562-570. doi: 10.1152/ajpcell.00336.2002
- Wang J, Silva JP, Gustafsson CM, Rustin P, Larsson NG (2001). Increased in vivo apoptosis in cells lacking mitochondrial DNA gene expression. Proc Natl Acad Sci U S A 98(7): 4038-4043. doi: 10.1073/pnas.061038798
- Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, Montminy M, Cantley LC (2005). The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 310(5754): 1642-1646. doi: 10.1126/science.1120781
- Madiraju AK, Erion DM, Rahimi Y, Zhang XM, Braddock DT, Albright RA, Prigaro BJ, Wood JL, Bhanot S, MacDonald MJ, Jurczak MJ, Camporez JP, Lee HY, Cline GW, Samuel VT, Kibbey RG, Shulman GI (2014). Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 510(7506): 542-546. doi: 10.1038/nature13270
- Caballero AE, Delgado A, Aguilar-Salinas CA, Herrera AN, Castillo JL, Cabrera T, Gomez-Perez FJ, Rull JA (2004). The differential effects of metformin on markers of endothelial activation and inflammation in subjects with impaired glucose tolerance: a placebo-controlled, randomized clinical trial. J Clin Endocrinol Metab 89(8): 3943-3948. doi: 10.1210/jc.2004-0019
- Dandona P, Aljada A, Ghanim H, Mohanty P, Tripathy C, Hofmeyer D, Chaudhuri A (2004). Increased plasma concentration of macrophage migration inhibitory factor (MIF) and MIF mRNA in mononuclear cells in the obese and the suppressive action of metformin. J Clin Endocrinol Metab 89(10): 5043-5047. doi: 10.1210/jc.2004-0436
- Isoda K, Young JL, Zirlik A, MacFarlane LA, Tsuboi N, Gerdes N, Schonbeck U, Libby P (2006). Metformin inhibits proinflammatory responses and nuclear factor-kappaB in human vascular wall cells. Arterioscler Thromb Vasc Biol 26(3): 611-617. doi: 10.1161/01.ATV.0000201938.78044.75
- Tousoulis D, Psarros C, Demosthenous M, Patel R, Antoniades C, Stefanadis C (2014). Innate and adaptive inflammation as a therapeutic target in vascular disease: the emerging role of statins. J Am Coll Cardiol 63(23): 2491-2502. doi: 10.1016/j.jacc.2014.01.054
- Schonbeck U, Libby P (2004). Inflammation, immunity, and HMG-CoA reductase inhibitors: statins as antiinflammatory agents? Circulation 109(21 Suppl 1): II18-26. doi: 10.1161/01.CIR.0000129505.34151.23
- Kwak B, Mulhaupt F, Myit S, Mach F (2000). Statins as a newly recognized type of immunomodulator. Nat Med 6(12): 1399-1402. doi: 10.1038/82219
- Weitz-Schmidt G, Welzenbach K, Brinkmann V, Kamata T, Kallen J, Bruns C, Cottens S, Takada Y, Hommel U (2001). Statins selectively inhibit leukocyte function antigen-1 by binding to a novel regulatory integrin site. Nat Med 7(6): 687-692. doi: 10.1038/89058
- Aktas O, Waiczies S, Smorodchenko A, Dorr J, Seeger B, Prozorovski T, Sallach S, Endres M, Brocke S, Nitsch R, Zipp F (2003). Treatment of relapsing paralysis in experimental encephalomyelitis by targeting Th1 cells through atorvastatin. J Exp Med 197(6): 725-733. doi: 10.1084/jem.20021425
- Diomede L, Albani D, Sottocorno M, Donati MB, Bianchi M, Fruscella P, Salmona M (2001). In vivo anti-inflammatory effect of statins is mediated by nonsterol mevalonate products. Arterioscler Thromb Vasc Biol 21(8): 1327-1332. doi: 10.1161/hq0801.094222
- Dunn SE, Youssef S, Goldstein MJ, Prod'homme T, Weber MS, Zamvil SS, Steinman L (2006). Isoprenoids determine Th1/Th2 fate in pathogenic T cells, providing a mechanism of modulation of autoimmunity by atorvastatin. J Exp Med 203(2): 401-412. doi: 10.1084/jem.20051129
- Kagami S, Owada T, Kanari H, Saito Y, Suto A, Ikeda K, Hirose K, Watanabe N, Iwamoto I, Nakajima H (2009). Protein geranylgeranylation regulates the balance between Th17 cells and Foxp3+ regulatory T cells. Int Immunol 21(6): 679-689. doi: 10.1093/intimm/dxp037
- Kim YC, Kim KK, Shevach EM (2010). Simvastatin induces Foxp3+ T regulatory cells by modulation of transforming growth factor-beta signal transduction. Immunology 130(4): 484-493. doi: 10.1111/j.1365-2567.2010.03269.x
- Gervois P, Kleemann R, Pilon A, Percevault F, Koenig W, Staels B, Kooistra T (2004). Global suppression of IL-6-induced acute phase response gene expression after chronic in vivo treatment with the peroxisome proliferator-activated receptor-alpha activator fenofibrate. J Biol Chem 279(16): 16154-16160. doi: 10.1074/jbc.M400346200
- Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, Red Eagle A, Vats D, Brombacher F, Ferrante AW, Chawla A (2007). Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature 447(7148): 1116-1120. doi: 10.1038/nature05894
- Kratz M, Coats BR, Hisert KB, Hagman D, Mutskov V, Peris E, Schoenfelt KQ, Kuzma JN, Larson I, Billing PS, Landerholm RW, Crouthamel M, Gozal D, Hwang S, Singh PK, Becker L (2014). Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab 20(4): 614-625. doi: 10.1016/j.cmet.2014.08.010
- Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, Shoelson SE, Benoist C, Mathis D (2012). PPAR-gamma is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature 486(7404): 549-553. doi: 10.1038/nature11132
- Mandard S, Patsouris D (2013). Nuclear control of the inflammatory response in mammals by peroxisome proliferator-activated receptors. PPAR Res 2013: 613864. doi: 10.1155/2013/613864
ACKNOWLEDGMENTS
FMB is the recipient of the British Heart Foundation Chair of Cardiovascular Immunology (CH/15/2/32064). DA acknowledges support from Queen Mary University of London, School of Biological and Chemical Sciences and Bart’s Charity (Grant Ref MRC0215).
COPYRIGHT
© 2019

Immunometabolic cross-talk in the inflamed heart by Morelli-Berg and Aksentijevic is licensed under a Creative Commons Attribution 4.0 International License.


