Reviews:
Cell Stress, Vol. 5, No. 7, pp. 99 - 118; doi: 10.15698/cst2021.07.253
Towards a better understanding of the neuro-developmental role of autophagy in sickness and in health

1 Centro de Investigaciones Biológicas Margarita Salas, CSIC, Madrid, Spain.
2 Universiteit van Amsterdam, Amsterdam, The Netherlands.
Keywords: neurodevelopment, developmental disorders, neuronal autophagy, autism spectrum disorder, neurogenesis.
Abbreviatons:
AMPK – AMP activated protein kinase;
ASD – autism spectrum disorder;
ATG – autophagy-related;
BPAN – β-propeller protein-associated neurodegeneration;
CMA – chaperone-mediated autophagy;
CNS – central nervous system;
ER – endoplasmic reticulum;
FXS – Fragile X syndrome; HSP – hereditary spastic paraplegia;
ID – intellectual disability;
IM – isolation membrane;
KO – knockout;
LAMP2 – lysosome-associated membrane protein 2;
LC – light chain;
LDD – Lhermitte-Duclos disease;
mTOR – mechanistic target of rapamycin;
mTORC – mTOR complex;
NSC – neuronal stem cell;
PI3K – phoshatidylinositol 3- kinase;
PI3P – phosphatidylinositol 3-phosphate;
POMC – proopiomelanocortin;
TSC – tuberous sclerosis complex;
ULK1 – UNC51-like kinase 1.
Received originally: 03/04/2021 Received in revised form: 31/05/2021
Accepted: 02/06/2021
Published: 29/06/2021
Correspondence:
Patricia Boya, Centro de Investigaciones Biológicas Margarita Salas, CSIC, Madrid, Spain; patricia.boya@csic.es
Conflict of interest statement: The author’s declare no conflict of interest.
Please cite this article as: Juan Zapata-Muñoz, Beatriz Villarejo-Zori, Pablo Largo-Barrientos and Patricia Boya (2021). Towards a better understand-ing of the neurodevelopmental role of autophagy in sickness and in health. Cell Stress 5(7): 99-118. doi: 10.15698/cst2021.07.253
Abstract
Autophagy is a critical cellular process by which biomolecules and cellular organelles are degraded in an orderly manner inside lysosomes. This process is particularly important in neurons: these post-mitotic cells cannot divide or be easily replaced and are therefore especially sensitive to the accumulation of toxic proteins and damaged organelles. Dysregulation of neuronal autophagy is well documented in a range of neurodegenerative diseases. However, growing evidence indicates that autophagy also critically contributes to neurodevelopmental cellular processes, including neurogenesis, maintenance of neural stem cell homeostasis, differentiation, metabolic reprogramming, and synaptic remodelling. These findings implicate autophagy in neurodevelopmental disorders. In this review we discuss the current understanding of the role of autophagy in neurodevelopment and neurodevelopmental disorders, as well as currently available tools and techniques that can be used to further investigate this association.
INTRODUCTION
All living organisms and their cells maintain an internal equilibrium between their components, a phenomenon known as ‘homeostasis'. Cellular homeostasis is maintained by tight regulation of the balance between the synthesis and degradation of different cellular components, from macromolecules (e.g. proteins, nucleic acids) to cellular organelles (e.g. ribosomes, mitochondria). Cell homeostasis relies on several catabolic or degradative mechanisms, including autophagy. Derived from the Greek for ‘self-eating', autophagy was first proposed by Christian de Duve at the 1963 Ciba Foundation Symposium on Lysosomes, after listening to a presentation on Alex Novikoff's recent work [1]. The previous decade, Sam L. Clark and Novikoff had observed mitochondria inside membrane-bound compartments with lysosomal enzymes in mouse kidneys [2][3], representing the first image of what we now know as autophagy.
–
Autophagy involves an ensemble of cellular mechanisms, all of which promote the degradation of cytosolic components (macromolecules or organelles) by the lysosome [4][5]. Three different types of autophagy are described in mammalian cells: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA). Only macroautophagy involves the participation of a double-membrane organelle called the autophagosome, which engulfs the cytosolic component (cargo) and delivers it to the lysosome for degradation [6]. In microautophagy, the cargo is directly taken up by the lysosome, where it is degraded through invagination of the lysosomal membrane [7]. CMA is a very specific type of autophagy described only in mammalian cells whereby cytosolic proteins are selectively identified by a chaperone protein that delivers them to the surface of the lysosome [8]. The chaperone interaction with the CMA receptor at the lysosomal membrane allows the receptor dimerization, and the unfolding and translocation of the protein inside the lysosome following degradation. This review will focus specifically on macroautophagy (henceforth referred to simply as “autophagy”) and its roles in mammalian neurodevelopment, neurogenesis, and associated diseases.
–
Molecular mechanisms of autophagy
Autophagy and the components involved are highly conserved from yeast to mammals. At the molecular level, autophagy is orchestrated by a cohort of proteins generically referred to as autophagy-related (ATG) proteins. The majority of these proteins were first characterized in yeast, and their respective mammalian orthologues were subsequently described. To date, 42 ATG proteins have been described in yeast, most of which are conserved in mammalian cells, where they mediate different roles at discrete stages of the autophagy process [9][10].
–
Macroautophagy differs from other forms of autophagy as it involves the autophagosome, a large double-membrane structure (mean diameter in mammalian cells, 0.5–1.5 µm) that is transiently formed upon autophagy induction. Despite intensive research, the precise origin of the autophagosome remains unclear. It is generally accepted that the autophagosome emerges from a pre-existing structure called the phagophore or isolation membrane (IM), but the source of the membrane that comprises the IM is still under debate. It appears that the IM originates from endoplasmic reticulum (ER), specifically from membrane compartments enriched in phosphatidylinositol 3-phosphate (PI3P), sometimes referred to as omegasomes [11]. However, other membrane sources also implicated in the autophagosome formation include the mitochondria, plasma membrane, lipid droplets, Golgi apparatus, and even endosomes. The current theory is that multiple membrane sources can supply lipids for autophagosomal membrane formation, a process in which ER-mitochondria contact sites appear to play an increasingly important role [12]. However, it is unclear how and when these different membrane compartments participate in autophagosome formation and whether this is stimulus- or even cell-type-dependent [13].
–
Regardless of the specific origin of the IM, initiation of autophagy and formation of the autophagosome involve the generation of two protein complexes. The first initiation complex, commonly known as the UNC51-like kinase-1 (ULK1) complex, comprises the following proteins: the unc-51-like autophagy activating kinase 1 (ULK1, mammalian orthologue of yeast Atg1); ATG13; ATG101; and focal adhesion kinase family interacting protein of 200 kD (FIP200, a mammalian orthologue of yeast Atg17). The ULK1 complex is assembled and activated at the IM, where it recruits and phosphorylates the transmembrane protein ATG9, which in turn is inserted into the IM [14]. Phosphorylation of ATG9 by the ULK1 complex triggers the recruitment of phospholipids from various sources, initiating elongation of the IM [15]. ULK1 also phosphorylates key components of the second key protein complex, the class III phosphatidylinositol 3-kinase (PI3K) complex, an action required for initiation of autophagy [16]. The PI3K complex comprises the following proteins: the PI3K catalytic subunit type 3 (PIK3C3, also known as vacuolar protein sorting 34 [VPS34]); PI3K regulatory subunit type 4 (PIK3KR4, also known as VPS15); ATG14; Beclin 1 (BECN1, mammalian orthologue of yeast Atg6); and the nuclear receptor binding factor 2 (NRBF2). The PI3K activity of this complex produces PI3P units, which also integrate into the IM and further contribute to its elongation [17]. Another critical event in autophagosome formation involves the activity of two ubiquitin-like conjugation systems. In the first of these systems, the E1-like activation enzyme ATG7 activates ATG12 and the E2-like conjugation enzyme ATG10 induces the formation of the ATG5-ATG12 conjugates. ATG5-ATG12 conjugates act as E3-like ligation enzymes, binding ATG16L to form the ATG5-ATG12-ATG16L complex, which in turn dimerizes and associates with the outer membrane of the nascent autophagosome [18]. In the other conjugation system, the cysteine protease ATG4 cleaves the microtubule-associated proteins 1 light chain 3 beta (MAP1LC3B, one of the mammalian orthologues of yeast Atg8 and commonly known as LC3). Cleaved LC3 (or LC3-I) is subsequently activated by ATG7 and lipidated by E2-like conjugation enzyme ATG3 and the ATG5-ATG12-ATG16L complex, which acts as an E3-like ligation enzyme [19]. Lipidated LC3, commonly referred to as LC3-II, is distributed on the outer and inner membranes of the forming autophagosome [20], contributing to its elongation and closure [21] while also recruiting autophagy receptors that bind the cargo targeted for degradation [16][22].
–
Autophagy has long been viewed as a non-selective degradative mechanism. While in CMA the cargo is selected by chaperones with a high degree of specificity, the autophagosome can engulf a broad range of cargo types. This appears to contradict the selective nature of autophagy. However, recent evidence indicates that several molecules that are recruited by Atg8 to the forming autophagosome can act as autophagy receptors, providing a degree of specificity in terms of the cargo selected for degradation. These autophagy receptors, the list of which is continuously growing, use the LIR (LC3-interacting region) to act as a bridge between Atg8 family members and the cargo [23]. An excellent review of selective autophagy receptors can be found elsewhere [24]. The most studied autophagy receptors are p62 and NBR1, which participate in the degradation of protein aggregates (aggrephagy) and damaged organelles; NDP52 and TAX1BP1, which are implicated in autophagy of damaged mitochondria (mitophagy) and bacteria (xenophagy); and OPTN, which plays important roles in aggrephagy, mitophagy and xenophagy [24].
–
The last stages of the autophagy process involve closure of the IM, resulting in engulfment of the cargo, and fusion of the autophagosome with a lysosome to give rise to the autolysosome, in which the cargo is degraded [25]. The process of fusion and formation of the autolysosome involves various proteins of the endocytic pathway, including the Ras-related protein RAB7A, soluble N-ethylmaleimide-sensitive factor activating protein receptor (SNARE) proteins, and the lysosome-associated membrane protein 2 (LAMP2) [16]. Before fusing with the lysosome, the autophagosome can fuse with a late endosome resulting in the formation of an intermediate structure called amphisome, which in turn fuses with a lysosome to proceed with the degradation of the cargo [26]. In both cases, macromolecules and organelles are degraded due to the action of acidic hydrolases in the lysosomal lumen. Finally, as the process of cargo degradation is completed, the inner membrane of the autolysosome is disassembled and the resulting biomolecules are released and recycled in the cytosol.
–
Regulation and function
Given its importance for cell homeostasis, it is not surprising that autophagy is tightly regulated. The induction of autophagy in mammalian cells is under the control of multiple signaling pathways, most of which converge on the mechanistic target of rapamycin (mTOR), a central regulator of multiple cellular processes, including cell growth and differentiation, that is localized in lysosomes [27]. Nutrient starvation is the best characterized autophagy-inducing stressor. Under nutrient rich conditions (resting state), initiation of autophagy is prevented by mTOR complex 1 (mTORC1), which inactivates the ULK1 complex. During starvation, the decrease in lysosome amino acid levels triggers mTOR release from lysosomes, resulting in reduced phosphorylation of its targets [28]. mTORC1 consists of the regulatory-associated protein of mTOR (RAPTOR), the proline-rich AKT1 substrate of 40 kDa (PRAS40), the mammalian lethal with SEC13 protein 8 (MLST8), and the DEP domain-containing mTOR-interacting protein (DEPTOR). Active mTORC1 inactivates ULK1 through phosphorylation of Ser757 [29], preventing initiation of autophagy. Conversely, decrease in adenosine triphosphate (ATP) results in increased AMP levels in the cell, which in turn promote autophagy via AMP activated protein kinase (AMPK), which inactivates mTORC1 [30].
–
The role of AMPK in promoting the initiation of autophagy goes beyond its inactivation of mTORC1. Recent evidence shows that AMPK activity phosphorylates TFEB and TFE3, two key transcription factors that regulate the expression of many autophagy and lysosomal proteins after their nuclear translocation [31]. Furthermore, under conditions of reduced ATP and increased AMP levels, active AMPK also promotes autophagy by activating ULK1 and class III PI3K complexes. AMPK directly activates ULK1 through phosphorylation of Ser317 and Ser777 [29], and the regulatory subunit BECN1 through phosphorylation of Ser91 and Ser94 [32]. The class III PI3K complex is also activated at the level of BECN1 by ULK1, which directly phosphorylates BECN1 at Ser14 [15], and by the activating molecule in BECN1-regulated autophagy (AMBRA1) [33]. By contrast, initiation of autophagy is prevented by the antiapoptotic protein BCL2, which binds to and inhibits BECN1 [34]. In summary, multiple signaling pathways regulate autophagy by interacting with different components, resulting in an intricate regulatory network that promotes or prevents the initiation of autophagy depending on cellular conditions.
–
The main result of autophagy is degradation of macromolecules and cellular organelles. Under resting conditions initiation of autophagy is generally blocked by mTORC1, whereas under low ATP conditions autophagy is promoted by AMPK. Typically, autophagy can be induced in vitro and in vivo by nutrient deprivation or starvation; the lack of any type of essential nutrient (e.g. glucose, amino acids) in growth medium effectively induces autophagy as the decrease in the metabolic rate reduces ATP levels [35]. Under these circumstances, the utility of autophagy lies in the reutilization of small biomolecules produced by degradation of macromolecules and organelles (e.g. amino acids resulting from protein degradation can be used as an energy source through the tricarboxylic acid cycle). However, it is important to note that autophagy also occurs constitutively (i.e. without nutrient deprivation) in all cells. Another important role of autophagy is to eliminate abnormal or excess cytosolic components in order to prevent their accumulation. In autophagy-deficient cells aberrant proteins and organelles accumulate in the absence of any external stressor [36]. Other recently proposed roles of autophagy not directly related to its degradative function include transport of proteins from the cytoplasm to the lysosome [37] and unconventional secretion of proteins that lack the typical signal peptide [38]. Many autophagy proteins have also non-autophagy-related functions that are often overlooked, and can confound the interpretation of their functions [4]. It is therefore advisable to validate phenotypes by knockdown of several autophagy regulators [39][40].
–
Autophagy and neuronal diseases
Autophagy has been implicated in multiple diseases, especially neurodegenerative and inflammatory diseases, autoimmune disorders, and cancer [41]. Research conducted over years has probed the connection between autophagy and neurodegeneration. Because the accumulation of unfolded or misfolded proteins and damaged organelles is a hallmark of most neurodegenerative disorders, the link between neurodegeneration and dysregulation of neuronal autophagy has been proposed [42].
–
Autophagy provides a mechanism by which neurons eliminate intracellular damaged components. Therefore, defective autophagy could reasonably result in the accumulation of altered proteins and organelles, leading to neurodegeneration. Over a decade ago, massive accumulation of proteins and progressive loss of neurons were described in mice in which the essential autophagy factors ATG5 [43] and ATG7 [44] were specifically absent in the central nervous system (CNS). Although these conditional knockout (KO) mice are born viable, they eventually die prematurely [43][44]. However, analyses of the embryos revealed diffuse ubiquitinated proteins and inclusions in dorsal root ganglion neurons, indicating that alterations in proteostasis occur as early as embryonic development. Moreover, depletion of the essential autophagy factor FIP200 in mouse neuronal brain cells results in neuronal loss preceded by accumulation of ubiquitinated protein aggregates, damage to mitochondria, and axonal degeneration [45]. The results of these studies clearly indicate that autophagy is crucial to prevent neurodegeneration and that deficient autophagy may contribute to the development of neurodegenerative disorders, starting during development.
–
A second link is implied by the recently established role of autophagy in protein homeostasis and neuronal activity in the presynaptic terminal (see following section). In this context, deficits in autophagy would be expected to impair synaptic function, leading to synaptic loss and neurodegeneration. Several studies have shown that neuronal stimulation induces upregulation of autophagy and accumulation of autophagosomes in synaptic terminals and axons [46][47][48][49]. Additionally, the loss of neuronal autophagic function leads to abnormal accumulation of ER structures in the axon, resulting in elevated calcium release and abnormal synaptic neurotransmission [50]. These evidences highlight the importance of autophagy to maintain neuronal function in a healthy brain.
–
Multiple studies have shown that mutations in several autophagy-related genes are risk factors for human neurodegenerative disorders. For instance, the most common mutations associated with Parkinson's disease affect two proteins, PTEN-induced kinase 1 (PINK) and PARKIN, both of which are essential for mitophagy [51][52]. In Alzheimer's disease, mutations in PRESENELIN 1 result in increased lysosomal pH [53], while mutations in the amyloid precursor protein indirectly lead to RAB7A activation [54], in both cases resulting in impaired lysosomal activity and altered autophagic function [55]. Finally, mutations in genes that participate in the molecular mechanisms of autophagy, including TBK1, VAPB, ATP13A2 and SQSTM1 among others, are also implicated in neurodegenerative diseases [41].
AUTOPHAGY IN NEURODEVELOPMENT AND NEUROGENESIS
Autophagy and the neuron: a lifelong relationship
Neurons are highly specialized cells that form the nervous system. These post-mitotic cells cannot divide or be easily replaced, and are therefore particularly sensitive to any loss of homeostatic balance. It thus seems logical that autophagy may be even more essential for neurons than for other cells. As early as the 1960s, studies of the rat sciatic nerve revealed the presence of autophagosomes in neurons [56]. However, despite years of research, the role of autophagy in neurons is less well understood compared with other mammalian cells and tissues. One of the difficulties that arises when studying autophagy in neurons is that starvation, which is widely used to induce autophagy in several in vivo and in vitro models, does not clearly upregulate autophagy in either the mouse brain or primary cultures of mouse neurons. The first studies in mice revealed no detectable upregulation of autophagy in the brain after 48 hours of starvation, in contrast with other tissues such as liver, muscle, and heart, in which autophagy was upregulated [57]. However, subsequent studies using novel approaches demonstrated starvation-induced upregulation of autophagy in mouse cortical neurons and Purkinje cells [58]. Our group has also shown that food starvation in mice increases autophagic flux in the retina [59]. Nonetheless, starvation may not be the best inducer of autophagy in the brain given the brain's unique capacity to resist and adapt to nutrient deprivation. The insulin-glucagon hormonal system, which is modulated by the neurons of the hypothalamus, ensures that the brain receives an adequate supply of nutrients, for instance by inducing the catabolism of glycogen or adipose fat from peripheral sources. Starvation-dependent autophagy may also be regulated differently in distinct neuronal cell types and in glia [60]. A recent in vitro study reported that primary astrocytes are much more responsive to starvation than primary hippocampal neurons. Intriguingly, the study also showed that the buffer used in the cell culture media (bicarbonate versus HEPES) affected the response of astrocytes, but not primary neurons, to metabolic stress [61]. Further studies are needed to determine how neurons and glia activate autophagy in different settings and how their interactions modulate homeostatic mechanisms. Neurons (at least when studied in situ inside the brain) may not need to resort to upregulate autophagy under conditions of acute starvation, but this does not mean that autophagy is not important for neuronal function.
–
Other important pathways also involved in regulating neuronal and glial starvation-induced autophagy include the JNK/p38 pathway[62], calcium signalling [63], and the Hippo pathway [70]. JNK has been implicated in the phosphorylation of the Ser70, Ser87, and Thr69 residues of BCL2 [64], resulting in the dissociation of BCL2 and BECN1 and inducing autophagy under starvation conditions [34][64][65]. p38 also contributes to autophagy regulation: in response to lipopolysaccharide stimulation, p38α MAPK directly phosphorylates ULK1 in microglia, inhibiting ULK1 activity, disrupting its interaction with ATG13 in the autophagy initiation complex, and thereby reducing autophagic flux [66]. Ca2+ signalling also appears to influence autophagy regulation, as starvation induces lysosomal Ca2+ release and the formation of a lysosomal Ca2+ microdomain that activates calcineurin, resulting in subsequent activation of TFEB [67]. Ca2+ upregulation also regulates autophagy in the brain: CaMKK-b, a Ca2+-activated kinase, was recently identified as a direct activator of AMPK [63]. New data suggest that ER-located Bcl-2 inhibits autophagy induced by Ca2+ mobilizing agents by regulating Ca2+ homeostasis in a Beclin 1-independent manner [68]. Finally, the Hippo pathway, which primarily regulates tissue growth and organ size, is yet another signalling pathway implicated in autophagy regulation [69]. Glucose starvation increases AMPK-dependent phosphorylation of YAP [70], one of the main downstream transcription coactivators of the Hippo pathway. Moreover, recent findings have demonstrated a role of STK3/MST2 and STK4/MST1, the main components of the Hippo pathway, in the regulation of autophagy. Phosphorylation of LC3 at threonine 50 by these proteins is fundamental for autophagosome-lysosome fusion and subsequent elimination of the cargo [71].
–
Because neurons do not divide, as previously stated, they require strict quality control mechanisms to prevent the accumulation of damaged organelles and misfolded proteins. That basal autophagy is indeed an essential housekeeping mechanism is clearly demonstrated by the specific deletion of Atg5 and Atg7 in neuronal precursors, which results in death in three month old mice due to severe neurological defects including motor deficits and neurodegeneration [43][44]. Moreover, the neurons of these animals accumulate polyubiquitinated proteins and p62, giving rise to inclusion bodies that become more numerous with increasing age. These mice also exhibit retinal alterations including photoreceptor death and night vision loss [72]. That neurons have a higher basal level of autophagic flux than other cell types is well accepted, and underscores the importance of autophagy for the maintenance of homeostatic balance and neuronal function [73][74]. Neurons are polarized cells that in some cases have very long axons and neurites, and autophagosome formation and degradation has been shown to be differentially regulated in a compartment-specific manner, showing differential regulation in the soma vs. the axon for example [73]. How this correlates in vivo with neuronal function remains to be determined.
–
The many physiological alterations that accompany aging include macromolecular damage affecting all subcellular compartments. Lysosomal damage impairs the ability of aged tissues to eliminate damaged organelles and macromolecules [75]. In the CNS aging can also be accompanied by progressive mitochondrial dysfunction, resulting in chronic bioenergetic inefficiency and the generation of free radicals that damage cellular structures, including membranes [76][77]. Although failure of one of the cell's catabolic systems (e.g. the ubiquitin proteasome system or autophagy) is generally compensated for by another, aging is accompanied by a generalized decrease in catabolism, exacerbating cell and tissue damage. Finally, defects in the autophagy-lysosomal system favour the accumulation of misfolded proteins that contribute to the development of diseases typically associated with aging such as Alzheimer's disease, Parkinson's disease, and cataracts [78].
–
Autophagy in neural stem cell proliferation and differentiation
The processes of neuronal formation (neurogenesis) and differentiation (part of the broader process of neurodevelopment) have their own particularities and are especially important, as generally the mammalian brain produces very few novel neurons after development. During neurodevelopment, immature neurons are formed from neuronal stem cells (NSCs) and differentiate into mature, fully-functional neurons that are capable of establishing synaptic connections and producing excitatory or inhibitory action potentials, resulting in an intricate neuronal network that forms the nervous system. All these events are tightly regulated, as any failure in brain development can make the embryo unviable or cause mental retardation. Maintenance of homeostasis is therefore essential. Moreover, as described for other cell types and tissues, autophagy may play an essential role in neuronal formation and differentiation [79].
–
NSCs are multipotent cells that can self-renew and proliferate to produce progeny cells and terminally differentiate into neurons, a process commonly referred to as neurogenesis. In mammals, neurogenesis mainly occurs during embryonic development from NSCs located in the ventricular zone of the developing CNS [80]. However, it has been now accepted for years that neurogenesis is also present in the adult brain in the subgranular zone in the dentate gyrus of the hippocampus and in the subventricular zone of the lateral ventricles [81]. Like all stem cells, NSCs are maintained in a pluripotent state that allows them to divide continuously in order to self-renew and generate new neurons and glial cells. Accordingly, maintenance of homeostasis, elimination of protein aggregates, and degradation of organelles by autophagy are essential for NSC survival during development and adulthood [82].
–
A study using Atg16L1 hypomorph mice and primary neurons showed that NOTCH, a plasma membrane receptor and master regulator of neuronal development, is taken up into ATG16L1-positive autophagosome-precursor vesicles and modulates neurogenesis, indicating that neuronal differentiation can be controlled by the selective autophagic degradation of membrane receptors [83]. Thus, in addition to its classical quality-control function, autophagy plays a variety of other roles in controlling the maintenance and differentiation of NSCs (Figure 1) [84].
–
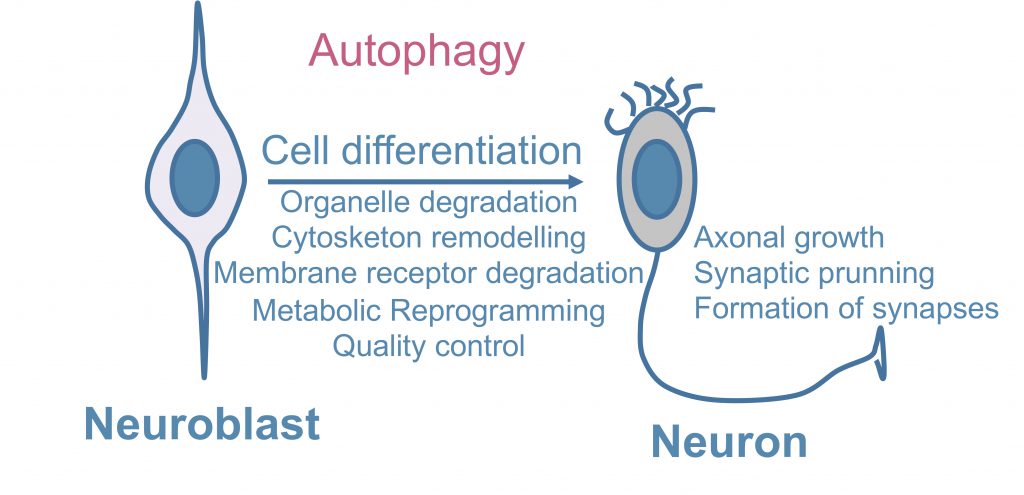 | FIGURE 1: Main roles of autophagy for neuronal differentiation. During neurodevelopment, neural stem cells and neuroblasts proliferate and differentiate to give rise to postmitotic neurons. Autophagy has a crucial role in differentiation by promoting cell remodelling including cytoskeleton and membrane receptor degradation as well as organelle degradation. Other functions include metabolic reprogramming and quality control functions. Moreover, other processes that occur during development, such as axonal growth, formation of synapses and synaptic pruning are regulated by autophagy. |
–
Autophagy and metabolic control in neurodevelopment
Autophagy is an important regulatory component of metabolic function because it supplies metabolic substrates obtained by degradation of cytosolic macromolecules and organelles. In fact, autophagy is typically described as a compensatory metabolic mechanism, activated in response to starvation or nutrient deprivation. However, as mentioned before, this function of autophagy appears to be limited in the brain and during neurodevelopment.
–
Recent studies have indicated additional roles of autophagy in regulating the proliferation and differentiation of NSCs. For example, several findings suggest important roles of autophagy in sustaining metabolic reprogramming during stem cell differentiation [84]. Our group has demonstrated that the expression of Atg7, Beclin1, LC3, and Ambra1 is markedly increased in cultured embryonic mouse olfactory bulb-derived NSCs during the initial period of neuronal differentiation [85]. In this setting Ambra1 and Atg5 deficiency decreases neurogenesis, a phenotype that can be reversed by methylpyruvate supplementation, suggesting that progenitor cells activate autophagy to meet their high energy demands [85].
–
Autophagy plays an additional metabolic role via the process of mitophagy [86]. Mitochondria, referred to as the “powerhouses of the cell”, are organelles that contain all the machinery required for oxidative phosphorylation and produce most of the cellular supply of ATP, the cell's main energy source [87]. Although mitophagy is commonly associated with the elimination of damaged or dysfunctional mitochondria in processes such as ageing and neurodegenerative disorders [88], recent evidence indicates a key role in neurodevelopment.
–
During neurodevelopment NSCs and immature postmitotic neurons undergo multiple changes. One such change is the neuron-glial switch, which is accompanied by increased levels of cell death, and another is a switch in their principal source of energy. Like all the other stem cell types, including cancer cells, NSCs and immature postmitotic neurons initially obtain the majority of their ATP via oxidative phosphorylation, which occurs in the mitochondria. After the aforementioned metabolic switch, glycolysis and lactate fermentation, which occur in the cytosol, become the main sources of energy. This phenomenon has been reported in a neuronal context in olfactory bulb NSCs [85] and in retinal ganglion cells [89]. Interestingly, studies conducted in retinal ganglion cells showed that the metabolic switch from oxidative phosphorylation to glycolysis and lactate fermentation is mediated by the selective elimination of mitochondria via mitophagy. Blockade of autophagy in general and mitophagy in particular not only prevents this metabolic reprogramming but also blocks neuronal differentiation of retinal ganglion cells [89]. This strongly suggests that autophagy participates in the metabolic switch that occurs during neurodevelopment by contributing to the elimination of mitochondria, the organelles responsible for oxidative phosphorylation.
–
What is the ultimate function of this metabolic reprogramming in neurodevelopment? It seems likely that the switch towards glycolytic and fermentative production of ATP is related to the proliferative capacity of NSCs. In fact, embryonic stem cells and hyperproliferative cancer cells generally depend on glycolytic metabolism rather than oxidative phosphorylation [90]. Proliferation (and subsequent neuronal differentiation) of NSCs entails high energy requirements and it may be more beneficial for NSCs to obtain the majority of this energy from glycolysis and lactate fermentation rather than from oxidative phosphorylation, even though the latter, which takes place in the mitochondria, is normally more efficient. This apparently paradoxical situation is comparable to the Warburg effect, which has been widely studied in cancer cells: these cells are also highly reliant on glycolysis for energy production, possibly because this route generates ATP faster than oxidative phosphorylation [91]. Hence, it is possible that NSCs also undergo a transient metabolic switch towards glycolysis and lactate fermentation during neurodevelopment in order to sustain their high level of protein synthesis for cytoplasm remodelling and axon growth, and that this switch is dependent on autophagy, which eliminates mitochondria and thereby prevents oxidative phosphorylation. Further research is required to confirm this hypothesis, especially in vivo studies demonstrating that mitophagy and metabolic reprogramming are indeed essential for proper mammalian neurodevelopment in other contexts besides the retina. In summary, while the metabolic transition from aerobic glycolysis to oxidative phosphorylation is regarded as a hallmark of neuronal differentiation, there are specific situations in which neuronal differentiation is controlled by the selective degradation of mitochondria via autophagy.
–
Autophagy in axonal growth, synapse formation, and synaptic pruning
The complex morphology of neurons poses a unique challenge for proteostasis [73]. The results of genetic studies conducted in hypothalamic proopiomelanocortin (POMC) neurons suggest that autophagy is essential for axonal growth in vivo[92]. Loss of ATG7 in POMC neurons results in metabolic dysregulation caused by developmental defects in POMC neurons, including accumulation of ubiquitinated proteins and loss of axonal projections [92]. Moreover, in vitro studies have shown that mTOR signaling inhibitors and autophagy activators tuberous sclerosis complex 1 and 2 (TSC1 and TSC2) localize in axons in developing cultured neurons and are important for axonal growth [93]. Regardless, further in vivo studies of different brain areas are needed to confirm a specific requirement of autophagy for axonal growth.
–
In vivo studies of Drosophila larvae have shown that autophagy promotes synaptic formation and development at neuromuscular junctions [94]. Specifically, autophagy reduces the levels of Highwire, an E3 ubiquitin ligase that prevents overgrowth of neuromuscular junctions. A growing body of evidence also suggests that proper autophagy levels are key for neuronal plasticity, which is essential for the formation and elimination of synapses during neurodevelopment [95]. ATG5-deficient mice display increased excitatory neurotransmission in CA3-CA1 synapses as early as two months of age. This increase is due to the accumulation of tubular ER, which is caused by the abnormal autophagic function in the absence of ATG5 and results in increased calcium release from the ER lumen. Thus, neuronal autophagy is essential to regulate synaptic plasticity via control of calcium efflux from ER stores [50][96].
–
Furthermore, autophagy may play a role in the elimination of synapses, a process of critical importance during neurodevelopment commonly referred to as synaptic pruning. This consists of the elimination of redundant, unnecessary, or inappropriate synapses that form during the early stages of neurodevelopment [97]. In vitro studies of ATG7-deficient hippocampal neurons have shown that these neurons are capable of developing dendritic spines and forming normal synapses, but are unable to subsequently prune the same number of spines as wild type neurons [98]. It will be essential to explore this phenomenon in mammalian models to confirm the requirement of autophagy for both the formation and elimination of synapses during neurodevelopment. Alterations in synaptic pruning are an underlying cause of multiple neurodevelopmental disorders. Interestingly, post-mortem analysis of brains from autism spectrum disorder (ASD) patients has shown that reduced synaptic pruning in certain brain regions is inversely associated with the levels of the autophagy marker LC3-II [98]. In the following section, we will discuss more thoroughly the association between autophagy and neurodevelopmental disorders.
AUTOPHAGY AND NEURODEVELOPMENTAL DISORDERS
Regardless of existing knowledge gaps, available evidence highlights the importance of autophagy in neurodevelopmental processes. Therefore, deficits in autophagy could feasibly lead to defective neurodevelopment and the appearance of neurodevelopmental disorders. Indeed, evidence published in recent years points to an important role of autophagy in several neurodevelopmental disorders.
–
Distinguishing neurodevelopmental from neurodegenerative diseases poses a challenge: both concepts imply neuronal demise, albeit over different time frames. According to the fifth edition of the Diagnostic and Statistical Manual of Mental Disorders, the term “neurodevelopmental disorders” describes a group of conditions with onset in the developmental period, usually before five years of age, that cause impairment of personal, social, academic, or occupational functioning, ranging from specific limitations (e.g. motor control deficits) to global impairment (e.g. deficits in social skills or intelligence) [99]. Mutations in a variety of genes related directly or indirectly to the autophagic process have been identified in different neurodevelopmental disorders (Table 1). In our discussion below on the consequences of autophagic dysfunction during neurodevelopment, neurodevelopmental disorders are grouped according to the stage of autophagy affected by the mutation and the clinical similarities. In broad terms, we can distinguish between neurodevelopmental disorders involving mTOR hyperactivity and those in which autophagy genes are dysregulated.
–
TABLE 1. Main neurodevelopmental disorders where mutations in autophagy proteins impacts disease. |
| [91][95][96][97][98][99][100][122][123][125][127][132][136][138][147][154][155][156][166][167][169][170][174] |
–
Neurodevelopmental disorders associated with mTOR hyperactivity
mTOR is one of the central suppressors of autophagy. Hyperactivation of mTOR therefore decreases autophagic flux. If this effect is prolonged it can have detrimental effects on cell physiology. Mutations in the mTOR inhibitor genes tuberous sclerosis complex 1/2 (TSC1/TSC2), neurofibromatosis type 1 (NF1), phosphatase and tensin homolog deleted on chromosome ten (PTEN), and fragile X mental retardation 1 (FMR1) lead to tuberous sclerosis complex (TSC), neurofibromatosis, Lhermitte-Duclos disease (LDD), and Fragile X syndrome (FXS), respectively [100][101], Figure 2. These genes negatively regulate mTOR activity in different ways. TSC1/2 acts as a GTPase-activating protein, deactivating Rheb-GTP by converting it into Rheb-GDP and thereby preventing the activation of mTOR kinase activity by Rheb-GTP [102]. NF1 supresses Ras, an upstream regulator of ERK signaling and, consequently, of the TSC1/2 complex [103]. Finally, PTEN and FMR1 regulate mTOR activity through PI3/Akt signaling [104] (Figure 2). These disorders are known as mTORopathies as their common biochemical feature is mTOR hyperactivity.
–
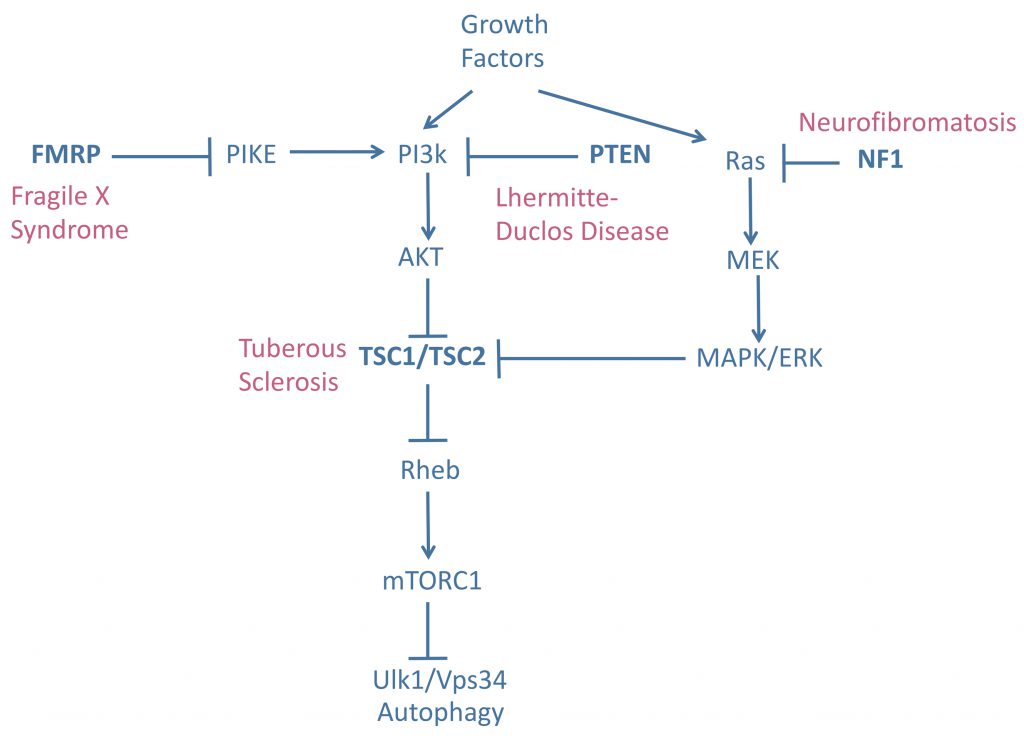 | FIGURE 2: Upstream mTOR regulation and associated mutations causing mTORpathies. The TSC1/TSC2 complex, FMRP, PTEN and NF1 are negative regulators of mTORC1. Loss of function-mutations in these genes lead to an increased mTORC1 activity and a reduced autophagy induction and produce a subset of neurodevelopmental disorders known as mTORopathies. TSC1/2: Tuberous sclerosis complex 1/2, FMRP: Fragile X mental retardation protein, PTEN: Phosphatase and tensin homolog deleted on chromosome ten and NF1: Neurofibromatosis type 1. |
–
TSC manifests clinically with the appearance of benign tumours in multiple organs and is caused by mutations affecting the activity of the TSC1/2 complex [105]. Neural disorders are common in TSC patients: 90% have epilepsy and 50% have ASD [106]. FXS patients are severely intellectually disabled and are usually autistic, making FXS the most common inherited cause of ASD. Other characteristic clinical signs of FXS include seizures, hypersensitivity, attention deficit, hyperactivity, and growth and craniofacial abnormalities [107]. FXS is caused by the expansion of a poly-CGG trinucleotide repeat in the 5'UTR of the FMR1 gene, which causes gene silencing and reduced expression of fragile X mental retardation protein (FMRP) [108]. Neurofibromatosis is a neurocutaneous disorder characterized by café-au-lait spots and benign and malignant tumours, with variable organ involvement. Epilepsy affects 10% of neurofibromatosis patients and ASD is a common manifestation of the disease [101][109]. Finally, LDD is caused by mutations in PTEN and is part of a group of disorders known as PTEN hamartoma tumour syndrome. LDD is characterized by the presence of gangliocytomas in the cerebellum, ataxia, and seizures [101]. PTEN mutations have also been identified as a cause of ASD with macrocephaly [110]. In summary, mTORopathies are a group of genetic diseases that confer a high degree of susceptibility to ASD and epilepsy [100][101], highlighting the importance of mTOR in epileptogenesis and the development of autism.
–
ASD is one of the most important neurodevelopmental disorders worldwide. It is characterized by restricted and repetitive behaviour and impairment of social interactions and verbal and non-verbal communication [111]. Autism is a multi-factorial disorder resulting from the interaction between genetic and non-genetic risk factors. Genes linked with the development of autism include mTOR genes as well as those implicated in modulating the balance between excitatory and inhibitory neurotransmission, cerebral connections, and synaptic plasticity [112]. These are all events in which autophagy may play a major role, as described previously. Epilepsy is a common neurological disorder that affects 1% of the world's population [101], and is characterized by spontaneous bursts of neuronal activity resulting in seizures [113]. Epileptogenesis, the process by which the brain becomes epileptic, can be the result of various causes, including structural brain lesions, congenital malformations, alterations in neuronal signaling, and defects in neuronal maturation and plasticity. In the neurodevelopmental context, the most common causes of epileptogenesis are malformations of the cortical layer and a reduction in the arrival of inhibitory interneurons to the cortical plate [113].
–
mTOR appears to play an important role in ASD and epileptogenesis. Although one of the key suppressors of autophagy, mTOR also participates in other processes (e.g. microtubule organization and protein synthesis [114]) that may be implicated in the autistic and epileptic phenotype. Studies performed over the last decade have reinforced the link between impaired autophagy and these disorders. A study of human post-mortem temporal lobe tissue from autistic patients revealed increased dendritic spine density and reduced developmental synaptic pruning associated with mTOR hyperactivation and impaired autophagy, as well as low levels of LC3-II and high levels of p62 [98]. These results indicate a strong correlation between mTOR hyperactivation (and therefore autophagy dysfunction) and defective synaptic pruning in autistic patients, but do not confirm a key role of autophagy in this process.
–
Animal models research strongly supports a central role of autophagy in social interactions. Pten mutant mice show impaired social behaviour and abnormal neuronal arborization, a phenotype that can be prevented by treatment with the mTOR inhibitor rapamycin [115]. A similar study of Tsc2+/- mice showed that rapamycin treatment corrects ASD-like behaviours and ameliorates spine-pruning defects, but does not rescue the phenotype in double mutant Tsc2+/-:Atg7CKO mice, supporting the view that autophagy is necessary to reverse the pathology [98]. Mice with Atg7-deficient microglia also show impaired synaptic pruning, abnormal social interactions, and repetitive behaviours [116], highlighting the importance of this process in a specific non-neuronal cell type. However, the potential role of synaptic pruning in the development of autism should be considered with caution. Hui et al. recently reported that Atg7 deletion in GABAergic interneurons and excitatory neurons of the medial prefrontal cortex of adolescent mice, after completion of synaptic refinement, causes behavioural abnormalities including reduced social interaction, altered nesting behaviour, and increased anxiety, suggesting that autophagy continues to play a role in social behaviour after postnatal development. This observation points out to the idea that behavioural deficits arising from autophagy deficiency may not be related to neurodevelopment and synaptic pruning, but to the disruption of neurotransmission processes independent of age [117]. The authors also described a novel pathogenic mechanism based on defective autophagy-dependent neurotransmission whereby suppression of autophagy, either due to mTOR hyperactivation or mutation of autophagy genes, disrupts GABAA receptor trafficking via sequestration of GABARAPs by p62-positive aggregates. The decrease in GABAA receptor expression results in a reduction of inhibitory input and consequent neuronal hyperexcitability [117]. Autophagy therefore constitutes a link between mTOR hyperactivation, the neuronal excitation/inhibition balance, and seizures and ASD-like behaviour [118].
–
This mechanism proposed by Hui et al. is reinforced by previous research. The WDFY3 gene (or ALFY), which participates in the clearance of p62-positive aggregates [119][120], has been detected as an ASD risk gene [121][122]. The mechanism is also in agreement with findings in a mouse model of TSC, in which loss of TSC1 in pyramidal neurons leads to deficits in inhibitory synaptic function [123], possibly due to reduced GABAA membrane translocation. Further evidence supporting a key role of GABA neurotransmission in TSC patients comes from the observation that TSC-related infantile spasms show a rapid and sustained response to treatment with vigabatrin [124], an irreversible inhibitor of the GABA degrading enzyme GABA transaminase. Finally, analysis of tissues from TSC patients has demonstrated reduced cortical expression of the GABAA receptor subunit [125]. Reduced efficiency of GABAergic neurotransmission has also been described in mouse models of FXS [126], which show reduced mRNA expression of GAD67, the main GABA synthesizing enzyme [127]. However, while GABAergic transmission appears to be altered, it remains to be determined whether autophagy impairment is causally linked to this defect in FSX. In conclusion, it is tempting to speculate that decreased GABAA membrane translocation may underlie the imbalance of excitatory and inhibitory synapses, and hence epilepsy and ASD, in patients with mTORopathies. However, the precise role of synaptic pruning in these pathologies remains to be elucidated.
–
Neurodevelopmental disorders associated with impaired autophagy
mTORopathies are not the sole cause of neurodevelopmental defects, which can also be caused by mutations in several genes involved in autophagy. These can be divided into disorders resulting from mutations affecting the early stages of autophagy, such as neurodegeneration with brain iron accumulation and hereditary spastic paraplegia 49 (SPG49), and those caused by mutations affecting later stages of the autophagy pathway, including hereditary spastic paraplegias 11 (SPG11) and 15 (SPG15), Vici Syndrome, and SNX14-associated ataxia. Here we describe the main phenotypes associated with these mutations (Figure 3).
–
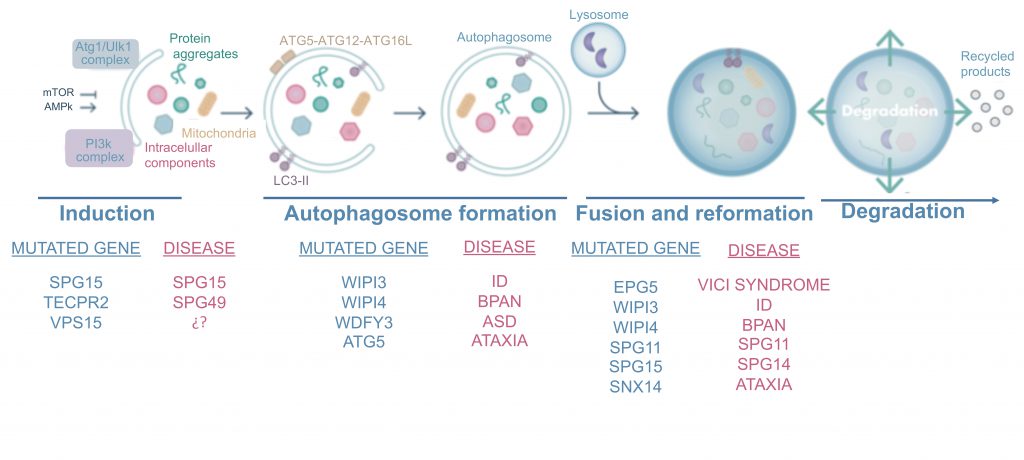 |
FIGURE 3: The autophagy process and most common mutated proteins in neurodevelopmental disorders. Autophagy is a highly conserved process that consists on the formation of a double membrane-vesicle engulfing cytosolic material and organelles, which finally fuses with lysosomes, where the cargo is degraded. ULK1 and PI3K complex participate in the autophagy induction, when the isolation membrane is formed. This membrane is elongated and specific materials are recognized by selective autophagic receptors until the closing and formation of autophagosome. The next step consists on the fusion with lysosomes and/or late endosomes, where cargo degradation takes place by lysosomal hydrolases. Finally, the resulting products are recycled and lysosomes can be reused through the process of lysosome reformation. Neurodevelopmental disorders described with mutations in autophagic genes are compiled according to the autophagic step affected. Defects in WIPI3 and WIPI4 have been found to correlate with ID and BPAN, respectively. The selective autophagic receptor WDFY3 gene, which participates in aggregates removal, is related with high susceptibility of ASD. Rare mutations have been found in the induction gene VPS15 and autophagosome elongating gene ATG5 in ataxic patients. Mutations in SPG11, SPG15 and TECPR2 produce hereditary spastic paraplegias. Finally, the EPG5 and SNX14 genes participating in the autophagosome/lysosome fusion are responsible for the Vici syndrome and ataxic disorders. |
–
β-Propeller protein-associated neurodegeneration (BPAN) and intellectual disability (ID)
–
Neurodegeneration with brain iron accumulation is a heterogeneous group of genetic disorders characterized by altered iron metabolism and iron accumulation in the basal ganglia [128], prominent extrapyramidal movement disorder, and intellectual deterioration [129]. Six different subtypes are described, depending on the gene affected [130], but here we will focus primarily on the subtype caused by mutations in WDR45, known as BPAN or static encephalopathy of childhood with neurodegeneration in adulthood. The course of BPAN has two distinct phases, beginning with ID, psychomotor retardation, febrile seizures, and even autistic features in childhood [129][130][131]. These clinical signs remain relatively stable until the second phase of the disease, in which progressive cognitive decline, dementia, dystonia, and parkinsonism are the predominant features [132]. It should be noted that mutations in WDR45B, a gene that shares a high level of homology with WDR45, are also associated with ID phenotypes [133].
–
WDR45 and WDR45B encode the proteins WDR45 and WDR45B (also known as WIPI4 and WIPI3, respectively), which belong to the PROPPINs (β-propellers that bind polyphosphoinositides) family, a group of proteins that bind phosphatidylinositol 3,5-bisphosphate and PI3P via a phosphoinositide-binding motif [134][135]. WIPI4 binds to the autophagic protein ATG2 and WIPI3 to FIP200, and both participate in the early stages of autophagosome formation [135][136], probably acting upstream of PI3P production and downstream of LC3 to control the size of the nascent autophagosome [135]. WIPI3 and WIPI4 may participate in autophagosome-lysosome fusion by promoting lysosomal localization of the protein EPG5 [137], which is mutated in Vici syndrome, as discussed later in this review.
–
Depletion of WIPI4 in U2OS cells results in an increase in autophagosome number, without blocking autophagic flux [138]. Studies of lymphoblasts from BPAN patients indicate partial blockade of autophagic flux [129]. While ATG9A is found only transiently in the autophagosome under normal conditions, these lymphoblasts also contain enlarged structures positive for ATG9A and LC3, indicating improper autophagosome formation [129]. This observation suggests that the absence of WIPI4 affects autophagosome maturation, resulting in autophagosome accumulation before fusion with the lysosomal machinery. Primary cells from CNS-specific Wdr45 KO mice also show defects in autophagic flux and the accumulation of protein aggregates. These mice partially recapitulate the BPAN phenotype, exhibiting extensive axon swelling, impaired motor coordination, and deficits in cognitive function [139]. Wdr45b KO mice present a more marked phenotype, characterized by severe motor deficits not present in Wdr45 KO mice, including limb-clasping reflexes, ataxia, and much more severe neural defects [140]. Double KO (Wdr45b-/-; Wdr45-/Y) mice exhibit the same suckling defect seen in Atg5 and Atg7 KO mice, and more severe autophagy defects than single KO mice [140], confirming the role of both proteins in autophagy.
–
A key question is whether WIPI4 mutations cause global attenuation of autophagy or if they only affect a subset of selective autophagy. Recent studies in Wdr45 KO mice and cells support a role of ER stress and the unfolded protein response in BPAN [141][142]. On the other hand, defects in ferritinophagy could also explain the toxic accumulation of iron. Ferritinophagy is the selective autophagic degradation of ferritin, the main iron storage protein in cells [143]. Whether the absence of WIPI4 results in altered iron homeostasis due to defective ferritinophagy will need to be further investigated: while ferritin levels in fibroblasts from patients with WIPI4 mutations are reduced compared with controls, no such difference has been observed in neurons [144]. WIPI4 and WIP3 are therefore proteins participating in autophagosome formation and in the fusion step and mutations of them are related with two neurodevelopmental affections, BPAN and ID (Figure 3).
–
Hereditary spastic paraplegia (HSP)
–
HSP is a group of heterogeneous neurological disorders characterized by upper motor neuron degeneration resulting in spasticity of the lower limbs, spastic gait, and pyramidal weakness. Complex forms of this disease include additional neurological features such as epilepsy, ataxia, mental retardation, and extraneurological signs including cataracts, retinal degeneration, and skeletal abnormalities [145], and in some cases CNS structural abnormalities including thin corpus callosum (the connection between both hemispheres), white matter hyperintensities (as observed by magnetic resonance imaging), and cortical and cerebellar atrophy [145]. More than 50 spastic paraplegia genes have been identified to date [132]. Here, we discuss the three forms in which autophagy impairment has been described: HSP caused by mutations in SPG11, SPG15, and SPG49. Almost one third of complex HSP cases involve alterations in the corpus callosum, and 70% of these are due to mutations in SPG15/ZFYVE26 or SPG11/KIAA1840[146]. SPG11 and SPG15 encode SPATACSIN and SPASTIZIN, respectively. SPASTIZIN is a large protein that participates in autophagosome maturation through interaction with the RUBICON-UVRAG-BECN1 complex. Cells without SPASTIZIN show reduced colocalization of LAMP1 and LC3, although autophagic flux is not completely blocked, as evidenced by increases in LC3-II levels following lysosomal inhibition with bafilomycin A1 [147]. It has been proposed that SPATACSIN and SPASTIZIN may participate in autophagic lysosome reformation, the process of lysosome biogenesis by which lysosomes are generated from pre-existing autolysosomes [148][149]. In fact, recent research in mice points toward phosphatidylinositol 4-kinase type 2 alpha accumulation to be responsible for autophagic lysosome reformation disturbance in mice models of the disease [150]. In zebrafish, knockdown of spatacsin and spastizin results in motor impairment and defective branching of spinal cord motoneurons at neuromuscular junctions [151]. While no structural alterations have been reported in the brains of Spg15 knockout mice, these animals develop a progressive spastic and ataxic gait disorder, and show neuronal loss in the motor cortex and cerebellum, resembling human HSP [152]. Before degeneration, these neurons accumulate large deposits of autofluorescent particles that colocalize with the lysosomal marker LAMP1, suggesting a link between lysosomal impairment and neurodegeneration [152]. Two different Spg11 KO mouse models have been developed. The first was created by inserting a genetrap cassette into the first intron of the SPG11 gene, resulting in the absence of SPATACSIN and consequently impairment of autolysosome reformation. This model reproduces the neurodegeneration in the motor cortex and cerebellum observed in SPG11 patients, but shows no early motor, anatomical, or cognitive deficits [153]. The second model, generated by Branchu et al.[154], simulates the most typical mutation found in SPG11 patients, and presents early-onset motor and cognitive deficits as well as lipid accumulation in lysosomes, as previously reported in SPG11 patients [155].
–
In 2012 Oz-Levi et al. identified a new form of complex HSP (SPG49) characterized by a thin corpus callosum and cerebral and cerebellar atrophy [156]. Patients with this complex form carry mutations in TECPR2, which encodes a protein that interacts with the six human Atg8 homologs and is a positive regulator of autophagosome accumulation [138]. TECPR2 regulates autophagy by maintaining functional endoplasmic exit sites, which serve as scaffolds for autophagosome formation [157]. However, recent studies also suggest a role for TECPR2 in the lysosomal targeting of autophagosomes [158][159].
–
Other forms of HSP that involve dysregulation of lysosomal activity, and therefore of autophagy, are those caused by mutations in AP-5 subunits, including SPG48 (AP-5 ζ), and in AP-4 subunits, including SPG47 (AP-4 β1), SPG50 (AP-4 µ1), SPG51 (AP-4 σ1), and SPG52 (AP-4 σ1) [160]. AP-5 is the most recently identified of the five adaptor complexes and has been proposed to form a complex with SPATACSIN and SPASTIZIN that participates in protein sorting [161]. AP-4 is thought to participate in autophagosome biogenesis by regulating ATG9 export to autophagosomes [162]. These findings support the view that alterations throughout the entire autophagic pathway are implicated in corticospinal tract degeneration and the development of HSPs, as these disorders can be caused by mutations in genes involved in different stages of the autophagy process (Figure 3).
–
Vici Syndrome
–
Vici syndrome is a multisystem disease first described by Carlo Dionisi Vici in 1988 [163]. This hereditary disease is classically defined based on the presence of five characteristic features: agenesis of the corpus callosum; cataracts; cardiomyopathy; immunodeficiency; and hypopigmentation [132]. Muscle and neurogenic anomalies were later described in Vici syndrome patients [164][165][166], and in the last decade recessive mutations in EPG5 were identified as cause of the disease [167]. Vici syndrome is a severe disease: most patients present developmental delay and die before the age of three [168]. EPG5 is the homolog of the Caenorhabditis elegans gene epg-5, a specific autophagy gene that promotes fusion of autophagosomes with late endosomes/lysosomes during autophagy [169][170]. Analysis of fibroblasts from Vici syndrome patients has revealed the accumulation of p62 and NBR1, levels of which do not increase in response to treatment with rapamycin and bafilomycin, suggesting impairment of autophagosomal cargo clearance in these cells [167]. However, colocalization of LC3 with LAMP1 is decreased in these fibroblasts, suggesting dysfunctional autophagosome-lysosome fusion. Epg5-/- mice also show blockade of autophagic flux as a consequence of impaired proteolytic activity of autolysosomes [171]. A phenotype common to Vici syndrome patients and Epg5-/- mice is agenesis of the corpus callosum, although the mouse model only partially reproduces the clinical features of the human syndrome [168]. Specifically, Epg5-/- mice present selective degeneration of cortical layer 5 pyramidal neurons and spinal cord motor neurons, features reminiscent of the key clinical signs of amyotrophic lateral sclerosis [171]. This mouse also shows a decrease in the thickness of the outer nuclear layer of the retina from six months of age, and consequent reduction in electroretinogram responses, suggesting that it may constitute a useful model of retinitis pigmentosa [172]. Vici syndrome patients develop cataracts, and while retinal degeneration cannot be ruled out it is difficult to demonstrate owing to the short lifespan of these patients.
–
Vici syndrome is another example of a pathology caused by disturbed embryonic CNS development and aberrant autophagy. It features anatomical defects including callosal agenesis, which entails the ablation of white matter connecting the two brain hemispheres, and abnormal neuronal migration [167], in addition to behavioural and physiological defects. In fact, around two-thirds of Vici syndrome patients present severe epilepsy with different types of seizures, suggesting the inclusion of the syndrome in the context of mTOR-related disorders [166]. In addition to the neurodevelopmental features, loss of learned skills and microcephaly in children with Vici syndrome also suggest a neurodegenerative component [132]. This neurodegeneration has also been described in Drosophila following downregulation of epg5 in the adult stage [166]. The neurodegeneration also observed in BPAN further reinforces the aforementioned functional connection between WIPI3, WIPI4, and EPG5.
–
SNX14-associated autosomal-recessive cerebellar ataxia
–
Hereditary cerebellar ataxia is a heterogeneous group of clinical conditions mainly characterized by imbalance and poor coordination with frequent cerebellar atrophy and Purkinje cell loss [173]. Two separate research groups recently identified mutations in the sorting nexin gene SNX14 as the cause of a specific subtype of cerebellar ataxia known as autosomal recessive spinocerebellar ataxia 20 (SCAR20) [174][175]. This clinical syndrome, which accounts for around 10% of early onset cerebellar atrophy and ataxia cases, is characterized by delayed development, cerebellar atrophy, motor ataxia, mental retardation and seizures [132][176]. Some degree of overlap with lysosomal storage diseases is observed in certain cases [132], suggesting that cerebellar cells are highly sensitive to lysosomal impairment [174]. Sorting nexin (SNX) proteins are a large family of proteins characterized by the presence of a PX domain, which binds membrane phosphatidylinositol phospholipids, and a GTPase activating domain. Most SNX proteins (except SNX19) use this GTPase activating domain to regulate the G-protein signaling domain, thereby playing an important role in decreasing G-protein-coupled receptor signaling [177]. SNX14 is thought to bind to PI-enriched autophagosomes and mediate fusion with lysosomes, although this is yet to be definitively demonstrated [132][177]. Animal models reproduce key features of SCAR20. Knockdown of SNX14 in zebrafish results in loss of cerebellar parenchyma and decreases in Purkinje cell number, autophagosome accumulation, and apoptosis [174]. Studies in mice suggest an important role of SNX14 in mouse neuronal development, as it is expressed at high levels in the brain, testes, and lungs and its expression increases further during development and maturation [178]. In fact, mice with Nestin-Cre-mediated Snx14 deletion in neurons and glia recapitulate the pathological features of SCAR20 and present Purkinje cell loss and cerebellar degeneration [176]. Snx14 knockdown in mouse cortical neurons also reduces intrinsic neuronal excitability, which correlates with decreased input resistance, underscoring the importance of SNX14 in neuronal excitability and synaptic function [178].
–
Cells from patients with SNX14 mutations contain large lysosomes suggestive of lysosomal alterations and show impaired autophagosome clearance [174][175]. These findings support a central role of lysosomal function in Purkinje cell viability and, therefore, the onset of cerebellar ataxia.
FUTURE PERSPECTIVES
In this review we have summarized the key findings indicating a connection between autophagy, neurodevelopment, and neurodevelopmental disorders. Despite clear gaps in our knowledge of the relationships between these processes, the available evidence strongly supports a central role of autophagy in multiple neurodevelopmental processes.
–
The contribution of autophagy to neurodevelopment has been investigated both in vitro and in vivo in a range of animal models, the most common of which are Drosophila and mouse or rat models in which autophagy is either impaired by deletion of one or more autophagy-related genes or is directly inhibited using pharmacological approaches. A better understanding of the role of autophagy in neurodevelopment will require in vivo verification of in vitro findings and replication of the results obtained in mouse models through deletion of different autophagy-related genes. Several discrepancies arise between studies, depending on the experimental approach.
–
Mutations in genes that participate in a given stage of the autophagy pathway do not always give rise to the same neurologic phenotype, likely because the affected proteins can interact with different counterparts and participate in distinct cellular processes that influence the final phenotype. It is also possible that a given affected protein may play multiple, as-yet-unknown roles in the autophagy process. For example, WIPI4 and SPATACSIN both participate in early and late stages of autophagy. In addressing whether the impacted stage of autophagy can determine the neurodevelopmental disorder, some general conclusions can be drawn based on analyses of different disorders. First, it is important to note that epilepsy and seizures are features of almost every neurodevelopmental disorder described in this review (with the exception of HSPs), regardless of the stage of autophagy affected. This supports a role of autophagy in modulating the balance between excitatory and inhibitory synaptic connections. In fact, uncontrolled induction of autophagy also produces an epileptic phenotype: loss-of-function mutations in the mTOR activator gene TBCK give rise to a neurodevelopmental condition with ID, coarse facial features, hypotonia, leukoencephalopathy, neuronopathy, and seizures [179]. Therefore, epileptogenesis is described in patients with both impaired and hyperactivated autophagy. Second, autistic features are correlated with mTOR upregulation and defects in the early stages of the autophagy pathway, and are very common in mTORopathies. Finally, corpus callosum involvement and cerebellar atrophy appear to be more common consequences of defects in the later stages of autophagy. Alterations in the corpus callosum are observed in Vici syndrome and SPG patients, while cerebellar involvement is mainly observed in SCAR20. However, it is important to note that mutations in the gene encoding ATG5, which participates in the autophagosome elongation, have also been detected in ataxic patients [180], and that a mutation in VPS15 has also been reported in a child with different neurodevelopmental features: cortical and optic nerve atrophy, intellectual impairment, spasticity, ataxia, psychomotor affection and epilepsy [181]. These last two mutations are highly less frequent, but increment the complexity of our current understanding of neurodevelopmental disorders.
–
In this review we have proposed some correlations between alterations in specific stages of autophagy and discrete neurodevelopmental disorders. However, in order to acquire a more complete perspective it will be essential to further investigate how autophagy is differentially regulated in selected neuronal types, as this could provide crucial insights into the aetiology and pathogenesis of different neurodevelopmental disorders. Finally, it will be important to breach the species barrier and investigate the role of autophagy in human neurodevelopment. Human cerebral organoids and humanized brains or chimeric brains constitute valuable and valid models in which the contributions of autophagy to human neurodevelopment can be studied [182][183].
REFERENCES
- Klionsky DJ (2008). Autophagy revisited: A conversation with Christian de Duve. Autophagy 4(6): 740–743. 10.4161/auto.6398
- Clark S. (1957). Cellular differentiation in the kidneys of newborn mice studied with the electron mircroscope. Biophys Biochem Cytol 3(3): 349–362. 10.1083/jcb.3.3.349
- B. Novikoff A (1959). The Proximal Tubule Cell in Experimental Hydronephrosis. J Biophys Biochem Cytol 6(1):136-8. 10.1083/jcb.6.1.136
- Boya P, Reggiori F, and Codogno P (2013). Emerging regulation and functions of autophagy. Nat Cell Biol 15(7): 713–720. 10.1038/ncb2788
- Morishita H, and Mizushima N (2019). Diverse cellular roles of autophagy. Annu Rev Cell Dev Biol 35: 453–475. 10.1146/annurev-cellbio-100818-125300
- Yim WWY, and Mizushima N (2020). Lysosome biology in autophagy. Cell Discov 6(1): 6. 10.1038/s41421-020-0141-7
- Schuck S (2020). Microautophagy – distinct molecular mechanisms handle cargoes of many sizes. J Cell Sci 133(17): jcs246322. 10.1242/jcs.246322
- Kaushik S, and Cuervo AM (2018). The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol 19(6): 365–381. 10.1038/s41580-018-0001-6
- Sheng R, and Qin Z-H (2019). History and Current Status of Autophagy Research. In: Qin Z-H, editor Autophagy Biol. Dis. Springer Singapore, Singapore; pp 3–37.
- Mizushima N (2020). The ATG conjugation systems in autophagy. Curr Opin Cell Biol 63: 1–10. 10.1016/j.ceb.2019.12.001
- Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, Griffiths G, and Ktistakis NT (2008). Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol 182(4): 685–701. 10.1083/jcb.200803137
- Melia TJ, Lystad AH, and Simonsen A (2020). Autophagosome biogenesis: From membrane growth to closure. J Cell Biol 219(6): 1–18. 10.1083/JCB.202002085
- Tooze SA, and Yoshimori T (2010). The origin of the autophagosomal membrane. Nat Cell Biol 12(9): 831–835. 10.1038/ncb0910-831
- Papinski D, Schuschnig M, Reiter W, Wilhelm L, Barnes CA, Maiolica A, Hansmann I, Pfaffenwimmer T, Kijanska M, Stoffel I, Lee SS, Brezovich A, Lou JH, Turk BE, Aebersold R, Ammerer G, Peter M, and Kraft C (2014). Early Steps in Autophagy Depend on Direct Phosphorylation of Atg9 by the Atg1 Kinase. Mol Cell 53(3): 471–483. 10.1016/j.molcel.2013.12.011
- Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, Kim H, Neufeld TP, Dillin A, and Guan KL (2013). ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol 15(7): 741–750. 10.1038/ncb2757
- Walker SA, and Ktistakis NT (2020). Autophagosome Biogenesis Machinery. J Mol Biol 432(8): 2449–2461. 10.1016/j.jmb.2019.10.027
- Jaber N, Dou Z, Chen JS, Catanzaro J, Jiang YP, Ballou LM, Selinger E, Ouyang X, Lin RZ, Zhang J, and Zong WX (2012). Class III PI3K Vps34 plays an essential role in autophagy and in heart and liver function. Proc Natl Acad Sci U S A 109(6): 2003–2008. 10.1073/pnas.1112848109
- Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, Klionsky DJ, Ohsumi M, and Ohsumi Y (1998). A protein conjugation system essential for autophagy. Nature 395(6700): 395–398. 10.1038/26506
- Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, Mizushima N, Tanida I, Kominami E, Ohsumi M, Noda T, and Ohsumi Y (2000). A ubiquitin-like system mediates protein lipidation. Nature 408(6811): 488–492. 10.1038/35044114
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, and Yoshimori T (2000). LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 19(21): 5720–5728. 10.1093/emboj/19.21.5720
- Fujita N, Hayashi-Nishino M, Fukumoto H, Omori H, Yamamoto A, Noda T, and Yoshimori T (2008). An Atg4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol Biol Cell 19(11): 4651–4659. 10.1091/mbc.E08-03-0312
- Shvets E, Fass E, Scherz-Shouval R, and Elazar Z (2008). The N-terminus and Phe52 residue of LC3 recruit p62/SQSTM1 into autophagosomes. J Cell Sci 121(16): 2685–2695. 10.1242/jcs.026005
- Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Øvervatn A, Stenmark H, and Johansen T (2005). p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 171(4): 603–614. 10.1083/jcb.200507002
- Johansen T, and Lamark T (2020). Selective Autophagy: ATG8 Family Proteins, LIR Motifs and Cargo Receptors. J Mol Biol 432(1): 80–103. 10.1016/j.jmb.2019.07.016
- Deter RL, Baudhuin P, and De Duve C (1967). Participation of lysosomes in cellular autophagy induced in rat liver by glucagon. J Cell Biol 35(2): C11-6. 10.1083/jcb.35.2.c11
- Gordon PB, and Seglen PO (1988). Prelysosomal convergence of autophagic and endocytic pathways. Biochem Biophys Res Commun 151(1): 40–47. 10.1016/0006-291X(88)90556-6
- Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, and Sabatini1 DM (2011). mTORC1 Senses Lysosomal Amino Acids. Science 334: 678–683. 10.1126/science.1207056
- Kawabata T, and Yoshimori T (2020). Autophagosome biogenesis and human health. Cell Discov 6(1): 33. 10.1038/s41421-020-0166-y
- Kim J, Kundu M, Viollet B, and Guan KL (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13(2): 132–141. 10.1038/ncb2152
- Inoki K, Li Y, Zhu T, Wu J, and Guan KL (2002). TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 4(9): 648–657. 10.1038/ncb839
- Paquette M, El-Houjeiri L, Zirden LC, Puustinen P, Blanchette P, Jeong H, Dejgaard K, Siegel PM, and Pause A (2021). AMPK-dependent phosphorylation is required for transcriptional activation of TFEB/TFE3 Authors/Affiliation. bioRxiv 00(00): 2021.01.27.428292. 10.1080/15548627.2021.1898748
- Kim J, Kim YC, Fang C, Russell RC, Kim JH, Fan W, Liu R, Zhong Q, and Guan KL (2013). Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell 152(1–2): 290–303. 10.1016/j.cell.2012.12.016
- Maria Fimia G, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R, Corazzari M, Fuoco C, Ucar A, Schwartz P, Gruss P, Piacentini M, Chowdhury K, and Cecconi F (2007). Ambra1 regulates autophagy and development of the nervous system. Nature 447(7148): 1121–1125. 10.1038/nature05925
- Pattingre S, Tassa A, Qu X, Garuti R, Xiao HL, Mizushima N, Packer M, Schneider MD, and Levine B (2005). Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122(6): 927–939. 10.1016/j.cell.2005.07.002
- Mizushima N (2007). Autophagy: Process and function. Genes Dev 21(22): 2861–2873. 10.1101/gad.1599207
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, Kominami E, Tanaka K, and Chiba T (2005). Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 169(3): 425–434. 10.1083/jcb.200412022
- Lynch-Day MA, and Klionsky DJ (2010). The Cvt pathway as a model for selective autophagy. FEBS Lett 584(7): 1359–66. 10.1016/j.febslet.2010.02.013
- Ponpuak M, Mandell MA, Kimura T, Chauhan S, Cleyrat C, and Deretic V (2015). Secretory autophagy. Curr Opin Cell Biol 35: 106–16. 10.1016/j.ceb.2015.04.016
- Yeo SK, and Guan JL (2020). Regulation of immune checkpoint blockade efficacy in breast cancer by FIP200: A canonical-autophagy-independent function. Cell Stress 4(8): 216–217. 10.15698/cst2020.08.229
- Klionsky DJ et al. (2021). Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 1–382. 10.1080/15548627.2020.1797280
- Mizushima N, and Levine B (2020). Autophagy in Human Diseases. N Engl J Med 383(16): 1564–1576. 10.1056/nejmra2022774
- Menzies FM, Fleming A, Caricasole A, Bento CF, Andrews SP, Ashkenazi A, Füllgrabe J, Jackson A, Jimenez Sanchez M, Karabiyik C, Licitra F, Lopez Ramirez A, Pavel M, Puri C, Renna M, Ricketts T, Schlotawa L, Vicinanza M, Won H, Zhu Y, Skidmore J, and Rubinsztein DC (2017). Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 93(5): 1015–1034. 10.1016/j.neuron.2017.01.022
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, and Mizushima N (2006). Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441(7095): 885–889. 10.1038/nature04724
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata JI, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, and Tanaka K (2006). Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441(7095): 880–884. 10.1038/nature04723
- Liang CC, Wang C, Peng X, Gan B, and Guan JL (2010). Neural-specific deletion of FIP200 leads to cerebellar degeneration caused by increased neuronal death and axon degeneration. J Biol Chem 285(5): 3499–3509. 10.1074/jbc.M109.072389
- Soukup SF, Kuenen S, Vanhauwaert R, Manetsberger J, Hernández-Díaz S, Swerts J, Schoovaerts N, Vilain S, Gounko N V., Vints K, Geens A, De Strooper B, and Verstreken P (2016). A LRRK2-Dependent EndophilinA Phosphoswitch Is Critical for Macroautophagy at Presynaptic Terminals. Neuron 92(4): 829–844. 10.1016/j.neuron.2016.09.037
- Wang T, Martin S, Papadopulos A, Harper CB, Mavlyutov TA, Niranjan D, Glass NR, Cooper-White JJ, Sibarita JB, Choquet D, Davletov B, and Meunier FA (2015). Control of autophagosome axonal retrograde flux by presynaptic activity unveiled using botulinum neurotoxin type A. J Neurosci 35(15): 6179–6194. 10.1523/JNEUROSCI.3757-14.2015
- Cheng XT, Zhou B, Lin MY, Cai Q, and Sheng ZH (2015). Axonal autophagosomes recruit dynein for retrograde transport through fusion with late endosomes. J Cell Biol 209(3): 377–386. 10.1083/jcb.201412046
- Soykan T, Haucke V, and Kuijpers M (2021). Mechanism of synaptic protein turnover and its regulation by neuronal activity. Curr Opin Neurobiol 69: 76–83. 10.1016/j.conb.2021.02.006
- Kuijpers M, Kochlamazashvili G, Stumpf A, Puchkov D, Swaminathan A, Lucht MT, Krause E, Maritzen T, Schmitz D, and Haucke V (2021). Neuronal Autophagy Regulates Presynaptic Neurotransmission by Controlling the Axonal Endoplasmic Reticulum. Neuron 109(2): 299-313.e9. 10.1016/j.neuron.2020.10.005
- Dagda RK, Cherra SJ, Kulich SM, Tandon A, Park D, and Chu CT (2009). Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J Biol Chem 284(20): 13843–13855. 10.1074/jbc.M808515200
- Kawajiri S, Saiki S, Sato S, Sato F, Hatano T, Eguchi H, and Hattori N (2010). PINK1 is recruited to mitochondria with parkin and associates with LC3 in mitophagy. FEBS Lett 584(6): 1073–1079. 10.1016/j.febslet.2010.02.016
- Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, Wolfe DM, Martinez-Vicente M, Massey AC, Sovak G, Uchiyama Y, Westaway D, Cuervo AM, and Nixon RA (2010). Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 141(7): 1146–1158. 10.1016/j.cell.2010.05.008
- Jiang Y, Mullaney KA, Peterhoff CM, Che S, Schmidt SD, Boyer-Boiteau A, Ginsberg SD, Cataldo AM, Mathews PM, and Nixon RA (2010). Alzheimer's-related endosome dysfunction in Down syndrome is Aβ-independent but requires APP and is reversed by BACE-1 inhibition. Proc Natl Acad Sci U S A 107(4): 1630–1635. 10.1073/pnas.0908953107
- Nixon RA, and Yang DS (2011). Autophagy failure in Alzheimer's disease-locating the primary defect. Neurobiol Dis 43(1): 38–45. 10.1016/j.nbd.2011.01.021
- Holtzman E, and Novikoff AB (1965). Lysomes in the rat sciatic nerve following crush. J Cell Biol 27(3): 651–669. 10.1083/jcb.27.3.651
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, and Ohsumi Y (2004). In Vivo Analysis of Autophagy in Response to Nutrient Starvation Using Transgenic Mice Expressing a Fluorescent Autophagosome Marker. Mol Biol Cell 15(3): 1101–1111. 10.1091/mbc.E03-09-0704
- Alirezaei M, Kemball CC, Flynn CT, Wood MR, Whitton JL, and Kiosses WB (2010). Short-term fasting induces profound neuronal autophagy. Autophagy 6(6): 702–710. 10.4161/auto.6.6.12376
- Esteban-Martínez L, and Boya P (2015). Autophagic flux determination in vivo and ex vivo. Methods 75: 79–86. 10.1016/j.ymeth.2015.01.008
- Nikoletopoulou V, Sidiropoulou K, Kallergi E, Dalezios Y, and Tavernarakis N (2017). Modulation of Autophagy by BDNF Underlies Synaptic Plasticity. Cell Metab 26(1): 230-242.e5. 10.1016/j.cmet.2017.06.005
- Kulkarni A, Dong A, Kulkarni VV, Chen J, Laxton O, Anand A, and Maday S (2020). Differential regulation of autophagy during metabolic stress in astrocytes and neurons. Autophagy 16(9): 1651–1667. 10.1080/15548627.2019.1703354
- Guo C, Hao LJ, Yang ZH, Chai R, Zhang S, Gu Y, Gao HL, Zhong ML, Wang T, Li JY, and Wang ZY (2016). Deferoxamine-mediated up-regulation of HIF-1α prevents dopaminergic neuronal death via the activation of MAPK family proteins in MPTP-treated mice. Exp Neurol 280: 13–23. 10.1016/j.expneurol.2016.03.016
- Nakanishi A, Hatano N, Fujiwara Y, Sha'ri A, Takabatake S, Akano H, Kanayama N, Magari M, Nozaki N, and Tokumitsu H (2017). AMP-activated protein kinase–mediated feedback phosphorylation controls the Ca2+/calmodulin (CaM) dependence of Ca2+/CaM-dependent protein kinase kinase β. J Biol Chem 292(48): 19804–19813. 10.1074/jbc.M117.805085
- Wei Y, Pattingre S, Sinha S, Bassik M, and Levine B (2008). JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell 30(6): 678–688. 10.1016/j.molcel.2008.06.001
- Maejima Y, Kyoi S, Zhai P, Liu T, Li H, Ivessa A, Sciarretta S, P. Del Re D, K. Zablocki D, Hsu C-P, Lim D-S, Isobe M, and Sadoshima J (2014). Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Physiol Behav 19(11): 1478–1488. 10.1038/nm.3322
- He Y, She H, Zhang T, Xu H, Cheng L, Yepes M, Zhao Y, and Mao Z (2018). p38 MAPK inhibits autophagy and promotes microglial inflammatory responses by phosphorylating ULK1. J Cell Biol 217(1): 315–328. 10.1083/jcb.201701049
- Medina DL, Di Paola S, Peluso I, Armani A, De Stefani D, Venditti R, Montefusco S, Scotto-Rosato A, Prezioso C, Forrester A, Settembre C, Wang W, Gao Q, Xu H, Sandri M, Rizzuto R, De Matteis MA, and Ballabio A (2015). Lysosomal calcium signaling regulates autophagy via calcineurin and TFEB. M Nat Cell Biol 17(3): 288–299. 10.1038/ncb3114.Lysosomal
- Høyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T, Bianchi K, Fehrenbacher N, Elling F, Rizzuto R, Mathiasen IS, and Jäättelä M (2007). Control of Macroautophagy by Calcium, Calmodulin-Dependent Kinase Kinase-β, and Bcl-2. Mol Cell 25(2): 193–205. 10.1016/j.molcel.2006.12.009
- Varelas X (2014). The hippo pathway effectors TAZ and YAP in development, homeostasis and disease. Dev 141(8): 1614–1626. 10.1242/dev.102376
- Wang W, Xiao Z-D, Li X, Aziz KE, Gan B, Johnson RL, and Chen J (2015). AMPK modulates Hippo pathway activity to regulate energy homeostasis. Nat Cell Biol 17(4): 490–499. 10.1038/ncb3113.AMPK
- Shrestha BK, Rasmussen MS, Abudu YP, Bruun JA, Larsen KB, Alemu EA, Sjøttem E, Lamark T, and Johansen T (2020). NIMA-related kinase 9–mediated phosphorylation of the microtubule-associated LC3B protein at Thr-50 suppresses selective autophagy of p62/sequestosome 1. J Biol Chem 295(5): 1240–1260. 10.1074/jbc.RA119.010068
- Rodriguez-Muela N, Koga H, García-Ledo L, Villa P de la, De La Rosa EJ, Cuervo AM, and Boya (2013). Balance between autophagic pathways preserves retinal homeostasis. Natl Inst Heal 23(1): 1–7. 10.1111/acel.12072.Balance
- Stavoe AKH, and Holzbaur ELF (2019). Autophagy in Neurons. Annu Rev Cell Dev Biol 176(1): 100–106. 10.1146/annurev-cellbio-100818-125242.Autophagy
- Mizushima N, and Komatsu M (2011). Autophagy: Renovation of cells and tissues. Cell 147(4): 728–741. 10.1016/j.cell.2011.10.026
- Gómez-Sintes R, Ledesma MD, and Boya P (2016). Lysosomal cell death mechanisms in aging. Ageing Res Rev 32: 150–168. 10.1016/j.arr.2016.02.009
- López-Otín C, Blasco MA, Partridge L, Serrano M, and Kroemer G (2013). The Hallmarks of Aging. Cell 153(6): 1194–1217. 10.1016/j.cell.2013.05.039
- Rubinsztein DC, Mariño G, and Kroemer G (2011). Autophagy and aging. Cell 146(5): 682–695. 10.1016/j.cell.2011.07.030
- Boland B, Yu WH, Corti O, Mollereau B, Henriques A, Bezard E, Pastores GM, Rubinsztein DC, Nixon RA, Duchen MR, Mallucci GR, Kroemer G, Levine B, Eskelinen EL, Mochel F, Spedding M, Louis C, Martin OR, and Millan MJ (2018). Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nat Rev Drug Discov 17(9): 660–688. 10.1038/nrd.2018.109
- Guan JL, Simon AK, Prescott M, Menendez JA, Liu F, Wang F, Wang C, Wolvetang E, Vazquez-Martin A, and Zhang J (2013). Autophagy in stem cells. Autophagy 9(6): 830–849. 10.4161/auto.24132
- Gage FH (2000). Mammalian neural stem cells. Science 287(5457): 1433–8. 10.1126/science.287.5457.1433
- Ming G-L, and Song H (2011). Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron 70(4): 687–702. 10.1016/j.neuron.2011.05.001
- Llamas E, Alirzayeva H, Loureiro R, and Vilchez D (2020). The intrinsic proteostasis network of stem cells. Curr Opin Cell Biol 67: 46–55. 10.1016/j.ceb.2020.08.005
- Wu X, Fleming A, Ricketts T, Pavel M, Virgin H, Menzies FM, and Rubinsztein DC (2016). Autophagy regulates Notch degradation and modulates stem cell development and neurogenesis. Nat Commun 7: 1–17. 10.1038/ncomms10533
- Boya P, Codogno P, and Rodriguez-Muela N (2018). Autophagy in stem cells: repair, remodelling and metabolic reprogramming. Development 145(4): 1–14. 10.1242/dev.146506
- Vázquez P, Arroba AI, Cecconi F, De La Rosa EJ, Boya P, and De Pablo F (2012). Atg5 and Ambra1 differentially modulate neurogenesis in neural stem cells. Autophagy 8(2): 187–199. 10.4161/auto.8.2.18535
- Yoke Wui Ng M, Wai T, and Simonsen A (2021). Quality control of the mitochondrion. Dev Cell 97: 1–25. 10.1016/j.devcel.2021.02.009
- Lane N (2006). Mitochondrial disease: Powerhouse of disease. Nature 440(7084): 600–602. 10.1038/440600a
- Palikaras K, and Tavernarakis N (2012). Mitophagy in neurodegeneration and aging. Front Genet 3: 297. 10.3389/fgene.2012.00297
- Esteban-Martínez L, Sierra-Filardi E, McGreal RS, Salazar-Roa M, Mariño G, Seco E, Durand S, Enot D, Graña O, Malumbres M, Cvekl A, Cuervo AM, Kroemer G, and Boya P (2017). Programmed mitophagy is essential for the glycolytic switch during cell differentiation. EMBO J 36(12): 1688–1706. 10.15252/embj.201695916
- Chandel NS, Jasper H, Ho TT, and Passegué E (2016). Metabolic regulation of stem cell function in tissue homeostasis and organismal ageing. Nat Cell Biol 18(8): 823–832. 10.1038/ncb3385
- Liberti M V., and Locasale JW (2016). The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem Sci 41(3): 211–218. 10.1016/j.tibs.2015.12.001
- Coupé B, Ishii Y, Dietrich MO, Komatsu M, Horvath TL, and Bouret SG (2012). Loss of autophagy in pro-opiomelanocortin neurons perturbs axon growth and causes metabolic dysregulation. Cell Metab 15(2): 247–255. 10.1016/j.cmet.2011.12.016
- Choi YJ, Di Nardo A, Kramvis I, Meikle L, Kwiatkowski DJ, Sahin M, and He X (2008). Tuberous sclerosis complex proteins control axon formation. Genes Dev 22(18): 2485–2495. 10.1101/gad.1685008
- Shen W, and Ganetzky B (2009). Autophagy promotes synapse development in Drosophila. J Cell Biol 187(1): 71–79. 10.1083/jcb.200907109
- Fleming A, and Rubinsztein DC (2020). Autophagy in Neuronal Development and Plasticity. Trends Neurosci 43(10): 767–779. 10.1016/j.tins.2020.07.003
- Kuijpers M, and Haucke V (2021). Neuronal autophagy controls the axonal endoplasmic reticulum to regulate neurotransmission in healthy neurons. Autophagy 17(4): 1049-1051. 10.1080/15548627.2021.1893569
- Waites CL, Craig AM, and Garner CC (2005). Mechanisms of vertebrate synaptogenesis. Annu Rev Neurosci 28(1): 251–74. 10.1146/annurev.neuro.27.070203.144336
- Tang G, Gudsnuk K, Kuo S-H, Cotrina ML, Rosoklija G, Sosunov A, Sonders MS, Kanter E, Castagna C, Yamamoto A, Yue Z, Arancio O, Peterson BS, Champagne F, Dwork AJ, Goldman J, and Sulzer D (2014). Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 83(5): 1131–43. 10.1016/j.neuron.2014.07.040
- Association AP (2013). Diagnostic and statistical manual of mental disorders., 5th ed. American Psychiatric Association.
- Bourgeron T (2009). A synaptic trek to autism. Curr Opin Neurobiol 19(2): 231–234. 10.1016/j.conb.2009.06.003
- Lipton JO, and Sahin M (2014). The Neurology of mTOR. Neuron 84(2): 275–291. 10.1016/j.neuron.2014.09.034
- Long X, Lin Y, Ortiz-Vega S, Yonezawa K, and Avruch J (2005). Rheb binds and regulates the mTOR kinase. Curr Biol 15(8): 702–713. 10.1016/j.cub.2005.02.053
- Lee K-M, Hwang S-K, and Lee J-A (2013). Neuronal Autophagy and Neurodevelopmental Disorders. Exp Neurobiol 22(3): 133–142. 10.5607/en.2013.22.3.133
- Zhou J, and Parada LF (2012). PTEN signaling in autism spectrum disorders. Curr Opin Neurobiol 22(5): 873–879. 10.1016/j.conb.2012.05.004
- Han JM, and Sahin M (2011). TSC1/TSC2 signaling in the CNS. FEBS Lett 585(7): 973–980. 10.1016/j.febslet.2011.02.001
- Fu C, Cawthon B, Clinkscales W, Bruce A, Winzenburger P, and Ess KC (2012). GABAergic interneuron development and function is modulated by the Tsc1 gene. Cereb Cortex 22(9): 2111–2119. 10.1093/cercor/bhr300
- Bhakar AL, Dolen G, and Bear MF (2012). The pathophysiology of fragile X (and what it teaches us about synapses). Annu Rev Neurosci 35: 417–443. 10.1146/annurev-neuro-060909-153138
- Zheng-Hong Q (2019). Autophagy: biology and diseases, 3rd ed. Springer.
- Garg S, Green J, Leadbitter K, Emsley R, Lehtonen A, Evans G, and Huson SM (2013). Neurofibromatosis type 1 and autism spectrum disorder. Pediatrics 132(6). 10.1542/peds.2013-1868
- Buxbaum JD, Cai G, Chaste P, Nygren G, Goldsmith J, Reichert J, Anckarsäter H, Rastam M, Smith CJ, Silverman JM, Hollander E, Leboyer M, Gillberg C, Verloes A, and Betancur C (2007). Mutation screening of the PTEN gene in patients with autism spectrum disorders and macrocephaly. Am J Med Genet Part B Neuropsychiatr Genet 144(4): 484–491. 10.1002/ajmg.b.30493
- Park HR, Lee JM, Moon HE, Lee DS, Kim BN, Kim J, Kim DG, and Paek SH (2016). A short review on the current understanding of autism spectrum disorders. Exp Neurobiol 25(1): 1–13. 10.5607/en.2016.25.1.1
- Geschwind DH (2008). Autism: Many Genes, Common Pathways? Cell 135(3): 391–395. 10.1016/j.cell.2008.10.016
- Bozzi Y, Casarosa S, and Caleo M (2012). Epilepsy as a neurodevelopmental disorder. Front Psychiatry 3: 1–14. 10.3389/fpsyt.2012.00019
- Laplante M, and Sabatini DM (2012). mTOR signaling. Cold Spring Harb Perspect Biol 4(2): 10–13. 10.1101/cshperspect.a011593
- Zhou J, Blundell J, Ogawa S, Kwon CH, Zhang W, Sinton C, Powell CM, and Parada LF (2009). Pharmacological inhibition of mTORCl suppresses anatomical, cellular, and behavioral abnormalities in neural-specific PTEN knock-out mice. J Neurosci 29(6): 1773–1783. 10.1523/JNEUROSCI.5685-08.2009
- Kim HJ, Cho MH, Shim WH, Kim JK, Jeon EY, Kim DH, and Yoon SY (2017). Deficient autophagy in microglia impairs synaptic pruning and causes social behavioral defects. Mol Psychiatry 22(11): 1576–1584. 10.1038/mp.2016.103
- Hui KK, Takashima N, Watanabe A, Chater TE, Matsukawa H, Nekooki-Machida Y, Nilsson P, Endo R, Goda Y, Saido TC, Yoshikawa T, and Tanaka M (2019). GABARAPs dysfunction by autophagy deficiency in adolescent brain impairs GABAA receptor trafficking and social behavior. Sci Adv 5(4): 1–16. 10.1126/sciadv.aau8237
- Hui KK, and Tanaka M (2019). Autophagy links MTOR and GABA signaling in the brain. Autophagy 15(10): 1848–1849. 10.1080/15548627.2019.1637643
- Ichimura Y, Takagi K, Yang Y, Pankiv S, Kanegae Y, Kageyama S, Suzuki M, Saito I, Mizushima T, Komatsu M, and Simonsen A (2014). Structural determinants in GABARAP required for the selective binding and recruitment of ALFY to LC3B-positive structures. EMBO Rep 15(5): 557–565. 10.1002/embr.201338003
- Clausen TH, Lamark T, Isakson P, Finley K, Larsen KB, Brech A, Øvervatn A, Stenmark H, Bjørkøy G, Simonsen A, and Johansen T (2010). p62/SQSTM1 and ALFY interact to facilitate the formation of p62 bodies/ALIS and their degradation by autophagy. Autophagy 6(3): 330–344. 10.4161/auto.6.3.11226
- Iossifov I et al. (2012). De Novo Gene Disruptions in Children on the Autistic Spectrum. Neuron 74(2): 285–299. 10.1016/j.neuron.2012.04.009
- Wang T, Guo H, Xiong B, Stessman HAF, Wu H, Coe BP, Turner TN, Liu Y, Zhao W, Hoekzema K, Vives L, Xia L, Tang M, Ou J, Chen B, Shen Y, Xun G, Long M, Lin J, Kronenberg ZN, Peng Y, Bai T, Li H, Ke X, Hu Z, Zhao J, Zou X, Xia K, and Eichler EE (2016). De novo genic mutations among a Chinese autism spectrum disorder cohort. Nat Commun 7(13): 1–10. 10.1038/ncomms13316
- Bateup HS, Johnson CA, Denefrio CL, Saulnier JL, Kornacker K, and Sabatini BL (2013). Excitatory / Inhibitory Synaptic Imbalance Leads to Hippocampal Hyperexcitability in Mouse Models of Tuberous Sclerosis. Neuron 78(3): 510–522. 10.1016/j.neuron.2013.03.017
- Curatolo P, Verdecchia M, and Bombardieri R (2001). Vigabatrin for tuberous sclerosis complex. Brain Dev 23(7): 649–653. 10.1016/S0387-7604(01)00290-X
- Talos DM, Sun H, Kosaras B, Joseph A, Folkerth RD, Poduri A, Madsen JR, Black PM, and Jensen FE (2012). Altered inhibition in tuberous sclerosis and type IIb cortical dysplasia. Ann Neurol 71(4): 539–551. 10.1002/ana.22696
- D'Antuono M, Merlo D, and Avoli M (2003). Involvement of cholinergic and gabaergic systems in the fragile X knockout mice. Neuroscience 119(1): 9–13. 10.1016/S0306-4522(03)00103-9
- D'Hulst C, Heulens I, Brouwer JR, Willemsen R, De Geest N, Reeve SP, De Deyn PP, Hassan BA, and Kooy RF (2009). Expression of the GABAergic system in animal models for fragile X syndrome and fragile X associated tremor/ataxia syndrome (FXTAS). Brain Res 1253: 176–183. 10.1016/j.brainres.2008.11.075
- Horvath R (2013). Brain iron takes off: A new propeller protein links neurodegeneration with autophagy. Brain 136(6): 1687–1689. 10.1093/brain/awt098
- Saitsu H, Nishimura T, Muramatsu K, Kodera H, Kumada S, Sugai K, Kasai-Yoshida E, Sawaura N, Nishida H, Hoshino A, Ryujin F, Yoshioka S, Nishiyama K, Kondo Y, Tsurusaki Y, Nakashima M, Miyake N, Arakawa H, Kato M, Mizushima N, and Matsumoto N (2013). De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat Genet 45(4): 445–449. 10.1038/ng.2562
- Nishioka K, Oyama G, Yoshino H, Li Y, Matsushima T, Takeuchi C, Mochizuki Y, Mori-Yoshimura M, Murata M, Yamasita C, Nakamura N, Konishi Y, Ohi K, Ichikawa K, Terada T, Obi T, Funayama M, Saiki S, and Hattori N (2015). High frequency of beta-propeller protein-associated neurodegeneration (BPAN) among patients with intellectual disability and young-onset parkinsonism. Neurobiol Aging 36(5): 2004.e9-2004.e15. 10.1016/j.neurobiolaging.2015.01.020
- Yoganathan S, Arunachal G, Sudhakar SV, Rajaraman V, Thomas M, and Danda S (2016). Beta Propellar Protein-Associated Neurodegeneration: A Rare Cause of Infantile Autistic Regression and Intracranial Calcification. Neuropediatrics 47(2): 123–127. 10.1055/s-0035-1571189
- Ebrahimi-Fakhari D, Saffari A, Wahlster L, Lu J, Byrne S, Hoffmann GF, Jungbluth H, and Sahin M (2016). Congenital disorders of autophagy: An emerging novel class of inborn errors of neuro-metabolism. Brain 139(2): 317–337. 10.1093/brain/awv371
- Suleiman J, Allingham-Hawkins D, Hashem M, Shamseldin HE, Alkuraya FS, and El-Hattab AW (2018). WDR45B-related intellectual disability, spastic quadriplegia, epilepsy, and cerebral hypoplasia: A consistent neurodevelopmental syndrome. Clin Genet 93(2): 360–364. 10.1111/cge.13054
- Krick R, Busse RA, Scacioc A, Stephan M, Janshoff A, Thumm M, and Kühnel K (2012). Structural and functional characterization of the two phosphoinositide binding sites of PROPPINs, a β-propeller protein family. Proc Natl Acad Sci U S A 109(30): E2042-9. 10.1073/pnas.1205128109
- Bakula D, Müller AJ, Zuleger T, Takacs Z, Franz-Wachtel M, Thost AK, Brigger D, Tschan MP, Frickey T, Robenek H, Macek B, and Proikas-Cezanne T (2017). WIPI3 and WIPI4 β-propellers are scaffolds for LKB1-AMPK-TSC signalling circuits in the control of autophagy. Nat Commun 8: 15637. 10.1038/ncomms15637
- Hor CHH, and Tang BL (2019). Beta-propeller protein-associated neurodegeneration (BPAN) as a genetically simple model of multifaceted neuropathology resulting from defects in autophagy. Rev Neurosci 30(3): 261–277. 10.1515/revneuro-2018-0045
- Ji C, Zhao H, Chen D, Zhang H, and Zhao YG (2021). β-propeller proteins WDR45 and WDR45B regulate autophagosome maturation into autolysosomes in neural cells. Curr Biol 31(8):1666-1677.e6. 10.1016/j.cub.2021.01.081
- Behrends C, Sowa ME, Gygi SP, and Harper JW (2010). Network organization of the human autophagy system. Nature 466(7302): 68–76. 10.1038/nature09204
- Zhao YG, Sun L, Miao G, Ji C, Zhao H, Sun H, Miao L, Yoshii SR, Mizushima N, and Zhang H (2015). The autophagy gene Wdr45/Wipi4 regulates learning and memory function and axonal homeostasis. Autophagy 11(6): 881–890. 10.1080/15548627.2015.1047127
- Ji C, Zhao H, Li D, Sun H, Hao J, Chen R, Wang X, Zhang H, and Zhao YG (2020). Role of Wdr45b in maintaining neural autophagy and cognitive function. Autophagy 16(4): 615–625. 10.1080/15548627.2019.1632621
- Mollereau B, and Walter L (2019). Is WDR45 the missing link for ER stress-induced autophagy in beta-propeller associated neurodegeneration? Autophagy 15(12): 2163–2164. 10.1080/15548627.2019.1668229
- Wan H, Wang Q, Chen X, Zeng Q, Shao Y, Fang H, Liao X, Li HS, Liu MG, Xu T Le, Diao M, Li D, Meng B, Tang B, Zhang Z, and Liao L (2020). WDR45 contributes to neurodegeneration through regulation of ER homeostasis and neuronal death. Autophagy 16(3): 531–547. 10.1080/15548627.2019.1630224
- Santana-Codina N, and Mancias JD (2018). The role of NCOA4-mediated ferritinophagy in health and disease. Pharmaceuticals 11(4): 114. 10.3390/ph11040114
- Seibler P, Burbulla LF, Dulovic M, Zittel S, Heine J, Schmidt T, Rudolph F, Westenberger A, Rakovic A, Münchau A, Krainc D, and Klein C (2018). Iron overload is accompanied by mitochondrial and lysosomal dysfunction in WDR45 mutant cells. Brain 141(10): 3052–3064. 10.1093/brain/awy230
- Goizet C, Boukhris A, Maltete D, Guyant-Maréchal L, Truchetto J, Mundwiller E, Hanein S, Jonveaux P, Roelens F, Loureiro J, Godet E, Forlani S, Melki J, Auer-Grumbach M, Fernandez JC, Martin-Hardy P, Sibon I, Sole G, Orignac I, Mhiri C, Coutinho P, Durr A, Brice A, and Stevanin G (2009). SPG15 is the second most common cause of hereditary spastic paraplegia with thin corpus callosum. Neurology 73(14): 1111–1119. 10.1212/WNL.0b013e3181bacf59
- Renvoisé B, Chang J, Singh R, Yonekawa S, FitzGibbon EJ, Mankodi A, Vanderver A, Schindler AB, Toro C, Gahl WA, Mahuran DJ, Blackstone C, and Pierson TM (2014). Lysosomal abnormalities in hereditary spastic paraplegia types SPG15 and SPG11. Ann Clin Transl Neurol 1(6): 379–389. 10.1002/acn3.64
- Vantaggiato C, Crimella C, Airoldi G, Polishchuk R, Bonato S, Brighina E, Scarlato M, Musumeci O, Toscano A, Martinuzzi A, Santorelli FM, Ballabio A, Bresolin N, Clementi E, and Bassi MT (2013). Defective autophagy in spastizin mutated patients with hereditary spastic paraparesis type 15. Brain 136(10): 3119–3139. 10.1093/brain/awt227
- Chang J, Lee S, and Blackstone C (2014). Spastic paraplegia proteins spastizin and spatacsin mediate autophagic lysosome reformation. J Clin Invest 124(12): 5249–5262. 10.1172/JCI77598
- Chen Y, and Yu L (2017). Recent progress in autophagic lysosome reformation. Traffic 18(6): 358–361. 10.1111/tra.12484
- Khundadze M, Ribaudo F, Hussain A, Stahlberg H, Brocke-Ahmadinejad N, Franzka P, Varga RE, Zarkovic M, Pungsrinont T, Kokal M, Ganley IG, Beetz C, Sylvester M, and Hübner CA (2021). Mouse models for hereditary spastic paraplegia uncover a role of PI4K2A in autophagic lysosome reformation. Autophagy 1–17. 10.1080/15548627.2021.1891848
- Martin E, Yanicostas C, Rastetter A, Naini SMA, Maouedj A, Kabashi E, Rivaud-Péchoux S, Brice A, Stevanin G, and Soussi-Yanicostas N (2012). Spatacsin and spastizin act in the same pathway required for proper spinal motor neuron axon outgrowth in zebrafish. Neurobiol Dis 48(3): 299–308. 10.1016/j.nbd.2012.07.003
- Khundadze M, Kollmann K, Koch N, Biskup C, Nietzsche S, Zimmer G, Hennings JC, Huebner AK, Symmank J, Jahic A, Ilina EI, Karle K, Schöls L, Kessels M, Braulke T, Qualmann B, Kurth I, Beetz C, and Hübner CA (2013). A Hereditary Spastic Paraplegia Mouse Model Supports a Role of ZFYVE26/SPASTIZIN for the Endolysosomal System. PLoS Genet 9(12). 10.1371/journal.pgen.1003988
- Varga RE, Khundadze M, Damme M, Nietzsche S, Hoffmann B, Stauber T, Koch N, Hennings JC, Franzka P, Huebner AK, Kessels MM, Biskup C, Jentsch TJ, Qualmann B, Braulke T, Kurth I, Beetz C, and Hübner CA (2015). In Vivo Evidence for Lysosome Depletion and Impaired Autophagic Clearance in Hereditary Spastic Paraplegia Type SPG11. PLoS Genet 11(8): 1–20. 10.1371/journal.pgen.1005454
- Branchu J, Boutry M, Sourd L, Depp M, Leone C, Corriger A, Vallucci M, Esteves T, Matusiak R, Dumont M, Muriel MP, Santorelli FM, Brice A, El Hachimi KH, Stevanin G, and Darios F (2017). Loss of spatacsin function alters lysosomal lipid clearance leading to upper and lower motor neuron degeneration. Neurobiol Dis 102: 21–37. 10.1016/j.nbd.2017.02.007
- Denora PS, Smets K, Zolfanelli F, Groote CC De, Casali C, Deconinck T, Sieben A, Gonzales M, Zuchner S, Darios F, Peeters D, Brice A, Malandrini A, De Jonghe P, Santorelli FM, Stevanin G, Martin JJ, and El Hachimi KH (2016). Motor neuron degeneration in spastic paraplegia 11 mimics amyotrophic lateral sclerosis lesions. Brain 139(6): 1723–1734. 10.1093/brain/aww061
- Oz-Levi D, Ben-Zeev B, Ruzzo EK, Hitomi Y, Gelman A, Pelak K, Anikster Y, Reznik-Wolf H, Bar-Joseph I, Olender T, Alkelai A, Weiss M, Ben-Asher E, Ge D, Shianna K V, Elazar Z, Goldstein DB, Pras E, and Lancet D (2012). Mutation in TECPR2 reveals a role for autophagy in hereditary spastic paraparesis. Am J Hum Genet 91(6): 1065–1072. 10.1016/j.ajhg.2012.09.015
- Stadel D, Millarte V, Tillmann KD, Huber J, Tamin-Yecheskel BC, Akutsu M, Demishtein A, Ben-Zeev B, Anikster Y, Perez F, Dötsch V, Elazar Z, Rogov V, Farhan H, and Behrends C (2015). TECPR2 Cooperates with LC3C to Regulate COPII-Dependent ER Export. Mol Cell 60(1): 89–104. 10.1016/j.molcel.2015.09.010
- Tamim-Yecheskel B-C, Fraiberg M, Kokabi K, Freud S, Shatz O, Marvaldi L, Subic N, Brenner O, Tsoory M, Eilam-Altstadter R, Biton I, Savidor A, Dezorella N, Heimer G, Behrends C, Ben-Zeev B, and Elazar Z (2020). A tecpr2 knockout mouse exhibits age-dependent neuroaxonal dystrophy associated with autophagosome accumulation. Autophagy 1–14. 10.1080/15548627.2020.1852724
- Fraiberg M, Tamim-Yecheskel BC, Kokabi K, Subic N, Heimer G, Eck F, Nalbach K, Behrends C, Ben-Zeev B, Shatz O, and Elazar Z (2020). Lysosomal targeting of autophagosomes by the TECPR domain of TECPR2. Autophagy 1-13. 10.1080/15548627.2020.1852727
- Edmison D, Wang L, and Gowrishankar S (2021). Lysosome function and dysfunction in hereditary spastic paraplegias. Brain Sci 11(2): 1–16. 10.3390/brainsci11020152
- Hirst J, Borner GHH, Edgar J, Hein MY, Mann M, Buchholz F, Antrobus R, and Robinson MS (2013). Interaction between AP-5 and the hereditary spastic paraplegia proteins SPG11 and SPG15. Mol Biol Cell 24(16): 2558–2569. 10.1091/mbc.E13-03-0170
- Ivankovic D, Drew J, Lesept F, White IJ, López Doménech G, Tooze SA, and Kittler JT (2020). Axonal autophagosome maturation defect through failure of ATG9A sorting underpins pathology in AP-4 deficiency syndrome. Autophagy 16(3): 391–407. 10.1080/15548627.2019.1615302
- Vici CD, Sabetta G, Gambarara M, Vigevano F, Bertini E, Boldrini R, Parisi SG, Quinti I, Aiuti F, and Fiorilli (1988). Agenesis of the corpus callosum, combined immunodeficiency, bilateral cataract, and hypopigmentation in two brothers. Am J Med Genet 29(1): 1–8. 10.1002/ajmg.1320290102
- McClelland V, Cullup T, Bodi I, Ruddy D, Buj-Bello A, Biancalana V, Boehm J, Bitoun M, Miller O, Jan W, Menson E, Amaya L, Trounce J, Laporte J, Mohammed S, Sewry C, Raiman J, and Jungbluth H (2010). Vici syndrome associated with sensorineural hearing loss and evidence of neuromuscular involvement on muscle biopsy. Am J Med Genet Part A 152(3): 741–747. 10.1002/ajmg.a.33296
- Al-Owain M, Al-Hashem A, Al-Muhaizea M, Humaidan H, Al-Hindi H, Al-Homoud I, and Al-Mogarri I (2010). Vici syndrome associated with unilateral lung hypoplasia and myopathy. Am J Med Genet Part A 152(7): 1849–1853. 10.1002/ajmg.a.33421
- Byrne S et al. (2016). EPG5-related Vici syndrome: A paradigm of neurodevelopmental disorders with defective autophagy. Brain 139(3): 765–781. 10.1093/brain/awv393
- Cullup T et al. (2013). Recessive mutations in EPG5 cause Vici syndrome, a multisystem disorder with defective autophagy. Nat Genet 45(1): 83–87. 10.1038/ng.2497
- Zhao YG, Zhao H, Sun H, and Zhang H (2013). Role of Epg5 in selective neurodegeneration and Vici syndrome. Autophagy 9(8): 1258–1262. 10.4161/auto.24856
- Tian Y, Li Z, Hu W, Ren H, Tian E, Zhao Y, Lu Q, Huang X, Yang P, Li X, Wang X, Kovács AL, Yu L, and Zhang H (2010). C. elegans Screen Identifies Autophagy Genes Specific to Multicellular Organisms. Cell 141(6): 1042–1055. 10.1016/j.cell.2010.04.034
- Wang Z, Miao G, Xue X, Guo X, Yuan C, Wang Z, Zhang G, Chen Y, Feng D, Hu J, and Zhang H (2016). The Vici Syndrome Protein EPG5 Is a Rab7 Effector that Determines the Fusion Specificity of Autophagosomes with Late Endosomes/Lysosomes. Mol Cell 63(5): 781–795. 10.1016/j.molcel.2016.08.021
- Zhao H, Zhao YG, Wang X, Xu L, Miao L, Feng D, Chen Q, Kovács AL, Fan D, and Zhang H (2013). Mice deficient in epg5 exhibit selective neuronal vulnerability to degeneration. J Cell Biol 200(6): 731–741. 10.1083/jcb.201211014
- Miao G, Zhao YG, Zhao H, Ji C, Sun H, Chen Y, and Zhang H (2016). Mice deficient in the Vici syndrome gene Epg5 exhibit features of retinitis pigmentosa. Autophagy 12(12): 2263–2270. 10.1080/15548627.2016.1238554
- Bird TD (2020). Hereditary Ataxia Overview. Available at https://www.ncbi.nlm.nih.gov/books/NBK1138/ [Accessed 03/22/2021].
- Akizu N et al. (2015). Biallelic mutations in SNX14 cause a syndromic form of cerebellar atrophy and lysosome-autophagosome dysfunction. Nat Genet 47(5): 528–534. 10.1038/ng.3256
- Thomas AC et al. (2014). Mutations in SNX14 cause a distinctive autosomal-recessive cerebellar ataxia and intellectual disability syndrome. Am J Hum Genet.95(5): 611–621. 10.1016/j.ajhg.2014.10.007
- Zhang H, Hong Y, Yang W, and Wang R (2021). SNX14 deficiency-induced defective axonal mitochondrial transport in Purkinje cells underlies cerebellar ataxia and can be reversed by valproate. Natl Sci Rev.10.1093/nsr/nwab024
- Mas C, Norwood SJ, Bugarcic A, Kinna G, Leneva N, Kovtun O, Ghai R, Yanez LEO, Davis JL, Teasdale RD, and Collins BM (2014). Structural basis for different phosphoinositide specificities of the PX domains of sorting nexins regulating G-protein signaling. J Biol Chem 289(41): 28554–28568. 10.1074/jbc.M114.595959
- Huang HS, Yoon BJ, Brooks S, Bakal R, Berrios J, Larsen RS, Wallace ML, Han JE, Chung EH, Zylka MJ, and Philpot BD (2014). Snx14 regulates neuronal excitability, promotes synaptic transmission, and is imprinted in the brain of mice. PLoS One 9(5):e98383. 10.1371/journal.pone.0098383
- Fassio A, Falace A, Esposito A, Aprile D, Guerrini R, and Benfenati F (2020). Emerging Role of the Autophagy/Lysosomal Degradative Pathway in Neurodevelopmental Disorders With Epilepsy. Front Cell Neurosci 14: 1–9. 10.3389/fncel.2020.00039
- Kim M, Sandford E, Gatica D, Qiu Y, Liu X, Zheng Y, Schulman BA, Xu J, Semple I, Ro S-H, Kim B, Mavioglu RN, Tolun A, Jipa A, Takats S, Karpati M, Li JZ, Yapici Z, Juhasz G, Lee JH, Klionsky DJ, and Burmeister M (2016). Mutation in ATG5 reduces autophagy and leads to ataxia with developmental delay. Elife 5: 1–18. 10.7554/elife.12245
- Gstrein T, Edwards A, Přistoupilová A, Leca I, Breuss M, Pilat-Carotta S, Hansen AH, Tripathy R, Traunbauer AK, Hochstoeger T, Rosoklija G, Repic M, Landler L, Stránecký V, Dürnberger G, Keane TM, Zuber J, Adams DJ, Flint J, Honzik T, Gut M, Beltran S, Mechtler K, Sherr E, Kmoch S, Gut I, and Keays DA (2018). Mutations in Vps15 perturb neuronal migration in mice and are associated with neurodevelopmental disease in humans. Nat Neurosci 21(2): 207–217. 10.1038/s41593-017-0053-5
- Kanton S, Boyle MJ, He Z, Santel M, Weigert A, Sanchís-Calleja F, Guijarro P, Sidow L, Fleck JS, Han D, Qian Z, Heide M, Huttner WB, Khaitovich P, Pääbo S, Treutlein B, and Camp JG (2019). Organoid single-cell genomic atlas uncovers human-specific features of brain development. Nature 574(7778): 418–422. 10.1038/s41586-019-1654-9
- Agoglia RM, Sun D, Birey F, Yoon S, Miura Y, Sabatini K, Paşca SP, and Fraser HB (2021). Primate cell fusion disentangles gene regulatory divergence in neurodevelopment. Nature 592(7854):421-427. 10.1038/s41586-021-03343-3
–
ACKNOWLEDGMENTS
Research in the P.B. lab is supported by grant (PGC2018-098557-B-I00) from Spain’s Minis-terio Ciencia e Innovación, the Agencia Estatal de Inves-tigación (AEI), the Fondo Europeo de Desarrollo Regional (FEDER), and funding from the European Union’s Horizon 2020 Research and Innovation Programme under grant agreement No. 765912, Fundación Tatiana Pérez de Guzmán el Bueno Proyectos en Neurociencia 2018, and Redes de BioMedicina de la Comunidad de Madrid, BMD-3813. JZM is a recipient of a FPU fellowship (Minis-terio de Universidades). We thank O. Howard for Eng-lish-language editing and the members of the Boya Lab for discussion.
COPYRIGHT
© 2021

Towards a better understanding of the neuro-developmental role of autophagy in sickness and in health by Zapata-Muñoz et al. is licensed under a Creative Commons Attribution 4.0 International License.


