Reviews:
Cell Stress, Vol. 2, No. 11, pp. 292 - 310; doi: 10.15698/cst2018.11.162
Making the final cut: pathogenic amyloid-β peptide generation by γ-secretase

1 Biomedical Center (BMC), Metabolic Biochemistry, Ludwig-Maximilians-University Munich, Germany.
2 German Center for Neurodegenerative Diseases (DZNE), Munich, Germany.
3 Department of Aging Neurobiology, National Center for Geriatrics and Gerontology, Obu & Department of Mental Health Promotion, Osaka University Graduate School of Medicine, Toyonaka, Japan.
4 Neuropsychiatry, Department of Integrated Medicine, Division of Internal Medicine, Osaka University Graduate School of Medicine, Suita, Japan.
Keywords: Alzheimer's disease, amyloid β-peptide, presenilin, γ-secretase.
Abbreviations:
Aβ – amyloid-β,
AD – Alzheimer’s disease,
AICD – APP intracellular domain,
APP – amyloid precursor protein,
CTF – C-terminal fragment,
EM – electron microscopy,
FAD – familial AD,
GSI – γ-secretase inhibitor,
GSM - γ-secretase modulator,
ICD – intracellular domain,
NCT – nicastrin,
NTF – N-terminal fragment,
TMD – transmembrane domain,
TSA – transition-stage analogue,
wt – wild-type.
Received originally: 09/07/2018 Received in revised form: 05/10/2018
Accepted: 08/10/2018
Published: 28/10/2018
Correspondence:
Harald Steiner, Biomedical Center (BMC), Metabolic Biochemistry, Ludwig-Maximilians-University Munich, Feodor-Lynen-Str. 17, 81377 Munich, Germany, Phone: +49 89 4400 46535, Fax: +49 89 4400 46546 harald.steiner@med.uni-muenchen.de
Conflict of interest statement: The authors declare no conflict of interest.
Please cite this article as: Harald Steiner, Akio Fukumori, Shinji Tagami and Masayasu Okochi (2018). Making the final cut: pathogenic amyloid-β peptide generation by γ-secretase. Cell Stress 2(11): 292-310. doi: 10.15698/cst2018.11.162
Abstract
Alzheimer´s disease (AD) is a devastating neurodegenerative disease of the elderly population. Genetic evidence strongly suggests that aberrant generation and/or clearance of the neurotoxic amyloid-β peptide (Aβ) is triggering the disease. Aβ is generated from the amyloid precursor protein (APP) by the sequential cleavages of β- and γ-secretase. The latter cleavage by γ-secretase, a unique and fascinating four-component protease complex, occurs in the APP transmembrane domain thereby releasing Aβ species of 37-43 amino acids in length including the longer, highly pathogenic peptides Aβ42 and Aβ43. The lack of a precise understanding of Aβ generation as well as of the functions of other γ-secretase substrates has been one factor underlying the disappointing failure of γ-secretase inhibitors in clinical trials, but on the other side also been a major driving force for structural and in depth mechanistic studies on this key AD drug target in the past few years. Here we review recent breakthroughs in our understanding of how the γ-secretase complex recognizes substrates, of how it binds and processes β-secretase cleaved APP into different Aβ species, as well as the progress made on a question of outstanding interest, namely how clinical AD mutations in the catalytic subunit presenilin and the γ-secretase cleavage region of APP lead to relative increases of Aβ42/43. Finally, we discuss how the knowledge emerging from these studies could be used to therapeutically target this enzyme in a safe way.
INTRODUCTION
Alzheimer´s disease (AD) is the most common form of dementia worldwide currently affecting about 47 millions people. The disease affects the elderly typically above an age of ~65 and manifests with progressive memory impairment and cognitive decline. About a third of the people at an age of 85 are diagnosed with AD. Aging is thus the most important risk factor for the disease. In very rare cases, AD can also be genetically inherited. These familial forms of AD (FAD) are typically characterized by a much earlier onset of the disease below 65 years, which can occur in very aggressive forms already in adolescence. To date, there is no therapy available that can prevent the disease or cure it [1].
–
Pathologically, AD is characterized by a massive deposition of abnormal protein aggregates present as extracellular plaques of amyloid-β peptide (Aβ) in the brain parenchyma and as neurofibrillary tangles, intraneuronal deposits composed of the microtubule binding protein tau [2]. In addition, dystrophic neurites and neuropil threads of tau are commonly observed in AD. It is widely believed that abnormal levels of Aβ trigger the disease [3]. Aβ is a small hydrophobic ~4 kDa peptide, which is derived by sequential proteolytic processing of the amyloid precursor protein (APP), a single pass transmembrane protein with type I topology which is expressed in neurons as 695 amino acid splice variant [4] (Fig. 1). In the first step, APP is cleaved by β-secretase. This cleavage, which is carried out by the membrane-bound aspartyl protease BACE (β-site APP-cleaving enzyme) [5], removes the bulk of the APP ectodomain and leaves a 99 amino acid C-terminal fragment (CTF) in the membrane. This C99 or APP CTFβ termed fragment is cleaved in the next step by a membrane protein complex termed γ-secretase [6]. This intramembrane-cleaving aspartyl protease complex cleaves C99 in the transmembrane domain (TMD) at the ε-site thereby releasing the APP intracellular domain (AICD) from the membrane into the cytosol. In a stepwise process, which will be explained in detail further below, a variety of Aβ forms are generated by additional γ-secretase cleavages at the ζ- and γ-sites that trim the TMD [7] (Fig. 1). Trimming occurs until the TMD is short enough to release predominantly 38-42 amino acid long Aβ forms from the membrane into the extracellular space or into the lumen of secretory pathway organelles. Aβ40 is generated as major product along minor amounts of the shorter Aβ38 and the longer Aβ42. Besides these species, very little amounts of Aβ37 and Aβ43 are generated. Generation of Aβ is prevented by an alternatively occurring APP cleavage by α-secretase [8], which cleaves within the Aβ region and generates the shorter APP CTF, C83.
–
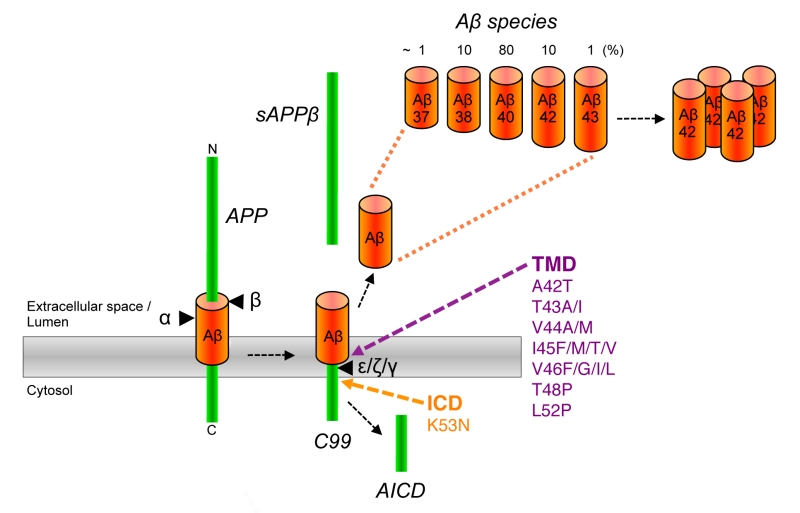 | FIGURE 1: APP processing and generation of Aβ. APP is first cleaved by β-secretase (β) in its ectodomain close to the extracellular/luminal membrane border thereby generating a 99 amino acid C-terminal APP fragment (C99). Consecutive cleavages of C99 by γ-secretase at ε-, ζ-, and γ-sites releases the APP intracellular domain (AICD) into the cytosol and 37-43 amino acid Aβ species into the extracellular space or lumen of secretory pathway organelles. Longer Aβ forms such as in particular Aβ42 are highly aggregation prone and ultimately deposit as plaques in AD patient brains. An alternative cleavage of APP by α-secretase (α) in the Aβ domain prevents the formation of Aβ. Pathogenic APP FAD mutations that have been shown or are likely to cause relative increases in the generation of Aβ42 species are located in the γ-secretase cleavage region of the APP TMD and in the AICD. |
–
Although Aβ42 is normally generated only in minor amounts, it is the initially and major Aβ form deposited in plaques [9]. It is more hydrophobic than Aβ40 and displays a considerably higher propensity to form neurotoxic aggregates and is thus believed to be the culprit of AD pathogenesis [2]. A central pathological role of Aβ42 is evident from the finding that mutations associated with FAD were identified in APP and presenilin [10][11][12], the catalytic subunit of γ-secretase [6][13]. They affect the cleavage of γ-secretase such that increased amounts of the pathogenic Aβ42 relative to Aβ40 are generated [14][15][16]. Missense mutations in APP were the first reported FAD mutations [17][18]. Most of these mutations are found within the APP TMD in the γ-secretase cleavage region and like presenilin FAD mutations increase the Aβ42/Aβ40 ratio [19] (Fig. 1). Mutations were also found that increase the total amounts of Aβ generated. A double mutation at the β-secretase cleavage site occurring in a Swedish family [20][21] and more recently, also a duplication of the APP gene could be linked to FAD [22]. Interestingly, another mutation close to the β-secretase cleavage site that lowers Aβ levels and protects against AD has been identified in the Icelandic population [23]. FAD mutations were also found in the N-terminal and mid-domain of Aβ [19]. These mutations increase the aggregation properties of Aβ. Also for the more common sporadic forms of AD, genetic components, which increase the risk of developing the disease, have been identified. These include the ε4 allele of the apolipoprotein E (APOE) gene, which affects Aβ aggregation and/or clearance [24] as well as the microglial protein TREM2 (triggering receptor expressed on myeloid cells 2) that is involved in the clearance of Aβ plaques [25]. Finally, a central role of Aβ in the etiology of AD is further supported by genome-wide association studies of AD, which indicate a role of genes involved in lipid metabolism and intracellular protein trafficking – all factors, which affect Aβ metabolism [26].
–
Since γ-secretase makes the final cut in the generation of Aβ, it is obvious that the enzyme is one of the major drug targets in AD. We will now have a closer look at its structure and functions and in particular at the mechanism of how it generates pathogenic, longer Aβ species in the following chapters.
STRUCTURE AND FUNCTION OF γ-SECRETASE IN AD AND BEYOND
γ-Secretase is a protein complex composed of four subunits [27][28][29], whose structure has recently been solved at atomic resolution by cryo-electron microscopy [30] (Fig. 2). The largest subunit nicastrin (NCT) is a type I membrane protein [31], whose large bilobar highly glycosylated extracellular domain covers the horseshoe-like transmembrane part of the complex [32]. The catalytic subunit of the complex, presenilin, is a polytopic membrane protein with nine TMDs. TMDs 6 and 7 carry the active site aspartate residues [33][34][35]. Upon assembly and maturation of the complex [36], presenilin is cleaved within the large cytoplasmic loop into two fragments, the N-terminal fragment (NTF) comprising TMDs 1-6 and the CTF comprising TMDs 7-9 [37]. Presenilin endoproteolysis is heterogeneous and occurs in a stepwise manner [38][39] by autoproteolysis [27][33][34][40]. The complex can be composed either of presenilin-1 (PS1) or its homolog presenilin-2 (PS2) [41]. Likewise, the subunit APH-1 (anterior pharynx-defective 1) [42][43][44], a polytopic membrane protein with seven TMDs, which serves a stabilizing, structural role in the complex, exists as APH-1a or APH-1b variant that are part of separate complexes [45][46]. Finally, there is the smallest subunit of the complex, PEN-2 (presenilin enhancer 2) [42][47]. Of its two hydrophobic domains, one forms a straight TMD, while the other one adopts a hairpin-like structure embedded in the membrane as two half-helices [48][49]. Low-resolution electron microscopy (EM)-structures indicate that the enzyme has substantial conformational flexibility and is compacted upon inhibitor binding [50][51]. The identification of three different apo-states in the atomic cryo-EM structure has shown that in particular presenilin together with PEN-2 shows considerable flexibility in the complex [52]. The different complexes display differential subcellular locations [53][54]. Careful cell biological experiments show that PS1-containing γ-secretase complexes are more broadly distributed within the secretory pathway, whereas PS2-containing γ-secretase complexes localize preferentially to the late endosome/lysosome. These different locations have an impact on the generation of longer Aβ species, as the lysosomal PS2 γ-secretase complexes are responsible for the generation of the bulk of an intracellular Aβ42 pool [54].
–
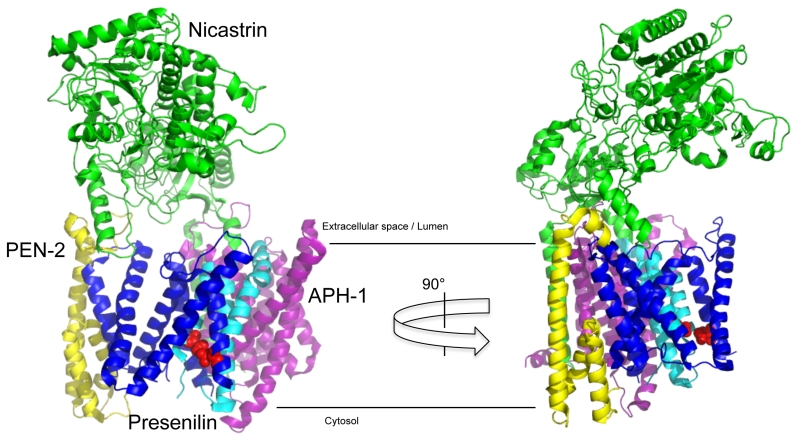 | FIGURE 2: Structure of γ-secretase. The atomic resolution structure of γ-secretase (PDB: 5FN3) shows a membrane embedded core containing the catalytic subunit presenilin-1 cleaved into NTF (blue) and CTF (cyan) flanked by the subunits PEN-2 (yellow) and APH-1a (purple), which is covered by the large bilobar extracellular domain of the nicastrin subunit (green). This domain allows nicastrin to serve as gatekeeper controlling substrate access to the active site by excluding proteins with too large (or sterically incompatible) ectodomains. Red spheres depict the active sites aspartate residues. |
–
Research over nearly two decades has shown that γ-secretase cleaves numerous other proteins besides APP, and the list of its substrates has grown to more than hundred since the last overview in 2011 [55]. A general feature that has emerged is that γ-secretase substrates are principally in type I membrane protein orientation, i.e. with an extracellular N-terminus and an intracellular C-terminus, and as an additional key feature have short ectodomains. These are typically generated by ectodomain shedding by proteases such as BACE or in probably most of the cases by metalloproteases of the ADAM (a disintegrin and metalloprotease) family, such as the α-secretases ADAM10 and 17 [56]. In case of APP, besides C99, the shorter CTFs C89 and C83 are generated by the cleavage of these sheddases [57]. However, naturally short substrates, which do not undergo ectodomain shedding such as BCMA (B-cell maturation antigen), have also recently been identified [58]. Apart from APP, at least one other major and critical substrate, which stands out is the cell surface receptor Notch1 [59][60]. Cleavage of Notch1 by γ-secretase is an essential function of the enzyme required for cell-fate decisions during embryonic development and remains also critical in adulthood [61]. As demonstrated first for this receptor, the intracellular domain (ICD) generated by γ-secretase cleavage translocates to the nucleus to activate transcription of target genes [62]. Similar signaling activities have been reported also for a number of other subsequently identified γ-secretase substrates and have also been suggested for APP [63], although this has remained controversial for this substrate [64]. While γ-secretase cleavage can lead to signaling pathway activation, examples for the opposite process have also been identified, where substrate cleavage abolishes signaling events [65][66]. The regulation of signaling pathways by the release of biologically active ICDs is thus clearly a vital function of γ-secretase events [67]. However, a more general and simple function of γ-secretase could be to clear the membrane from substrate CTFs that are left behind after ectodomain shedding [68]. Since an accumulation of these fragments could be toxic as inferable from observations with APP CTFs [69][70][71][72][73], this “membrane proteasome” function could serve to control toxic membrane protein abundance.
–
With the discovery of so many γ-secretase substrates it has become clear that the enzyme has multiple biological functions ranging from developmental processes of various organs to functions in the nervous system of the adult brain [55][67]. Besides AD, altered γ-secretase function can also be associated with other diseases including most prominently cancer, which has excellently been covered elsewhere [67]. It should also be noted here that γ-secretase-independent functions of presenilin have been identified which have recently been reviewed elsewhere [74].
–
A precise understanding of how the enzyme recognizes, selects and finally cleaves its substrates is crucial for the development of APP-selective γ-secretase-targeting drugs that should ideally not touch the cleavage of Notch1 and other critical substrates. Understanding this process is an area of active research where important developments have recently been achieved.
SUBSTRATE RECOGNITION BY γ-SECRETASE
At the very basic level of recognition, substrates of γ-secretase have to locate in the same subcellular compartment as the protease in order to be cleavable. The more restricted localization of PS2-containing γ-secretase complexes to late endosomes/lysosomes thus provides some selectivity in substrate recognition and cleavage as substrates that are targeted to these compartments will preferentially be cleaved by PS2 rather than PS1 [54]. Once substrate and enzyme encounter each other in the correct compartments, a crucial requirement for the selection of substrates by γ-secretase is that these have short ectodomains typically between 15-30 amino acids in length. The shorter these are the better substrates are cleavable [75][76]. Early studies showed that substrates become increasingly less well cleavable when their ectodomain lengths are increased above 50 amino acids [77]. This has subsequently been expanded by showing that NCT acts as a gatekeeper controlling whether type I membrane proteins getting in contact with the protease complex have appropriate ectodomain lengths that fit in size underneath the large globular extracellular domain of NCT [76] (Fig. 2). In agreement with such a substrate size control function of NCT are observations that antibodies against the NCT ectodomain can block substrate binding and catalysis [78][79]. Although experimentally not yet shown, it is likely that bound antibodies sterically hinder substrate access to the enzyme. This could occur either directly or by inducing a conformational change of the NCT ectodomain towards the membrane-embedded part of the complex thereby interfering with substrate recruitment. Flexibility of NCT has been inferred by lower resolution EM studies [50][51] and is supported by molecular dynamics computer simulations [80][81].
–
Following this initial recognition step, in which type I membrane proteins with too large ectodomains are prevented from recruitment to the protease by steric hinderance [76], substrates gain access to the catalytic site in a stepwise-manner by sequential interactions with exosites, substrate binding sites outside the active site [82]. Photoaffinity-labeling studies with C99 using site-specifically introduced p-benzoyl-phenylalanine as crosslinkable amino acid identified the PS1 NTF as major substrate-binding site. Prominent contact points of C99 occurred in the cleavage site region and included among other residues V44 between the γ- and ζ-sites and around the ε-cleavage sites L49 that contact the PS1 NTF as well as residues M51 and L52 that predominantly contact the PS1 CTF [82] (Fig. 3A). In addition, NCT, PEN-2 and the PS1 NTF were identified as exosite-bearing γ-secretase subunits. Here, the most prominent contact points of C99 were H6 with NCT, A30 with PEN-2 and E3 with the PS1 NTF (Fig. 3A). Furthermore, as demonstrated for C99 by the photocrosslinking experiments, exosites in NCT and PEN-2 mediate the first interactions with γ-secretase (stage 1). Upon release from these sites, C99 binds to exosites in the PS1 NTF (stage 2). Following this stage, C99 can engage the catalytic site for cleavage (stage 3) (Fig. 3B). Binding of C99 to NCT at stage 1 is consistent with earlier findings that suggested an active role of this subunit in substrate recognition as a substrate receptor [83]. Although the contact points of C99 in the enzyme are currently not known, the initially proposed model that NCT interacts with the free N-terminus of substrates via an electrostatic interaction with E333 in its DAP domain (DYIGS and peptidase; residues 261-502) [83] has been controversial [76][84][85] and ultimately seems unlikely (Fig. 3C). As seen in the structure, E333 located in the large lobe cannot directly be accessed by a substrate because it is covered by a “lid” region in the small lobe, which would need to be displaced by a conformational change involving rotation around a pivot [30][86]. Since mutations of lid and pivot residues did not interfere with substrate cleavage [87], the proposed mechanism cannot apply and suggests that the substrate may contact NCT at another region. Interestingly, L571, another residue of the large lobe in α-helix 16 is less buried than E333 and was found to be critically required for substrate binding and catalysis [79], indicating that the substrate´s N-terminal extracellular domain may contact the protease in this region (Fig. 3C). Another candidate interaction site could be the loop of residues 240–244 of NCT, which is in close proximity to a co-purified potential substrate-mimic that adopts a kinked transmembrane α-helix in the class 1 apo state of the enzyme [52] (Fig. 3C). For PEN-2, the candidate-binding region of C99, which interacts with the extracellular juxta-membrane region of C99 including the N-terminal TMD end, is likely its corresponding extracellular juxtamembrane region starting from the C-terminal end of TMD 2. Whether C99 interacts with NCT or PEN-2 at stage 1 sequentially or simultaneously is not yet clear.
–
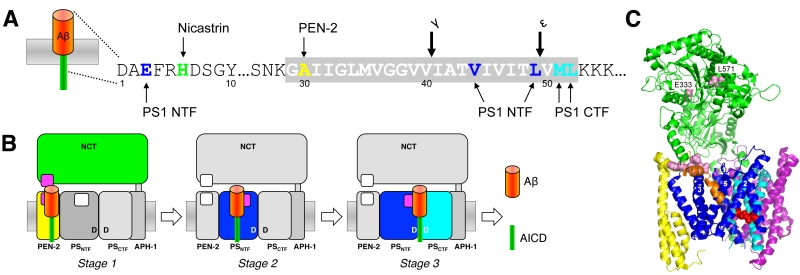 | FIGURE 3: Substrate recruitment of C99 by γ-secretase. (A) Schematic representation of the most prominent C99 residues interacting with γ-secretase subunits as determined by site-directed photocrosslinking using the unnatural amino acid p-benzoyl-phenylalanine. (B) Model depicting the stepwise translocation of C99 from exosites (purple) in NCT and PEN-2 (stage 1) and the PS1 NTF (stage 2) to the active site (stage 3). (C) Structure of γ-secretase (5FN3) with a co-isolated α-helix (orange), which might represent a substrate-mimic. Red spheres depict the active site aspartate residues. Light pink spheres represent candidate sites for substrate interactions with NCT. Although not visible in this view, L571 is less buried than E333. Numbers indicate TMDs of the PS1 NTF that surround the substrate-mimicking α-helix. |
–
It is highly likely that the substrate movements of C99 from the exosites encountered in stages 1 and 2 to the active site are associated with substantial conformational changes of the PEN-2 and presenilin TMD core of the complex. Lateral movement of PEN-2 towards the active site, potentially together with NCT and rearrangements of PS1 NTF TMDs 2-6 could translocate exosite-bound substrates to the active site. Based on the cryo-EM class 1 structure with the kinked substrate mimic, substrate entry to the active site may occur through the cavity formed by TMD2, TMD3 and TMD5 [52] (Fig. 3C). The PS1 hydrophilic loop 1 may play an important role in this process by allowing movements of TMD2, which like TMD6 is highly dynamic [30][48][52]. This would be consistent with mutational analysis that implicated this region in substrate binding [88] and supported by the clustering of many FAD mutations in this loop. Ultimately, as suggested by mutational analysis, a final substrate selection step likely takes place close to the catalytic aspartates and involves the GxGD-motif in TMD7. The conserved glycines are highly critical for γ-secretase activity [89][90] and L383 of PS1 at position x of the motif in PS1 provides substrate selectivity of APP over other substrates, such as Notch1 or CD44, that are not cleavable with certain substitutions of this residue [91][92]. This suggests that the GxGD motif is a critical region for the proper exposure of the substrate scissile bonds to the catalytic residues. Substrates not meeting the steric requirements may fall off from the enzyme at this stage. Similar observations have been made by mutational analysis of the conserved PAL motif, which is also located in close vicinity to the active site [93][94].
SEQUENTIAL CLEAVAGE OF THE APP TMD BY γ-SECRETASE
When C99 has reached the active site region, γ-secretase generates Aβ by consecutive intramembrane cleavages at the ε-, ζ-, and γ-cleavage sites (Fig. 4A). Following an initial endoproteolytic cleavage at the ε-49 or ε-48 cleavage sites [95][96][97][98], γ-secretase cleaves the remaining membrane-bound long Aβ counterparts by stepwise carboxyterminal trimming [99][100][101]. In the major pathway, Aβ40 is produced in a product line starting from Aβ49 via the intermediates Aβ46 and Aβ43. Aβ42 is generated likewise in a minor product line starting from Aβ48 via the intermediate Aβ45. Cleavages in the two lines can also continue further to the shorter species Aβ37 or Aβ38, respectively.
–
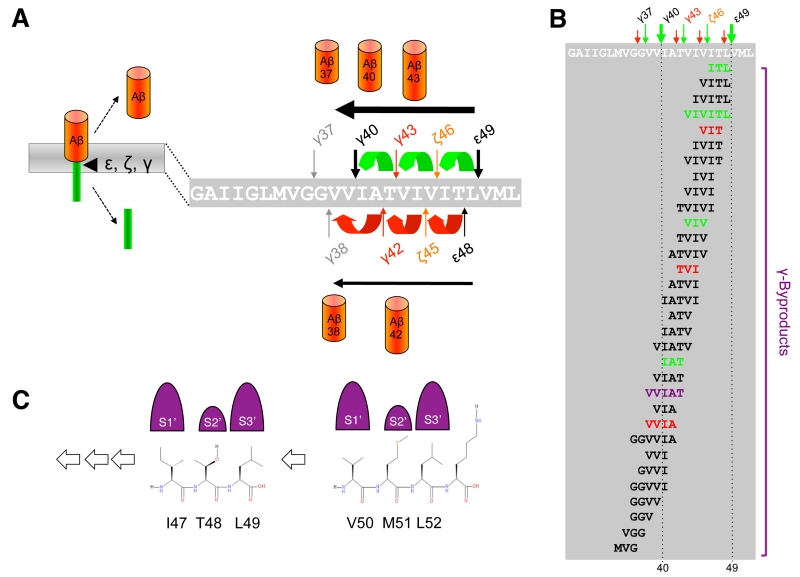 | FIGURE 4: Stepwise cleavage model of APP. (A) The two product lines leading to the generation of Aβ40 and Aβ42 are depicted. In the major product line Aβ40 is generated by consecutive tripeptide-releasing cleavages (green) at the ε-49, ζ-46 and γ-43 sites. In a minor product line, Aβ42 is generated in a similar manner by consecutive cleavages (red) at the ε-48 and ζ-45 sites. (B) Besides γ-byproducts from the two main product lines (green and red) including a hexapeptide resulting from direct cleavage of Aβ49 to Aβ43, multiple additional minor peptides have been identified suggesting multiple product line crossings. The pentapeptide resulting from cleavage of Aβ43 to Aβ38 is indicated in purple. (C) Sequential cleavage continuously requires sterically compatible interactions of P2´ residues with the S2´ enzyme subsite of γ-secretase; major product line shown. The adjacent P4´ residue K53 is additionally presented for the comparison of side chain proportions. |
–
Since the γ-cleavages occur in the middle of the TMD, while the ε-cleavages close to the cytosol border of the TMD, the C-termini of Aβ40/42 generated by γ-cleavage and the N-termini of AICD by ε-cleavage are distinct. It had thus initially been unclear whether ε-cleavage would precede γ-cleavage or the other way round or whether both cleavages may occur independently. A landmark discovery that provided a first evidence for a stepwise cleavage was the detection of C-terminally elongated Aβ species such as Aβ43, Aβ45/46, and Aβ48/49 in lysates of cultured cells [99][101][102]. Since corresponding N-terminally elongated AICD species (AICD-γ43 or AICD-ζ45/46) were also not found for the longer Aβ43 and Aβ45/46 species, it was hypothesized that ε-cleavage at the ε48/49 sites is followed by sequential cleavages proceeding to the N-terminus at every third or fourth residue, thereby resulting in generation of Aβ40 and Aβ42 [99]. Thus, assuming an α-helical conformation of the C99 TMD, small three to four amino acid peptides are produced every time the cleavage proceeds along the helix [99] (Fig. 4A). Accordingly, following the release of AICD-ε49 and AICD-ε48, the two Aβ production pathways outlined above in which Aβ40 is produced from Aβ49 and Aβ42 from Aβ48 were proposed [99].
–
Using a cell-free γ-secretase cleavage assay, in which cleavage of a recombinant C99 substrate by detergent-solubilized γ-secretase is directly analyzed, the putative tri- or tetrapeptides between the ε-, ζ- and γ-cleavage sites (G37GVVIATVIVITL49) were identified by liquid chromatography tandem-mass spectrometry [100]. Thus, ITL, VIV, and IAT peptides, of the Aβ40 product line, as well as VIT, TVI, and VVIA peptides of the Aβ42 product line were detected as “γ-byproducts” [103] of the cleavage reaction (Fig. 4B). In agreement with the sequential γ-cleavage model, the peptides were generated in a time-dependent manner [100]. Further strong evidence for this model was obtained by successfully measuring these small peptides also inside cultured cells [104] and in vivo in the brain of PS1 I213T knock-in mice overexpressing Swedish APP [103]. Interestingly, also a VVIAT pentapeptide resulting from a cleavage of Aβ43 to Aβ38 was detected both in the cell-free γ-secretase cleavage assay and in cultured cells [104] (Fig. 4B). Notably, ∼40% of Aβ38 was derived from Aβ43 in cells showing that Aβ38 can not only have Aβ42 as a precursor and that the Aβ40/42 product lines can overlap. In line with these findings, it could further be shown that both Aβ42 and Aβ43 itself can serve as γ-secretase substrates and be cleaved to Aβ38 [104]. Alternative minor production pathways with additional product line crossings were identified in other studies [105][106]. Altogether, the detailed analyses of the sequential cleavage mechanism of APP showed that it is much more complicated than it was initially considered (Fig. 4B). A critical implication of the sequential cleavage model for AD pathogenesis is that a processivity impairment in the Aβ product lines will lead to increases in the pathogenic Aβ42/43 species and an intracellular accumulation of longer Aβ species in membranes, which may also be pathologically relevant in AD for the origin of neuritic plaques as a consequence of neuronal loss [107].
–
The mechanism by which γ-secretase releases tripeptides in the sequential cleavage of C99 to Aβ was worked out by a recent study showing that γ-secretase has three distinct amino-acid-binding pockets in the active site region corresponding to the S1´, S2´ and S3´ subsites through which the enzyme forms a stable enzyme-substrate scission complex [108] (Fig. 4C). Fitting of the P1´- P3´ residues into these pockets brings the substrates into position for each catalytic cycle of the sequential cleavage. The S2´ pocket is smaller than the S1´ and S3´ pockets, which imposes steric requirements on the P2´ site of C99 and the ensuing Aβ substrates (Fig. 4C). It is currently unknown whether other substrates of γ-secretase follow the sequential cleavage model. However, this is not unlikely, since longer Aβ-like peptides such as APL1β28 produced from APLP1 or Nβ25 produced from Notch1, can serve as γ-secretase substrates and be cleaved in vitro into APL1β25 and Nβ21, respectively, indicating sequential cleavage mechanisms as well [104]. In addition, multiple cleavage sites have been identified in a number of substrates now that may possibly relate to consecutive cleavages along one or more product lines as well [109].
–
The molecular properties of substrates, which are recognized by γ-secretase differentiating them from nonsubstrates, are largely unknown. Since cleavages of C99 and Notch1 are kinetically extremely slow with very low turnover numbers kcat [76][110], it is likely that conformational flexibility of the substrate, in particular TMD helix dynamics, plays an important role to find the conformations that allow productive accommodation into the enzyme at the exosites and/or the active site [111][112]. Indeed, insertion of helix stabilizing and destabilizing residues in the cleavage domain has an inhibiting or promoting impact, respectively, on the cleavability of C99 [113][114].
MECHANISMS OF PRESENILIN AND APP FAD MUTATIONS UNDERLYING THE GENERATION OF PATHOGENIC Aβ SPECIES
By far the most mutations associated with FAD are found in the PS1 gene. To date over 210 pathogenic mutations have been identified covering ~25% of the residues (Fig. 5). Almost all of them are missense mutations that localize all over the protein clustering in the TMDs, in hydrophilic loop (HL) 1 and HL6 N-terminal to the endoproteolytic cleavage site within exon9. Mutations in PS2 show a similar broad distribution over the molecule but are much less frequent and have a later disease onset that is likely due to the lower expression of PS2. Although it had already been shown shortly after the discovery of presenilins that the mutations change the Aβ42/40 ratio [15], the mechanism(s) behind this phenomenon has remained obscure for a long time and only recently become more clear. Following the endoproteolytic cleavage at the ε-site, the carboxy-terminal trimming of the 42-product line becomes impaired such that the enzyme cannot efficiently convert Aβ42 to Aβ38 [115][116][117] (Fig. 6A). As a consequence, the generation of Aβ38 decreases causing an increase in the Aβ42/38 ratio. In addition, this leads to a relative increase of the generation of Aβ42 over that of Aβ40 and thus to an increased Aβ42/Aβ40 ratio. Likewise, trimming can also become impaired in the 40-product line manifesting in increased Aβ43/40 ratios. Depending on the mutation, these principal effects can also occur in combination, leading to an increased ratio of Aβ42/43 to Aβ40. Why these processivity changes occur for presenilin FAD mutations has been enigmatic and only recently convincing explanations could be offered. Thus, it could be shown that PS1 FAD mutations display an increase in the dissociation rate of Aβ42 from the enzyme [104]. In line with these findings, it was next found that FAD mutations destabilize the enzyme and as a consequence also the enzyme-substrate interactions [118]. Enzyme destabilization affected the initial enzyme-C99 interaction leading to impaired endopeptidase cleavage at the ε-site and continued to also weaken the interactions with the enzyme of the subsequently generated Aβ substrates. In addition to these findings, it was found that interactions of C99 with γ-secretase are altered by FAD mutants in the substrate cleavage domain, thereby changing the positioning of the substrate in the active site region [82] (Fig. 6B). Consistent with these recent results, earlier studies had suggested that FAD mutations affect the topography of the active site as shown by reduced inhibitor potencies [119][120][121] and altered interactions with active-site targeted inhibitors [122]. The model emerging from these studies that puts all observations together is that FAD mutations in presenilin cause structural alterations, which can, depending on the particular mutation, destabilize the enzyme to various extents. This leads to altered C99 binding and labile interactions of the subsequently generated longer Aβ species which dissociate faster from the enzyme than in the wild-type (wt) situation.
–
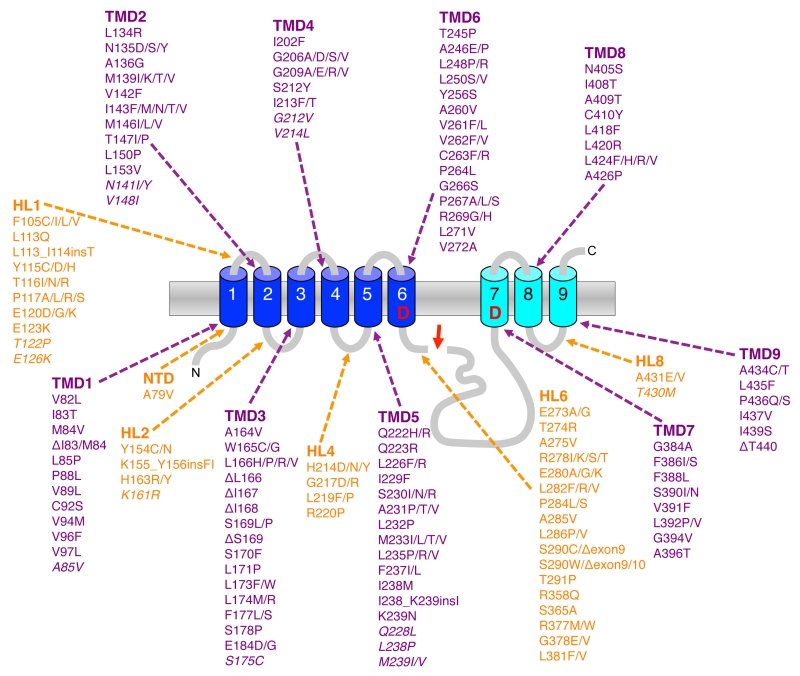 | FIGURE 5: FAD mutations in presenilin. Schematic representation of the nine TMD structure of presenilin in its cleaved form with the NTF (blue) and CTF (cyan). Pathogenic presenilin mutations (www.alzforum.org/mutations) are found in all TMDs and in some of the HLs. The compared to PS1 less frequent PS2 FAD mutations are represented in italics. The red arrow indicates the site of endoproteolysis. |
–
The mechanism of APP FAD mutations is less clear than that of the FAD mutations in presenilin described above. It had been reported that FAD mutations located in the γ-secretase cleavage region behave differently from those in presenilin that typically affect the efficiency of the conversions of Aβ42 to Aβ38 and of Aβ43 to Aβ40, and unlike these, cause a shift to the Aβ42 product line [115] (Fig. 7A). When this product line is entered, not only increased amounts of Aβ42 are generated. Remarkably, also increased Aβ38 levels, often comparable to those of Aβ42, have been observed [115], although not all studies showed consistent results [99][106][115][118][123][124][125]. Considering that the affinities of the sequentially generated Aβ species for the enzyme are decreasing from longer to shorter peptides [118], the accumulation of Aβ38 to similar levels as Aβ42 is surprising. Although Aβ38 can also be generated from Aβ43 [104], this should occur only to a very minor extent since the Aβ40 product line is not the preferred product line of APP FAD mutations. How APP FAD mutations cause an increase in the Aβ42/40 ratio can therefore not yet fully sufficiently be explained by these observations and additional mechanisms besides ε-site shifts have to be at play. Indeed, it has been demonstrated that the nature and the location of the FAD mutation will dictate which product line is selected [108]. It could be shown that the three residues locating C-terminal to the cleavage sites, i.e. residues P1´ – P3´, need to fit to the apparently large S1´, the small S2´, and the large S3´ subsite pockets of the en-zyme. Thus, the presence of aromatic amino acids at the P2´ position will clash with the small S2´ pocket, thereby blocking the respective Aβ40 or Aβ42 product line (Fig. 7B). For example, a phenylalanine occurring at the P2´ site of the γ-43 cleavage site as in the I45F FAD mutant, will cause a shift from the Aβ40 product line to the Aβ42 product line because the bulky phenylalanine will better accommodate to the large S3´ pocket of the γ-42 cleavage site (Fig. 7C). Interestingly, this happens without the typical ε-site shift [108]. However, the other so far known FAD mutant with a phenylalanine mutation, the V46F mutant, is not causing a product line block as the phenylalanine substitution does not clash with the small S2´ pocket in either of the two pathways. In this case, the increased Aβ42/40 ratio is caused by the product line shift at the ε-site.
–
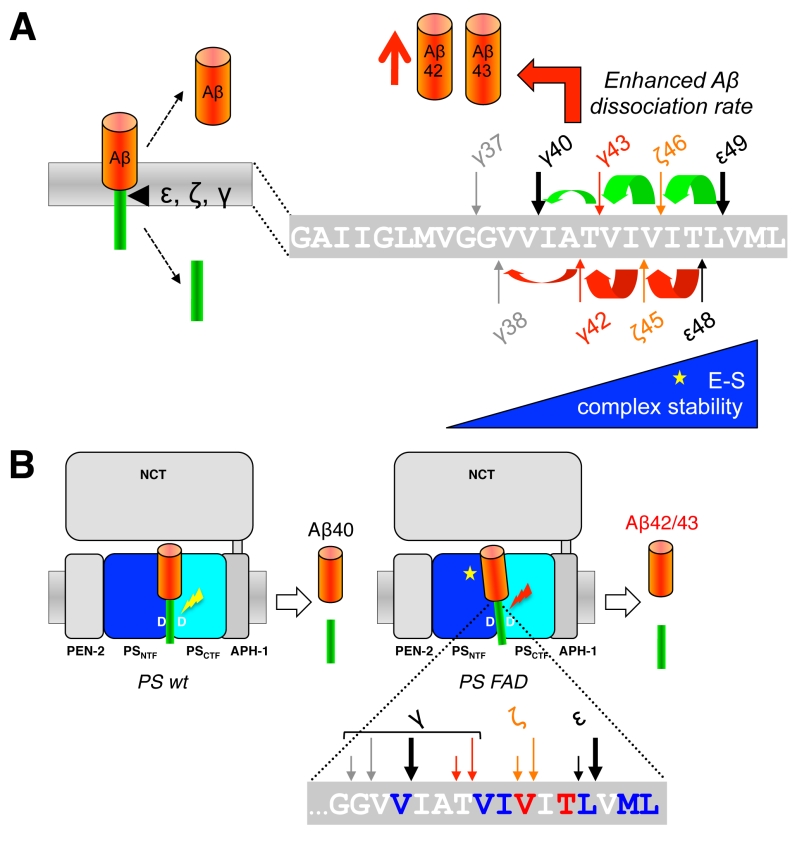 | FIGURE 6: Mechanism of presenilin FAD mutations. (A) Model depicting impaired processivity in the Aβ40 and Aβ42 product lines causing relative increases in the generation of the longer Aβ species Aβ42 and Aβ43. Structural instability of FAD mutant presenilin leads to impaired processivity manifested by faster dissociation rates and premature release of long Aβ such as Aβ42 from the enzyme. (B) Presenilin FAD mutations cause a mispositioning of C99 as shown by altered interactions of the substrate cleavage domain with the enzyme. Residues that were identified to show increased or decreased interactions with two different PS1 FAD mutants are shown in red and blue, respectively. Yellow asterisk indicates a FAD mutation. |
–
Taken together, while presenilin FAD mutations display reduced carboxy-terminal trimming within the two product lines and/or product line shifts, the APP FAD mutations seem to combine product line shifts with substrate side chain compatible occupancy of the S1´- S3´ pockets. A problem that is not yet solved with the currently available models is the generation of Aβ38. This species is generated for a number of PS1 FAD mutants at levels comparable to the wt enzyme and in increased amounts for at least some APP FAD mutants, although it should not be generated when Aβ42 and Aβ43 dissociate faster from the enzyme due to destabilized enzyme-substrate interactions in the mutant case [118]. Although exciting research of the past few years has thus provided increasing mechanistic insights, the models derived from these studies are still too simple to explain all experimental observations at the mechanistic level.
–
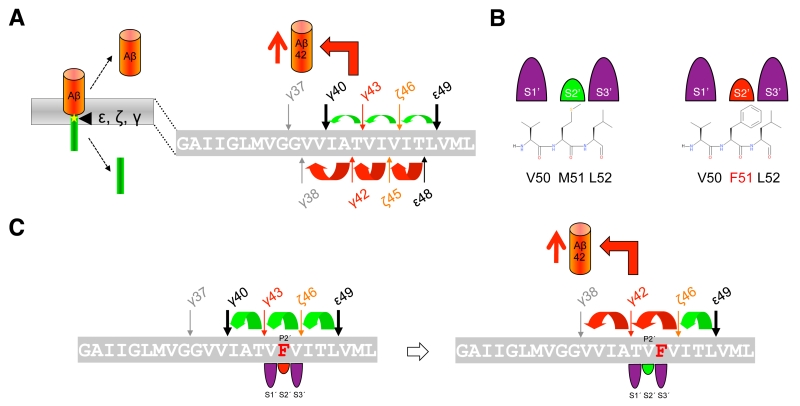 | FIGURE 7: Mechanism of APP FAD mutations. (A) Model depicting the shift in the Aβ40 and Aβ42 product lines by APP FAD mutations thereby causing relative increases in the generation of the longer Aβ42 species. Yellow asterisk indicates a FAD mutation. (B) Product line usage is governed by interactions with the S2´ enzyme subsite of γ-secretase. Sterically demanding aromatic amino acids at the P2´ position of C99 such as in the synthetic M51F mutant clash with the S2´ subsite of the Aβ40-line. (C) Pathway block by a clash of the aromatic P2´ with the S2´ subsite of the Aβ40-line in the FAD-associated I45F mutant causes a shift to the Aβ42-product line. Since this mutant does not alter the initial ε-site cleavage, Aβ42 is most likely generated from Aβ46. |
THE PRESENILIN LOSS OF FUNCTION HYPOTHESIS OF AD
Although the genetics strongly suggests that Aβ is central to AD pathogenesis, it has been challenging that neurodegeneration is not observed in transgenic mouse models that produce and extracellularly deposit aberrant amounts of pathogenic Aβ species by the expression of FAD mutant APP and/or presenilin variants (as an example see [126]). On the other hand, neurodegeneration has been observed in conditional PS1 knockout in mice in the absence of pathogenic Aβ production [127]. This has led to the hypothesis that a loss of presenilin function in cleaving crucial physiological substrates such as Notch1 is responsible for (F)AD rather than an absolute increase of total Aβ or a relative increase of Aβ42 [128]. An initially perplexing finding which seemed to support the “presenilin hypothesis” has been the biology of the PS1 L435F mutant, which causes early onset FAD although the mutant is virtually inactive and does not support the generation of Aβ40 and Aβ42 [129]. To explain the phenotype of this mutant, it was suggested that wt and FAD mutant presenilin form a dimeric complex in which the mutant could exert a dominant negative effect on the wt protein forcing it into a pathogenic Aβ42-generating conformation [129]. However, it has now been found that the PS1 L435F mutant generates and deposits the pathogenic Aβ43 species [130], which had been omitted from the Aβ analysis in the previous studies [131][132]. Importantly, this and other similar FAD mutants were shown to retain the capability to generate the pathogenic Aβ43 species in the absence of endogenous presenilins [131][132]. Potential trans-dominant effects of mutant presenilins on the wt protein that have been reported in the literature [133][134] can therefore not play a decisive role in the generation of pathogenic longer Aβ species and AD pathogenesis.
–
While presenilin FAD mutants can impair the cleavage of many substrates and thus potentially block or alter signaling pathways mediated by their ICDs, this seems unlikely to play a major role in vivo. The analysis of γ-secretase activity in human brain samples of PS1 FAD cases has shown that AICD formation is not affected whereas the carboxy-terminal processivity defects leading to increase Aβ42/40 ratios persist [117]. These data strongly suggest that potential effects on the “signaling cleavage” are compensated by the remaining wt PS alleles. A recent study investigating a total of 138 PS1 FAD mutations could not find a correlation with increased Aβ42 ratios and the age of onset while the vast majority of the mutants showed strongly reduced total γ-secretase activity [135]. This has been taken as another argument in favor of the presenilin hypothesis. However, the Aβ40 and Aβ42 levels and corresponding Aβ42/40 ratios of many PS1 FAD mutants that are well-characterized in cell-based assays were not recapitulated well in these assays and the pathogenic Aβ43 species had not been measured in the study. Moreover, predicting the age of onset of mutations from γ-secretase activities measured in cell-free in vitro assays using purified γ-secretase preparations in which loss of function effects are known to be more strongly pronounced [136] is problematic as this system differs dramatically from the heterozygous situation in FAD patient brain. Importantly, if loss of γ-secretase activity by presenilin mutations should be causative for AD, then haploinsufficiency of other γ-secretase subunits should also cause the disease. However, nonsense mutations that have been identified in PS1, NCT and PEN-2 are implicated in the cause of the rare skin disease acne inversa and not AD [137]. Finally, it is obvious that all types of APP FAD mutations as well as the protective Icelandic mutation or the presence of AD in Down syndrome patients with an APP gene triplication are not explainable by the presenilin hypothesis. Taken together, balancing the available evidence, the support of the amyloid cascade hypothesis remains overwhelming, whereas the problems of the presenilin hypothesis persist.
FAILURE OF γ-SECRETASE INHIBITION IN CLINICAL TRIALS
Since the amyloid cascade hypothesis predicts that lowering Aβ should be beneficial for AD, γ-secretase has been an obvious and major drug target. Thus, to inhibit its enzymatic activity and blocking Aβ generation, γ-secretase inhibitors (GSIs) were developed [138][139]. They fall into two principal classes, transition-state analogue (TSA) inhibitors, such as the prototypic L-685,458 [140] and related compounds as well as non-TSA compounds. The former compounds, which target the catalytic site of γ-secretase were the first highly potent inhibitors identified. Non-TSA inhibitors such as DAPT [141] or LY450139 (semagacestat) [142] are considered to bind nearby the active site. Unfortunately, since such pan-GSIs inhibit both Aβ and Notch ICD (NICD) production, severe side effects were observed when administered in vivo, mainly due to disturbance of Notch signaling [143][144][145][146]. While this is unwanted for AD therapy, treatment of certain cancers may benefit from Notch pathway inhibition by GSIs [147]. Several non-TSA GSIs were reported to have less inhibitory effects on NICD generation at a concentration range at which they sufficiently reduce Aβ secretion. Such Notch-sparing GSIs, as for example the promising candidate BMS-708163 (avagacestat) [148], were thus considered as improved AD therapeutics that should minimize side effects due to inhibition of Notch signaling. Altogether, many clinical studies have been conducted using non-TSA GSIs for the therapy of AD and/or are ongoing for cancers. Disappointingly, all of the AD clinical trials failed including a large phase 3 trial of semagacestat [149]. Even worse is that this trial and that of avagacestat [150] not only ended up with severe adverse effects but also with aggravated cognitive decline in patients. As semagacestat is a non-selective GSI and since follow-up studies suggested that the APP over Notch selectivity window of avagacestat was too narrow if present at all [115][151], the observed side effects can likely be attributed to the inhibition of Notch signaling. There has been no definitive answer yet on the cause of the aggravation of cognitive decline.
PSEUDOINHIBITION OF γ-SECRETASE BY SEMAGACESTAT AND RELATED GSIs
Using Aβ secretion as readout, GSIs were principally identified by high-throughput screening of small molecule libraries for compounds capable of lowering Aβ in conditioned media of cultured cells. However, since Aβ secretion is the result of several steps including its generation, liberation from membrane, trafficking, intracellular degradation and secretion, the levels of secreted Aβ do not necessarily accurately reflect the activity of γ-secretase. A further complicating and puzzling issue has been the observation that some GSIs, including DAPT, consistently increased the amounts of long intracellular Aβ species such as Aβ46 in APP-overexpressing cultured cells when used at concentrations that inhibit the formation of secreted Aβ [99][101][102]. The reasons had been unclear at the time, and clinical trials were performed without addressing this issue, although accumulation of such longer Aβ forms might be toxic to membranes. Considering that the γ-byproducts that are released by the sequential γ-secretase cleavages are not secreted and thus could serve as a more direct indicator of γ-secretase activity than secreted Aβ, the mechanism of action of non-TSA GSIs used in clinical trials was re-investigated by measuring their levels as readout [103]. Surprisingly, non-TSA GSIs, including semagacestat and avagacestat, did not decrease but rather increased the levels of γ-byproducts inside neurons derived from human iPS cells [103]. Along with the increased γ-byproducts levels, also an accumulation of long Aβ such as Aβ45 and Aβ46 was found inside neurons, although semagacestat and avagacestat decreased secreted Aβ. In contrast, L-685,458 decreased the levels of γ-byproducts and intracellular Aβ acting equivalently to a γ-secretase loss of function. The increased levels of γ-byproducts and the intracellular accumulation of long Aβ forms suggests that non-TSA GSIs, such as semagacestat and others, are in fact pseudoinhibitors of γ-secretase (Fig. 8). Apparently, these GSIs allow γ-secretase to initially cleave C99 but then γ-byproducts and/or inefficiently processed long Aβ intermediates may not efficiently be released from the enzyme and block access of further substrate eventually leading to a reduction of secreted Aβ as a secondary event. Interestingly, it was also found in this study that the γ-byproducts accumulate in the membrane [103]. This observation indicates that clearing the γ-byproducts from the membrane by facilitating their release into the hydrophilic space may be an additional function of γ-secretase. Semagacestat and other non-TSA GSIs may thus not inhibit the proteolytic activity of the enzyme but rather such a clearing function. One should note, however, that accumulation of γ-byproducts and long Aβ intermediates upon γ-secretase inhibition with DAPT has not been observed in reconstituted γ-secretase assays using detergent-solubilized enzyme [100][152]. This may be because such assays have only a simple and limited lipid composition compared to membrane-based cell-free assays with crude membrane fractions containing native γ-secretase [153][154] in which accumulation of γ-byproducts is observed [103], and/or because residual detergent present in these assays keeps the γ-byproducts soluble. Further analysis is necessary to elucidate whether γ-secretase has such a putative γ-byproduct clearance activity, and if so, whether and how a potential malfunction could be involved in AD pathogenesis as intracellular buildup of γ-byproducts or in particular of longer Aβ in neurons may potentially cause membrane toxicity.
–
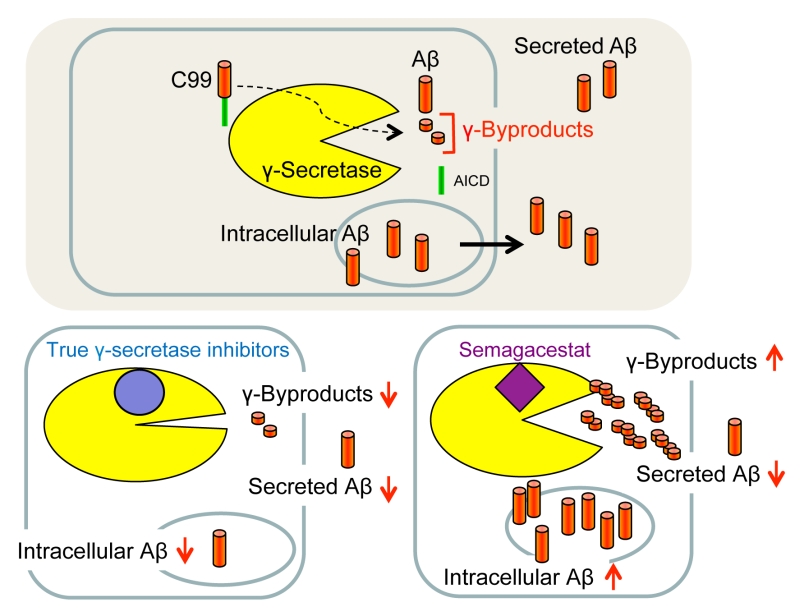 | FIGURE 8: Pseudoinhibiton of γ-secretase by semagacestat. Cleavage of C99 by γ-secretase causes the generation of secreted and intracellular Aβ pools as well as γ-byproducts from carboxy-terminal trimming of the C99 TMD. Unlike TSA-GSIs such as L-685,458, semagacestat causes an intracellular accumulation of long Aβ species and byproducts demonstrating a pseudo-inhibition of γ-secretase by this and related compounds. |
TARGETING γ-SECRETASE SAFELY
Unfortunately, the failure of GSIs in major clinical trials since 2010 caused a discouragement of pharmaceutical companies that lead to a stop in drug development for this AD target. However, GSIs that went into the trials were developed when our understanding of the many functions of γ-secretase and of its cleavage mechanism was very limited. Clearly, as it turned out, pan-inhibition of γ-secretase is highly problematic due to inhibition of critical substrates other than APP C99, potentially toxic C-terminal substrate fragment accumulation and Aβ rebound-effects from accumulated C99 [155] as well as cross-reactivities with signal peptide peptidase and related protease family members [139]. In addition, non-TSA GSIs that have been in clinical trials may paradoxically increase potentially toxic longer Aβ intermediates and γ-byproducts [103].
–
Rather than GSIs, γ-secretase modulators (GSMs) could be beneficial for AD. As shown first for a subset of NSAIDs (non-steroidal anti-inflammatory drugs), these compounds selectively lower the generation of Aβ42 while concomitantly increasing that of Aβ38, without affecting ε-site cleavages of γ-secretase substrates including Notch1 [156][157][158]. Moreover, as recently shown in animal models, short peptides such as Aβ38 are not only nontoxic but could even be protective by attenuating the toxicity of Aβ42 in vivo [159]. Mechanistically, GSMs enhance the processivity of γ-secretase by allowing prolonged residence time of Aβ42 at the enzyme [104], which is probably due to a stabilization of the enzyme-substrate transition state complex [118] through a conformational change in presenilin upon GSM binding [160][161], such that the longer Aβ can be processed to the shorter Aβ. Many highly potent GSMs of various structural classes have been developed [162] and a few of them entered clinical trials and successfully passed phase I. There is hope that proceeding with the development of GSMs could eventually allow safe inhibition of the enzyme without adverse effects [155]. Similarly, compounds that would stabilize presenilin in the γ-secretase complex are expected to generally activate the carboxy-peptidase activity in all product lines [118]. Certain GSMs such as the bridged aromates fulfill this property since they enhance processivity in both the Aβ40 and Aβ42 product lines leading to increases in both Aβ37 and Aβ38 [163].
–
Targeting the interaction of C99 with the γ-secretase complex might possibly represent another approach to selectively inhibit Aβ generation. It is conceivable that small compounds might be developable, which could interfere with binding of C99 at the γ-secretase exosites. Unlike the situation with classical GSIs, which bind at or near the catalytic site and are thus expected to provide little if any selectivity of APP over Notch or other substrates, such compounds could interfere with the initial recognition steps at more distant exosites used for substrate binding at stage I. As a consequence, substrates that cannot move to the catalytic site would be released back into the membrane bilayer. Modulating substrate binding at exosites rather than at the catalytic site might thus provide a way to improve substrate selectivity of GSIs. Interestingly, as implied by differences in the Aβ42/40 ratios and that of the corresponding shorter peptides generated from C89 and C83 [164], the length of the extracellular domain may be critical for substrate passage and presentation at the active site. Clearly, more mechanistic and structural studies on how γ-secretase recognizes and cleaves its substrates need to be performed but they are clearly warranted and could motivate the development of a new generation of truly substrate-selective γ-secretase inhibitors for clinical testing. It will also be necessary to rule out that GSMs, γ-secretase-stabilizing compounds, or exosite-interaction inhibitors, might, like non-TSA GSIs, cause a potentially unfavorable increase in the levels of the γ-byproducts and of (long) Aβ inside neurons.
–
Finally, should ongoing Aβ immunotherapy clinical trials with e.g. the promising aducanumab antibody that successfully removes patient brain amyloid [165] be beneficial in AD, a combination therapy with Aβ42 production modifiers or C99-selective inhibitors to maintain low amyloid levels, thereby preventing and/or shifting the onset of AD, might be an ideal and more cost-effective strategy for disease-modification than an immunotherapy alone.
References
- D.M. Holtzman, J.C. Morris, and A.M. Goate, "Alzheimer’s Disease: The Challenge of the Second Century", Science Translational Medicine, vol. 3, 2011. http://dx.doi.org/10.1126/scitranslmed.3002369
- D.J. Selkoe, "Alzheimer's Disease", Cold Spring Harbor Perspectives in Biology, vol. 3, pp. a004457-a004457, 2011. http://dx.doi.org/10.1101/cshperspect.a004457
- D.J. Selkoe, and J. Hardy, "The amyloid hypothesis of Alzheimer's disease at 25 years", EMBO Molecular Medicine, vol. 8, pp. 595-608, 2016. http://dx.doi.org/10.15252/emmm.201606210
- U.C. Müller, T. Deller, and M. Korte, "Not just amyloid: physiological functions of the amyloid precursor protein family", Nature Reviews Neuroscience, vol. 18, pp. 281-298, 2017. http://dx.doi.org/10.1038/nrn.2017.29
- R. Vassar, D.M. Kovacs, R. Yan, and P.C. Wong, "The β-Secretase Enzyme BACE in Health and Alzheimer's Disease: Regulation, Cell Biology, Function, and Therapeutic Potential", The Journal of Neuroscience, vol. 29, pp. 12787-12794, 2009. http://dx.doi.org/10.1523/JNEUROSCI.3657-09.2009
- B. De Strooper, T. Iwatsubo, and M.S. Wolfe, "Presenilins and -Secretase: Structure, Function, and Role in Alzheimer Disease", Cold Spring Harbor Perspectives in Medicine, vol. 2, pp. a006304-a006304, 2011. http://dx.doi.org/10.1101/cshperspect.a006304
- M. Morishima-Kawashima, "Molecular mechanism of the intramembrane cleavage of the β-carboxyl terminal fragment of amyloid precursor protein by γ-secretase", Frontiers in Physiology, vol. 5, 2014. http://dx.doi.org/10.3389/fphys.2014.00463
- S.F. Lichtenthaler, "Alpha-secretase in Alzheimer’s disease: molecular identity, regulation and therapeutic potential", Journal of Neurochemistry, vol. 116, pp. 10-21, 2010. http://dx.doi.org/10.1111/j.1471-4159.2010.07081.x
- T. Iwatsubo, A. Odaka, N. Suzuki, H. Mizusawa, N. Nukina, and Y. Ihara, "Visualization of Aβ42(43) and Aβ40 in senile plaques with end-specific Aβ monoclonals: Evidence that an initially deposited species is Aβ42(43)", Neuron, vol. 13, pp. 45-53, 1994. http://dx.doi.org/10.1016/0896-6273(94)90458-8
- E. Levy-Lahad, W. Wasco, P. Poorkaj, D.M. Romano, J. Oshima, W.H. Pettingell, C. Yu, P.D. Jondro, S.D. Schmidt, K. Wang, A.C. Crowley, Y. Fu, S.Y. Guenette, D. Galas, E. Nemens, E.M. Wijsman, T.D. Bird, G.D. Schellenberg, and R.E. Tanzi, "Candidate Gene for the Chromosome 1 Familial Alzheimer's Disease Locus", Science, vol. 269, pp. 973-977, 1995. http://dx.doi.org/10.1126/science.7638622
- E.I. Rogaev, R. Sherrington, E.A. Rogaeva, G. Levesque, M. Ikeda, Y. Liang, H. Chi, C. Lin, K. Holman, T. Tsuda, L. Mar, S. Sorbi, B. Nacmias, S. Piacentini, L. Amaducci, I. Chumakov, D. Cohen, L. Lannfelt, P.E. Fraser, J.M. Rommens, and P.H.S. George-Hyslop, "Familial Alzheimer's disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer's disease type 3 gene", Nature, vol. 376, pp. 775-778, 1995. http://dx.doi.org/10.1038/376775a0
- R. Sherrington, E.I. Rogaev, Y. Liang, E.A. Rogaeva, G. Levesque, M. Ikeda, H. Chi, C. Lin, G. Li, K. Holman, T. Tsuda, L. Mar, J. Foncin, A.C. Bruni, M.P. Montesi, S. Sorbi, I. Rainero, L. Pinessi, L. Nee, I. Chumakov, D. Pollen, A. Brookes, P. Sanseau, R.J. Polinsky, W. Wasco, H.A.R. Da Silva, J.L. Haines, M.A. Pericak-Vance, R.E. Tanzi, A.D. Roses, P.E. Fraser, J.M. Rommens, and P.H. St George-Hyslop, "Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease", Nature, vol. 375, pp. 754-760, 1995. http://dx.doi.org/10.1038/375754a0
- H. Steiner, "The Catalytic Core of γ-Secretase: Presenilin Revisited", Current Alzheimer Research, vol. 5, pp. 147-157, 2008. http://dx.doi.org/10.2174/156720508783954677
- D.R. Borchelt, G. Thinakaran, C.B. Eckman, M.K. Lee, F. Davenport, T. Ratovitsky, C. Prada, G. Kim, S. Seekins, D. Yager, H.H. Slunt, R. Wang, M. Seeger, A.I. Levey, S.E. Gandy, N.G. Copeland, N.A. Jenkins, D.L. Price, S.G. Younkin, and S.S. Sisodia, "Familial Alzheimer's Disease–Linked Presenilin 1 Variants Elevate Aβ1–42/1–40 Ratio In Vitro and In Vivo", Neuron, vol. 17, pp. 1005-1013, 1996. http://dx.doi.org/10.1016/S0896-6273(00)80230-5
- D. Scheuner, C. Eckman, M. Jensen, X. Song, M. Citron, N. Suzuki, T. Bird, J. Hardy, M. Hutton, W. Kukull, E. Larson, L. Levy-Lahad, M. Viitanen, E. Peskind, P. Poorkaj, G. Schellenberg, R. Tanzi, W. Wasco, L. Lannfelt, D. Selkoe, and S. Younkin, "Secreted amyloid β–protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease", Nature Medicine, vol. 2, pp. 864-870, 1996. http://dx.doi.org/10.1038/nm0896-864
- M. Citron, D. Westaway, W. Xia, G. Carlson, T. Diehl, G. Levesque, K. Johnson-wood, M. Lee, P. Seubert, A. Davis, D. Kholodenko, R. Motter, R. Sherrington, B. Perry, H. Yao, R. Strome, I. Lieberburg, J. Rommens, S. Kim, D. Schenk, P. Fraser, P. St George Hyslop, and D.J. Selkoe, "Mutant presenilins of Alzheimer's disease increase production of 42-residue amyloid β-protein in both transfected cells and transgenic mice", Nature Medicine, vol. 3, pp. 67-72, 1997. http://dx.doi.org/10.1038/nm0197-67
- M. Chartier-Harlin, F. Crawford, H. Houlden, A. Warren, D. Hughes, L. Fidani, A. Goate, M. Rossor, P. Roques, J. Hardy, and M. Mullan, "Early-onset Alzheimer's disease caused by mutations at codon 717 of the β-amyloid precursor protein gene", Nature, vol. 353, pp. 844-846, 1991. http://dx.doi.org/10.1038/353844a0
- A. Goate, M. Chartier-Harlin, M. Mullan, J. Brown, F. Crawford, L. Fidani, L. Giuffra, A. Haynes, N. Irving, L. James, R. Mant, P. Newton, K. Rooke, P. Roques, C. Talbot, M. Pericak-Vance, A. Roses, R. Williamson, M. Rossor, M. Owen, and J. Hardy, "Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease", Nature, vol. 349, pp. 704-706, 1991. http://dx.doi.org/10.1038/349704a0
- S. Weggen, and D. Beher, "Molecular consequences of amyloid precursor protein and presenilin mutations causing autosomal-dominant Alzheimer's disease", Alzheimer's Research & Therapy, vol. 4, pp. 9, 2012. http://dx.doi.org/10.1186/alzrt107
- M. Mullan, F. Crawford, K. Axelman, H. Houlden, L. Lilius, B. Winblad, and L. Lannfelt, "A pathogenic mutation for probable Alzheimer's disease in the APP gene at the N–terminus of β–amyloid", Nature Genetics, vol. 1, pp. 345-347, 1992. http://dx.doi.org/10.1038/ng0892-345
- M. Citron, T. Oltersdorf, C. Haass, L. McConlogue, A.Y. Hung, P. Seubert, C. Vigo-Pelfrey, I. Lieberburg, and D.J. Selkoe, "Mutation of the β-amyloid precursor protein in familial Alzheimer's disease increases β-protein production", Nature, vol. 360, pp. 672-674, 1992. http://dx.doi.org/10.1038/360672a0
- A. Rovelet-Lecrux, D. Hannequin, G. Raux, N.L. Meur, A. Laquerrière, A. Vital, C. Dumanchin, S. Feuillette, A. Brice, M. Vercelletto, F. Dubas, T. Frebourg, and D. Campion, "APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy", Nature Genetics, vol. 38, pp. 24-26, 2005. http://dx.doi.org/10.1038/ng1718
- T. Jonsson, J.K. Atwal, S. Steinberg, J. Snaedal, P.V. Jonsson, S. Bjornsson, H. Stefansson, P. Sulem, D. Gudbjartsson, J. Maloney, K. Hoyte, A. Gustafson, Y. Liu, Y. Lu, T. Bhangale, R.R. Graham, J. Huttenlocher, G. Bjornsdottir, O.A. Andreassen, E.G. Jönsson, A. Palotie, T.W. Behrens, O.T. Magnusson, A. Kong, U. Thorsteinsdottir, R.J. Watts, and K. Stefansson, "A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline", Nature, vol. 488, pp. 96-99, 2012. http://dx.doi.org/10.1038/nature11283
- J. Kim, J.M. Basak, and D.M. Holtzman, "The Role of Apolipoprotein E in Alzheimer's Disease", Neuron, vol. 63, pp. 287-303, 2009. http://dx.doi.org/10.1016/j.neuron.2009.06.026
- J.D. Ulrich, T.K. Ulland, M. Colonna, and D.M. Holtzman, "Elucidating the Role of TREM2 in Alzheimer’s Disease", Neuron, vol. 94, pp. 237-248, 2017. http://dx.doi.org/10.1016/j.neuron.2017.02.042
- C.M. Karch, and A.M. Goate, "Alzheimer’s Disease Risk Genes and Mechanisms of Disease Pathogenesis", Biological Psychiatry, vol. 77, pp. 43-51, 2015. http://dx.doi.org/10.1016/j.biopsych.2014.05.006
- D. Edbauer, E. Winkler, J.T. Regula, B. Pesold, H. Steiner, and C. Haass, "Reconstitution of γ-secretase activity", Nature Cell Biology, vol. 5, pp. 486-488, 2003. http://dx.doi.org/10.1038/ncb960
- W.T. Kimberly, M.J. LaVoie, B.L. Ostaszewski, W. Ye, M.S. Wolfe, and D.J. Selkoe, "γ-Secretase is a membrane protein complex comprised of presenilin, nicastrin, aph-1, and pen-2", Proceedings of the National Academy of Sciences, vol. 100, pp. 6382-6387, 2003. http://dx.doi.org/10.1073/pnas.1037392100
- N. Takasugi, T. Tomita, I. Hayashi, M. Tsuruoka, M. Niimura, Y. Takahashi, G. Thinakaran, and T. Iwatsubo, "The role of presenilin cofactors in the γ-secretase complex", Nature, vol. 422, pp. 438-441, 2003. http://dx.doi.org/10.1038/nature01506
- X. Bai, C. Yan, G. Yang, P. Lu, D. Ma, L. Sun, R. Zhou, S.H.W. Scheres, and Y. Shi, "An atomic structure of human γ-secretase", Nature, vol. 525, pp. 212-217, 2015. http://dx.doi.org/10.1038/nature14892
- G. Yu, M. Nishimura, S. Arawaka, D. Levitan, L. Zhang, A. Tandon, Y. Song, E. Rogaeva, F. Chen, T. Kawarai, A. Supala, L. Levesque, H. Yu, D. Yang, E. Holmes, P. Milman, Y. Liang, D.M. Zhang, D.H. Xu, C. Sato, E. Rogaev, M. Smith, C. Janus, Y. Zhang, R. Aebersold, L. Farrer, S. Sorbi, A. Bruni, P. Fraser, and P. St George-Hyslop, "Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and βAPP processing", Nature, vol. 407, pp. 48-54, 2000. http://dx.doi.org/10.1038/35024009
- P. Lu, X. Bai, D. Ma, T. Xie, C. Yan, L. Sun, G. Yang, Y. Zhao, R. Zhou, S.H.W. Scheres, and Y. Shi, "Three-dimensional structure of human γ-secretase", Nature, vol. 512, pp. 166-170, 2014. http://dx.doi.org/10.1038/nature13567
- M.S. Wolfe, W. Xia, B.L. Ostaszewski, T.S. Diehl, W.T. Kimberly, and D.J. Selkoe, "Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and γ-secretase activity", Nature, vol. 398, pp. 513-517, 1999. http://dx.doi.org/10.1038/19077
- H. Steiner, K. Duff, A. Capell, H. Romig, M.G. Grim, S. Lincoln, J. Hardy, X. Yu, M. Picciano, K. Fechteler, M. Citron, R. Kopan, B. Pesold, S. Keck, M. Baader, T. Tomita, T. Iwatsubo, R. Baumeister, and C. Haass, "A Loss of Function Mutation of Presenilin-2 Interferes with Amyloid β-Peptide Production and Notch Signaling", Journal of Biological Chemistry, vol. 274, pp. 28669-28673, 1999. http://dx.doi.org/10.1074/jbc.274.40.28669
- W. Kimberly, W. Xia, T. Rahmati, M.S. Wolfe, and D.J. Selkoe, "The Transmembrane Aspartates in Presenilin 1 and 2 Are Obligatory for γ-Secretase Activity and Amyloid β-Protein Generation", Journal of Biological Chemistry, vol. 275, pp. 3173-3178, 2000. http://dx.doi.org/10.1074/jbc.275.5.3173
- D. Dries, and G. Yu, "Assembly, Maturation, and Trafficking of the γ-Secretase Complex in Alzheimers Disease", Current Alzheimer Research, vol. 5, pp. 132-146, 2008. http://dx.doi.org/10.2174/156720508783954695
- G. Thinakaran, D.R. Borchelt, M.K. Lee, H.H. Slunt, L. Spitzer, G. Kim, T. Ratovitsky, F. Davenport, C. Nordstedt, M. Seeger, J. Hardy, A.I. Levey, S.E. Gandy, N.A. Jenkins, N.G. Copeland, D.L. Price, and S.S. Sisodia, "Endoproteolysis of Presenilin 1 and Accumulation of Processed Derivatives In Vivo", Neuron, vol. 17, pp. 181-190, 1996. http://dx.doi.org/10.1016/S0896-6273(00)80291-3
- M.B. Podlisny, M. Citron, P. Amarante, R. Sherrington, W. Xia, J. Zhang, T. Diehl, G. Levesque, P. Fraser, C. Haass, E.H. Koo, P. Seubert, P. St. George-Hyslop, D.B. Teplow, and D.J. Selkoe, "Presenilin Proteins Undergo Heterogeneous Endoproteolysis between Thr291and Ala299and Occur as Stable N- and C-Terminal Fragments in Normal and Alzheimer Brain Tissue", Neurobiology of Disease, vol. 3, pp. 325-337, 1997. http://dx.doi.org/10.1006/nbdi.1997.0129
- A. Fukumori, R. Fluhrer, H. Steiner, and C. Haass, "Three-Amino Acid Spacing of Presenilin Endoproteolysis Suggests a General Stepwise Cleavage of -Secretase-Mediated Intramembrane Proteolysis", Journal of Neuroscience, vol. 30, pp. 7853-7862, 2010. http://dx.doi.org/10.1523/JNEUROSCI.1443-10.2010
- D. Beher, J.D. Wrigley, A. Nadin, G. Evin, C.L. Masters, T. Harrison, J.L. Castro, and M.S. Shearman, "Pharmacological Knock-down of the Presenilin 1 Heterodimer by a Novel γ-Secretase Inhibitor", Journal of Biological Chemistry, vol. 276, pp. 45394-45402, 2001. http://dx.doi.org/10.1074/jbc.M103075200
- G. Yu, F. Chen, G. Levesque, M. Nishimura, D. Zhang, L. Levesque, E. Rogaeva, D. Xu, Y. Liang, M. Duthie, P.H.S. George-Hyslop, and P.E. Fraser, "The Presenilin 1 Protein Is a Component of a High Molecular Weight Intracellular Complex That Contains β-Catenin", Journal of Biological Chemistry, vol. 273, pp. 16470-16475, 1998. http://dx.doi.org/10.1074/jbc.273.26.16470
- R. Francis, G. McGrath, J. Zhang, D.A. Ruddy, M. Sym, J. Apfeld, M. Nicoll, M. Maxwell, B. Hai, M.C. Ellis, A.L. Parks, W. Xu, J. Li, M. Gurney, R.L. Myers, C.S. Himes, R. Hiebsch, C. Ruble, J.S. Nye, and D. Curtis, "aph-1 and pen-2 Are Required for Notch Pathway Signaling, γ-Secretase Cleavage of βAPP, and Presenilin Protein Accumulation", Developmental Cell, vol. 3, pp. 85-97, 2002. http://dx.doi.org/10.1016/S1534-5807(02)00189-2
- C. Goutte, M. Tsunozaki, V.A. Hale, and J.R. Priess, "APH-1 is a multipass membrane protein essential for the Notch signaling pathway in Caenorhabditis elegans embryos", Proceedings of the National Academy of Sciences, vol. 99, pp. 775-779, 2002. http://dx.doi.org/10.1073/pnas.022523499
- S. Lee, S. Shah, H. Li, C. Yu, W. Han, and G. Yu, "Mammalian APH-1 Interacts with Presenilin and Nicastrin and Is Required for Intramembrane Proteolysis of Amyloid-β Precursor Protein and Notch", Journal of Biological Chemistry, vol. 277, pp. 45013-45019, 2002. http://dx.doi.org/10.1074/jbc.M208164200
- S.S. Hébert, L. Serneels, T. Dejaegere, K. Horré, M. Dabrowski, V. Baert, W. Annaert, D. Hartmann, and B. De Strooper, "Coordinated and widespread expression of γ-secretase in vivo: evidence for size and molecular heterogeneity", Neurobiology of Disease, vol. 17, pp. 260-272, 2004. http://dx.doi.org/10.1016/j.nbd.2004.08.002
- K. Shirotani, D. Edbauer, S. Prokop, C. Haass, and H. Steiner, "Identification of Distinct γ-Secretase Complexes with Different APH-1 Variants", Journal of Biological Chemistry, vol. 279, pp. 41340-41345, 2004. http://dx.doi.org/10.1074/jbc.M405768200
- H. Steiner, E. Winkler, D. Edbauer, S. Prokop, G. Basset, A. Yamasaki, M. Kostka, and C. Haass, "PEN-2 Is an Integral Component of the γ-Secretase Complex Required for Coordinated Expression of Presenilin and Nicastrin", Journal of Biological Chemistry, vol. 277, pp. 39062-39065, 2002. http://dx.doi.org/10.1074/jbc.C200469200
- L. Sun, L. Zhao, G. Yang, C. Yan, R. Zhou, X. Zhou, T. Xie, Y. Zhao, S. Wu, X. Li, and Y. Shi, "Structural basis of human γ-secretase assembly", Proceedings of the National Academy of Sciences, vol. 112, pp. 6003-6008, 2015. http://dx.doi.org/10.1073/pnas.1506242112
- X. Zhang, C.J. Yu, and S.S. Sisodia, "The topology of pen-2, a γ-secretase subunit, revisited: evidence for a reentrant loop and a single pass transmembrane domain", Molecular Neurodegeneration, vol. 10, 2015. http://dx.doi.org/10.1186/s13024-015-0037-4
- Y. Li, S. Lu, C. Tsai, C. Bohm, S. Qamar, R. Dodd, W. Meadows, A. Jeon, A. McLeod, F. Chen, M. Arimon, O. Berezovska, B. Hyman, T. Tomita, T. Iwatsubo, C. Johnson, L. Farrer, G. Schmitt-Ulms, P. Fraser, and P. St George-Hyslop, "Structural Interactions between Inhibitor and Substrate Docking Sites Give Insight into Mechanisms of Human PS1 Complexes", Structure, vol. 22, pp. 125-135, 2014. http://dx.doi.org/10.1016/j.str.2013.09.018
- N. Elad, B. De Strooper, S. Lismont, W. Hagen, S. Veugelen, M. Arimon, K. Horré, O. Berezovska, C. Sachse, and L. Chávez-Gutiérrez, "The dynamic conformational landscape of γ-secretase", Journal of Cell Science, 2014. http://dx.doi.org/10.1242/jcs.164384
- X. Bai, E. Rajendra, G. Yang, Y. Shi, and S.H. Scheres, "Sampling the conformational space of the catalytic subunit of human γ-secretase", eLife, vol. 4, 2015. http://dx.doi.org/10.7554/eLife.11182
- X. Meckler, and F. Checler, "Presenilin 1 and Presenilin 2 Target γ-Secretase Complexes to Distinct Cellular Compartments", Journal of Biological Chemistry, vol. 291, pp. 12821-12837, 2016. http://dx.doi.org/10.1074/jbc.M115.708297
- R. Sannerud, C. Esselens, P. Ejsmont, R. Mattera, L. Rochin, A. Tharkeshwar, G. De Baets, V. De Wever, R. Habets, V. Baert, W. Vermeire, C. Michiels, A. Groot, R. Wouters, K. Dillen, K. Vints, P. Baatsen, S. Munck, R. Derua, E. Waelkens, G. Basi, M. Mercken, M. Vooijs, M. Bollen, J. Schymkowitz, F. Rousseau, J. Bonifacino, G. Van Niel, B. De Strooper, and W. Annaert, "Restricted Location of PSEN2/γ-Secretase Determines Substrate Specificity and Generates an Intracellular Aβ Pool", Cell, vol. 166, pp. 193-208, 2016. http://dx.doi.org/10.1016/j.cell.2016.05.020
- A. Haapasalo, and D.M. Kovacs, "The many substrates of presenilin/γ-secretase.", Journal of Alzheimer's disease : JAD, 2011. http://www.ncbi.nlm.nih.gov/pubmed/21335653
- B. Vincent, and F. Checler, "α-Secretase in Alzheimers Disease and Beyond: Mechanistic, Regulation and Function in the Shedding of Membrane Proteins", Current Alzheimer Research, vol. 9, pp. 140-156, 2012. http://dx.doi.org/10.2174/156720512799361646
- S.F. Lichtenthaler, C. Haass, and H. Steiner, "Regulated intramembrane proteolysis - lessons from amyloid precursor protein processing", Journal of Neurochemistry, vol. 117, pp. 779-796, 2011. http://dx.doi.org/10.1111/j.1471-4159.2011.07248.x
- S.A. Laurent, F.S. Hoffmann, P. Kuhn, Q. Cheng, Y. Chu, M. Schmidt-Supprian, S.M. Hauck, E. Schuh, M. Krumbholz, H. Rübsamen, J. Wanngren, M. Khademi, T. Olsson, T. Alexander, F. Hiepe, H. Pfister, F. Weber, D. Jenne, H. Wekerle, R. Hohlfeld, S.F. Lichtenthaler, and E. Meinl, "γ-secretase directly sheds the survival receptor BCMA from plasma cells", Nature Communications, vol. 6, 2015. http://dx.doi.org/10.1038/ncomms8333
- B. De Strooper, W. Annaert, P. Cupers, P. Saftig, K. Craessaerts, J.S. Mumm, E.H. Schroeter, V. Schrijvers, M.S. Wolfe, W.J. Ray, A. Goate, and R. Kopan, "A presenilin-1-dependent γ-secretase-like protease mediates release of Notch intracellular domain", Nature, vol. 398, pp. 518-522, 1999. http://dx.doi.org/10.1038/19083
- G. Struhl, and I. Greenwald, "Presenilin is required for activity and nuclear access of Notch in Drosophila", Nature, vol. 398, pp. 522-525, 1999. http://dx.doi.org/10.1038/19091
- C. Sato, G. Zhao, and M. Xenia G. Ilagan, "An Overview of Notch Signaling in Adult Tissue Renewal and Maintenance", Current Alzheimer Research, vol. 9, pp. 227-240, 2012. http://dx.doi.org/10.2174/156720512799361600
- E.H. Schroeter, J.A. Kisslinger, and R. Kopan, "Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain", Nature, vol. 393, pp. 382-386, 1998. http://dx.doi.org/10.1038/30756
- X. Cao, and T.C. Südhof, "A Transcriptively Active Complex of APP with Fe65 and Histone Acetyltransferase Tip60", Science, vol. 293, pp. 115-120, 2001. http://dx.doi.org/10.1126/science.1058783
- S.S. Hébert, L. Serneels, A. Tolia, K. Craessaerts, C. Derks, M.A. Filippov, U. Müller, and B. De Strooper, "Regulated intramembrane proteolysis of amyloid precursor protein and regulation of expression of putative target genes", EMBO reports, vol. 7, pp. 739-745, 2006. http://dx.doi.org/10.1038/sj.embor.7400704
- A.T. Parent, N.Y. Barnes, Y. Taniguchi, G. Thinakaran, and S.S. Sisodia, "Presenilin Attenuates Receptor-Mediated Signaling and Synaptic Function", The Journal of Neuroscience, vol. 25, pp. 1540-1549, 2005. http://dx.doi.org/10.1523/JNEUROSCI.3850-04.2005
- P. Marambaud, P.H. Wen, A. Dutt, J. Shioi, A. Takashima, R. Siman, and N.K. Robakis, "A CBP Binding Transcriptional Repressor Produced by the PS1/ϵ-Cleavage of N-Cadherin Is Inhibited by PS1 FAD Mutations", Cell, vol. 114, pp. 635-645, 2003. http://dx.doi.org/10.1016/j.cell.2003.08.008
- N. Jurisch-Yaksi, R. Sannerud, and W. Annaert, "A fast growing spectrum of biological functions of γ-secretase in development and disease", Biochimica et Biophysica Acta (BBA) - Biomembranes, vol. 1828, pp. 2815-2827, 2013. http://dx.doi.org/10.1016/j.bbamem.2013.04.016
- R. Kopan, and M.X.G. Ilagan, "γ-Secretase: proteasome of the membrane?", Nature Reviews Molecular Cell Biology, vol. 5, pp. 499-504, 2004. http://dx.doi.org/10.1038/nrm1406
- I. Lauritzen, R. Pardossi-Piquard, C. Bauer, E. Brigham, J. Abraham, S. Ranaldi, P. Fraser, P. St-George-Hyslop, O. Le Thuc, V. Espin, L. Chami, J. Dunys, and F. Checler, "The β-Secretase-Derived C-Terminal Fragment of βAPP, C99, But Not Aβ, Is a Key Contributor to Early Intraneuronal Lesions in Triple-Transgenic Mouse Hippocampus", The Journal of Neuroscience, vol. 32, pp. 16243-16255, 2012. http://dx.doi.org/10.1523/JNEUROSCI.2775-12.2012
- T. Bittner, M. Fuhrmann, S. Burgold, C.K.E. Jung, C. Volbracht, H. Steiner, G. Mitteregger, H.A. Kretzschmar, C. Haass, and J. Herms, "γ-Secretase Inhibition Reduces Spine DensityIn Vivovia an Amyloid Precursor Protein-Dependent Pathway", The Journal of Neuroscience, vol. 29, pp. 10405-10409, 2009. http://dx.doi.org/10.1523/JNEUROSCI.2288-09.2009
- Y. Mitani, J. Yarimizu, K. Saita, H. Uchino, H. Akashiba, Y. Shitaka, K. Ni, and N. Matsuoka, "Differential Effects between γ-Secretase Inhibitors and Modulators on Cognitive Function in Amyloid Precursor Protein-Transgenic and Nontransgenic Mice", The Journal of Neuroscience, vol. 32, pp. 2037-2050, 2012. http://dx.doi.org/10.1523/JNEUROSCI.4264-11.2012
- H. Acx, L. Serneels, E. Radaelli, S. Muyldermans, C. Vincke, E. Pepermans, U. Müller, L. Chávez‐Gutiérrez, and B. De Strooper, "Inactivation of γ‐secretases leads to accumulation of substrates and non‐Alzheimer neurodegeneration", EMBO Molecular Medicine, vol. 9, pp. 1088-1099, 2017. http://dx.doi.org/10.15252/emmm.201707561
- R. Tamayev, S. Matsuda, O. Arancio, and L. D'Adamio, "β‐ but not γ‐secretase proteolysis of APP causes synaptic and memory deficits in a mouse model of dementia", EMBO Molecular Medicine, vol. 4, pp. 171-179, 2012. http://dx.doi.org/10.1002/emmm.201100195
- S.P. Duggan, and J.V. McCarthy, "Beyond γ-secretase activity: The multifunctional nature of presenilins in cell signalling pathways", Cellular Signalling, vol. 28, pp. 1-11, 2016. http://dx.doi.org/10.1016/j.cellsig.2015.10.006
- S. Funamoto, T. Sasaki, S. Ishihara, M. Nobuhara, M. Nakano, M. Watanabe-Takahashi, T. Saito, N. Kakuda, T. Miyasaka, K. Nishikawa, T.C. Saido, and Y. Ihara, "Substrate ectodomain is critical for substrate preference and inhibition of γ-secretase", Nature Communications, vol. 4, 2013. http://dx.doi.org/10.1038/ncomms3529
- D.M. Bolduc, D.R. Montagna, Y. Gu, D.J. Selkoe, and M.S. Wolfe, "Nicastrin functions to sterically hinder γ-secretase–substrate interactions driven by substrate transmembrane domain", Proceedings of the National Academy of Sciences, vol. 113, 2015. http://dx.doi.org/10.1073/pnas.1512952113
- G. Struhl, and A. Adachi, "Requirements for Presenilin-Dependent Cleavage of Notch and Other Transmembrane Proteins", Molecular Cell, vol. 6, pp. 625-636, 2000. http://dx.doi.org/10.1016/S1097-2765(00)00061-7
- I. Hayashi, S. Takatori, Y. Urano, Y. Miyake, J. Takagi, M. Sakata-Yanagimoto, H. Iwanari, S. Osawa, Y. Morohashi, T. Li, P.C. Wong, S. Chiba, T. Kodama, T. Hamakubo, T. Tomita, and T. Iwatsubo, "Neutralization of the γ-secretase activity by monoclonal antibody against extracellular domain of nicastrin", Oncogene, vol. 31, pp. 787-798, 2011. http://dx.doi.org/10.1038/onc.2011.265
- X. Zhang, R.J. Hoey, G. Lin, A. Koide, B. Leung, K. Ahn, G. Dolios, M. Paduch, T. Ikeuchi, R. Wang, Y. Li, S. Koide, and S.S. Sisodia, "Identification of a tetratricopeptide repeat-like domain in the nicastrin subunit of γ-secretase using synthetic antibodies", Proceedings of the National Academy of Sciences, vol. 109, pp. 8534-8539, 2012. http://dx.doi.org/10.1073/pnas.1202691109
- J.Y. Lee, Z. Feng, X. Xie, and I. Bahar, "Allosteric Modulation of Intact γ-Secretase Structural Dynamics", Biophysical Journal, vol. 113, pp. 2634-2649, 2017. http://dx.doi.org/10.1016/j.bpj.2017.10.012
- R. Aguayo-Ortiz, C. Chávez-García, J.E. Straub, and L. Dominguez, "Characterizing the structural ensemble of γ-secretase using a multiscale molecular dynamics approach", Chemical Science, vol. 8, pp. 5576-5584, 2017. http://dx.doi.org/10.1039/c7sc00980a
- A. Fukumori, and H. Steiner, "Substrate recruitment of γ‐secretase and mechanism of clinical presenilin mutations revealed by photoaffinity mapping", The EMBO Journal, vol. 35, pp. 1628-1643, 2016. http://dx.doi.org/10.15252/embj.201694151
- S. Shah, S. Lee, K. Tabuchi, Y. Hao, C. Yu, Q. LaPlant, H. Ball, C.E. Dann, T. Südhof, and G. Yu, "Nicastrin Functions as a γ-Secretase-Substrate Receptor", Cell, vol. 122, pp. 435-447, 2005. http://dx.doi.org/10.1016/j.cell.2005.05.022
- L. Chávez-Gutiérrez, A. Tolia, E. Maes, T. Li, P.C. Wong, and B. de Strooper, "Glu332 in the Nicastrin Ectodomain Is Essential for γ-Secretase Complex Maturation but Not for Its Activity", Journal of Biological Chemistry, vol. 283, pp. 20096-20105, 2008. http://dx.doi.org/10.1074/jbc.M803040200
- D.R. Dries, S. Shah, Y. Han, C. Yu, S. Yu, M.S. Shearman, and G. Yu, "Glu-333 of Nicastrin Directly Participates in γ-Secretase Activity", Journal of Biological Chemistry, vol. 284, pp. 29714-29724, 2009. http://dx.doi.org/10.1074/jbc.M109.038737
- T. Xie, C. Yan, R. Zhou, Y. Zhao, L. Sun, G. Yang, P. Lu, D. Ma, and Y. Shi, "Crystal structure of the γ-secretase component nicastrin", Proceedings of the National Academy of Sciences, vol. 111, pp. 13349-13354, 2014. http://dx.doi.org/10.1073/pnas.1414837111
- X. Zhang, E. Sullivan, M. Scimeca, X. Wu, Y. Li, and S.S. Sisodia, "Evidence That the “Lid” Domain of Nicastrin Is Not Essential for Regulating γ-Secretase Activity", Journal of Biological Chemistry, vol. 291, pp. 6748-6753, 2016. http://dx.doi.org/10.1074/jbc.C115.701649
- P. Gong, K.S. Vetrivel, P.D. Nguyen, X. Meckler, H. Cheng, M.Z. Kounnas, S.L. Wagner, A.T. Parent, and G. Thinakaran, "Mutation Analysis of the Presenilin 1 N-terminal Domain Reveals a Broad Spectrum of γ-Secretase Activity toward Amyloid Precursor Protein and Other Substrates", Journal of Biological Chemistry, vol. 285, pp. 38042-38052, 2010. http://dx.doi.org/10.1074/jbc.M110.132613
- H. Steiner, M. Kostka, H. Romig, G. Basset, B. Pesold, J. Hardy, A. Capell, L. Meyn, M.L. Grim, R. Baumeister, K. Fechteler, and C. Haass, "Glycine 384 is required for presenilin-1 function and is conserved in bacterial polytopic aspartyl proteases", Nature Cell Biology, vol. 2, pp. 848-851, 2000. http://dx.doi.org/10.1038/35041097
- B.I. Pérez‐Revuelta, A. Fukumori, S. Lammich, A. Yamasaki, C. Haass, and H. Steiner, "Requirement for small side chain residues within the GxGD‐motif of presenilin for γ‐secretase substrate cleavage", Journal of Neurochemistry, vol. 112, pp. 940-950, 2010. http://dx.doi.org/10.1111/j.1471-4159.2009.06510.x
- A. Yamasaki, S. Eimer, M. Okochi, A. Smialowska, C. Kaether, R. Baumeister, C. Haass, and H. Steiner, "The GxGD Motif of Presenilin Contributes to Catalytic Function and Substrate Identification of γ-Secretase", The Journal of Neuroscience, vol. 26, pp. 3821-3828, 2006. http://dx.doi.org/10.1523/JNEUROSCI.5354-05.2006
-
B. Kretner, A. Fukumori, P. Kuhn, B.I. Pérez‐Revuelta, S.F. Lichtenthaler, C. Haass, and H. Steiner, "Important functional role of residue x of the presenilin Gx
GD protease active site motif forAPP substrate cleavage specificity and substrate selectivity of γ‐secretase", Journal of Neurochemistry, vol. 125, pp. 144-156, 2013. http://dx.doi.org/10.1111/jnc.12124 - J. Wang, D. Beher, A.C. Nyborg, M.S. Shearman, T.E. Golde, and A. Goate, "C‐terminal PAL motif of presenilin and presenilin homologues required for normal active site conformation", Journal of Neurochemistry, vol. 96, pp. 218-227, 2005. http://dx.doi.org/10.1111/j.1471-4159.2005.03548.x
- J. Wang, A.L. Brunkan, S. Hecimovic, E. Walker, and A. Goate, "Conserved “PAL” sequence in presenilins is essential for γ-secretase activity, but not required for formation or stabilization of γ-secretase complexes", Neurobiology of Disease, vol. 15, pp. 654-666, 2004. http://dx.doi.org/10.1016/j.nbd.2003.12.008
- M. Sastre, H. Steiner, K. Fuchs, A. Capell, G. Multhaup, M.M. Condron, D.B. Teplow, and C. Haass, "Presenilin‐dependent γ‐secretase processing of β‐amyloid precursor protein at a site corresponding to the S3 cleavage of Notch", EMBO reports, vol. 2, pp. 835-841, 2001. http://dx.doi.org/10.1093/embo-reports/kve180
- Y. Gu, H. Misonou, T. Sato, N. Dohmae, K. Takio, and Y. Ihara, "Distinct Intramembrane Cleavage of the β-Amyloid Precursor Protein Family Resembling γ-Secretase-like Cleavage of Notch", Journal of Biological Chemistry, vol. 276, pp. 35235-35238, 2001. http://dx.doi.org/10.1074/jbc.C100357200
- C. Yu, S. Kim, T. Ikeuchi, H. Xu, L. Gasparini, R. Wang, and S.S. Sisodia, "Characterization of a Presenilin-mediated Amyloid Precursor Protein Carboxyl-terminal Fragment γ", Journal of Biological Chemistry, vol. 276, pp. 43756-43760, 2001. http://dx.doi.org/10.1074/jbc.C100410200
- A. Weidemann, S. Eggert, F.B.M. Reinhard, M. Vogel, K. Paliga, G. Baier, C.L. Masters, K. Beyreuther, and G. Evin, "A Novel ε-Cleavage within the Transmembrane Domain of the Alzheimer Amyloid Precursor Protein Demonstrates Homology with Notch Processing", Biochemistry, vol. 41, pp. 2825-2835, 2002. http://dx.doi.org/10.1021/bi015794o
- Y. Qi-Takahara, M. Morishima-Kawashima, Y. Tanimura, G. Dolios, N. Hirotani, Y. Horikoshi, F. Kametani, M. Maeda, T.C. Saido, R. Wang, and Y. Ihara, "Longer Forms of Amyloid β Protein: Implications for the Mechanism of Intramembrane Cleavage by γ-Secretase", The Journal of Neuroscience, vol. 25, pp. 436-445, 2005. http://dx.doi.org/10.1523/JNEUROSCI.1575-04.2005
- M. Takami, Y. Nagashima, Y. Sano, S. Ishihara, M. Morishima-Kawashima, S. Funamoto, and Y. Ihara, "γ-Secretase: Successive Tripeptide and Tetrapeptide Release from the Transmembrane Domain of β-Carboxyl Terminal Fragment", The Journal of Neuroscience, vol. 29, pp. 13042-13052, 2009. http://dx.doi.org/10.1523/JNEUROSCI.2362-09.2009
- G. Zhao, M. Cui, G. Mao, Y. Dong, J. Tan, L. Sun, and X. Xu, "γ-Cleavage Is Dependent on ζ-Cleavage during the Proteolytic Processing of Amyloid Precursor Protein within Its Transmembrane Domain", Journal of Biological Chemistry, vol. 280, pp. 37689-37697, 2005. http://dx.doi.org/10.1074/jbc.M507993200
- G. Zhao, G. Mao, J. Tan, Y. Dong, M. Cui, S. Kim, and X. Xu, "Identification of a New Presenilin-dependent ζ-Cleavage Site within the Transmembrane Domain of Amyloid Precursor Protein", Journal of Biological Chemistry, vol. 279, pp. 50647-50650, 2004. http://dx.doi.org/10.1074/jbc.C400473200
- S. Tagami, K. Yanagida, T.S. Kodama, M. Takami, N. Mizuta, H. Oyama, K. Nishitomi, Y. Chiu, T. Okamoto, T. Ikeuchi, G. Sakaguchi, T. Kudo, Y. Matsuura, A. Fukumori, M. Takeda, Y. Ihara, and M. Okochi, "Semagacestat Is a Pseudo-Inhibitor of γ-Secretase", Cell Reports, vol. 21, pp. 259-273, 2017. http://dx.doi.org/10.1016/j.celrep.2017.09.032
- M. Okochi, S. Tagami, K. Yanagida, M. Takami, T. Kodama, K. Mori, T. Nakayama, Y. Ihara, and M. Takeda, "γ-Secretase Modulators and Presenilin 1 Mutants Act Differently on Presenilin/γ-Secretase Function to Cleave Aβ42 and Aβ43", Cell Reports, vol. 3, pp. 42-51, 2013. http://dx.doi.org/10.1016/j.celrep.2012.11.028
- N. Matsumura, M. Takami, M. Okochi, S. Wada-Kakuda, H. Fujiwara, S. Tagami, S. Funamoto, Y. Ihara, and M. Morishima-Kawashima, "γ-Secretase Associated with Lipid Rafts", Journal of Biological Chemistry, vol. 289, pp. 5109-5121, 2014. http://dx.doi.org/10.1074/jbc.M113.510131
- F. Olsson, S. Schmidt, V. Althoff, L.M. Munter, S. Jin, S. Rosqvist, U. Lendahl, G. Multhaup, and J. Lundkvist, "Characterization of Intermediate Steps in Amyloid Beta (Aβ) Production under Near-native Conditions", Journal of Biological Chemistry, vol. 289, pp. 1540-1550, 2014. http://dx.doi.org/10.1074/jbc.M113.498246
- A. Pensalfini, R. Albay, S. Rasool, J.W. Wu, A. Hatami, H. Arai, L. Margol, S. Milton, W.W. Poon, M.M. Corrada, C.H. Kawas, and C.G. Glabe, "Intracellular amyloid and the neuronal origin of Alzheimer neuritic plaques", Neurobiology of Disease, vol. 71, pp. 53-61, 2014. http://dx.doi.org/10.1016/j.nbd.2014.07.011
- D.M. Bolduc, D.R. Montagna, M.C. Seghers, M.S. Wolfe, and D.J. Selkoe, "The amyloid-beta forming tripeptide cleavage mechanism of γ-secretase", eLife, vol. 5, 2016. http://dx.doi.org/10.7554/eLife.17578
- Y. Ran, F. Hossain, A. Pannuti, C.B. Lessard, G.Z. Ladd, J.I. Jung, L.M. Minter, B.A. Osborne, L. Miele, and T.E. Golde, "γ‐Secretase inhibitors in cancer clinical trials are pharmacologically and functionally distinct", EMBO Molecular Medicine, vol. 9, pp. 950-966, 2017. http://dx.doi.org/10.15252/emmm.201607265
- F. Kamp, E. Winkler, J. Trambauer, A. Ebke, R. Fluhrer, and H. Steiner, "Intramembrane Proteolysis of β-Amyloid Precursor Protein by γ-Secretase Is an Unusually Slow Process", Biophysical Journal, vol. 108, pp. 1229-1237, 2015. http://dx.doi.org/10.1016/j.bpj.2014.12.045
- D. Langosch, C. Scharnagl, H. Steiner, and M. Lemberg, "Understanding intramembrane proteolysis: from protein dynamics to reaction kinetics", Trends in Biochemical Sciences, vol. 40, pp. 318-327, 2015. http://dx.doi.org/10.1016/j.tibs.2015.04.001
- D. Langosch, and H. Steiner, "Substrate processing in intramembrane proteolysis by γ-secretase – the role of protein dynamics", Biological Chemistry, vol. 398, pp. 441-453, 2016. http://dx.doi.org/10.1515/hsz-2016-0269
- M.A. Fernandez, K.M. Biette, G. Dolios, D. Seth, R. Wang, and M.S. Wolfe, "Transmembrane Substrate Determinants for γ-Secretase Processing of APP CTFβ", Biochemistry, vol. 55, pp. 5675-5688, 2016. http://dx.doi.org/10.1021/acs.biochem.6b00718
- T. Sato, T. Tang, G. Reubins, J.Z. Fei, T. Fujimoto, P. Kienlen-Campard, S.N. Constantinescu, J. Octave, S. Aimoto, and S.O. Smith, "A helix-to-coil transition at the ε-cut site in the transmembrane dimer of the amyloid precursor protein is required for proteolysis", Proceedings of the National Academy of Sciences, vol. 106, pp. 1421-1426, 2009. http://dx.doi.org/10.1073/pnas.0812261106
- L. Chávez-Gutiérrez, L. Bammens, I. Benilova, A. Vandersteen, M. Benurwar, M. Borgers, S. Lismont, L. Zhou, S. Van Cleynenbreugel, H. Esselmann, J. Wiltfang, L. Serneels, E. Karran, H. Gijsen, J. Schymkowitz, F. Rousseau, K. Broersen, and B. De Strooper, "The mechanism of γ-Secretase dysfunction in familial Alzheimer disease", The EMBO Journal, vol. 31, pp. 2261-2274, 2012. http://dx.doi.org/10.1038/emboj.2012.79
- M.A. Fernandez, J.A. Klutkowski, T. Freret, and M.S. Wolfe, "Alzheimer Presenilin-1 Mutations Dramatically Reduce Trimming of Long Amyloid β-Peptides (Aβ) by γ-Secretase to Increase 42-to-40-Residue Aβ", Journal of Biological Chemistry, vol. 289, pp. 31043-31052, 2014. http://dx.doi.org/10.1074/jbc.M114.581165
- M. Szaruga, S. Veugelen, M. Benurwar, S. Lismont, D. Sepulveda-Falla, A. Lleo, N.S. Ryan, T. Lashley, N.C. Fox, S. Murayama, H. Gijsen, B. De Strooper, and L. Chávez-Gutiérrez, "Qualitative changes in human γ-secretase underlie familial Alzheimer’s disease", Journal of Experimental Medicine, vol. 212, pp. 2003-2013, 2015. http://dx.doi.org/10.1084/jem.20150892
- M. Szaruga, B. Munteanu, S. Lismont, S. Veugelen, K. Horré, M. Mercken, T.C. Saido, N.S. Ryan, T. De Vos, S.N. Savvides, R. Gallardo, J. Schymkowitz, F. Rousseau, N.C. Fox, C. Hopf, B. De Strooper, and L. Chávez-Gutiérrez, "Alzheimer’s-Causing Mutations Shift Aβ Length by Destabilizing γ-Secretase-Aβn Interactions", Cell, vol. 170, pp. 443-456.e14, 2017. http://dx.doi.org/10.1016/j.cell.2017.07.004
- T. Ikeuchi, G. Dolios, S. Kim, R. Wang, and S.S. Sisodia, "Familial Alzheimer Disease-linked Presenilin 1 Variants Enhance Production of Both Aβ1–40 and Aβ1–42 Peptides That Are Only Partially Sensitive to a Potent Aspartyl Protease Transition State Inhibitor of “γ-Secretase”", Journal of Biological Chemistry, vol. 278, pp. 7010-7018, 2003. http://dx.doi.org/10.1074/jbc.M209252200
- E. Czirr, S. Leuchtenberger, C. Dorner-Ciossek, A. Schneider, M. Jucker, E.H. Koo, C.U. Pietrzik, K. Baumann, and S. Weggen, "Insensitivity to Aβ42-lowering Nonsteroidal Anti-inflammatory Drugs and γ-Secretase Inhibitors Is Common among Aggressive Presenilin-1 Mutations", Journal of Biological Chemistry, vol. 282, pp. 24504-24513, 2007. http://dx.doi.org/10.1074/jbc.M700618200
- W. Xia, B.L. Ostaszewski, W. Kimberly, T. Rahmati, C.L. Moore, M.S. Wolfe, and D.J. Selkoe, "FAD Mutations in Presenilin-1 or Amyloid Precursor Protein Decrease the Efficacy of a γ-Secretase Inhibitor: Evidence for Direct Involvement of PS1 in the γ-Secretase Cleavage Complex", Neurobiology of Disease, vol. 7, pp. 673-681, 2000. http://dx.doi.org/10.1006/nbdi.2000.0322
- D. Chau, C.J. Crump, J.C. Villa, D.A. Scheinberg, and Y. Li, "Familial Alzheimer Disease Presenilin-1 Mutations Alter the Active Site Conformation of γ-secretase", Journal of Biological Chemistry, vol. 287, pp. 17288-17296, 2012. http://dx.doi.org/10.1074/jbc.M111.300483
- R.M. Page, A. Gutsmiedl, A. Fukumori, E. Winkler, C. Haass, and H. Steiner, "β-Amyloid Precursor Protein Mutants Respond to γ-Secretase Modulators", Journal of Biological Chemistry, vol. 285, pp. 17798-17810, 2010. http://dx.doi.org/10.1074/jbc.M110.103283
- M. Dimitrov, J. Alattia, T. Lemmin, R. Lehal, A. Fligier, J. Houacine, I. Hussain, F. Radtke, M. Dal Peraro, D. Beher, and P.C. Fraering, "Alzheimer’s disease mutations in APP but not γ-secretase modulators affect epsilon-cleavage-dependent AICD production", Nature Communications, vol. 4, 2013. http://dx.doi.org/10.1038/ncomms3246
- J. Tan, G. Mao, M. Cui, S. Kang, B. Lamb, B. Wong, M. Sy, and X. Xu, "Effects of γ‐secretase cleavage‐region mutations on APP processing and Aβ formation: interpretation with sequential cleavage and α‐helical model", Journal of Neurochemistry, vol. 107, pp. 722-733, 2008. http://dx.doi.org/10.1111/j.1471-4159.2008.05643.x
- M.C. Irizarry, F. Soriano, M. McNamara, K.J. Page, D. Schenk, D. Games, and B.T. Hyman, "Aβ Deposition Is Associated with Neuropil Changes, but not with Overt Neuronal Loss in the Human Amyloid Precursor Protein V717F (PDAPP) Transgenic Mouse", The Journal of Neuroscience, vol. 17, pp. 7053-7059, 1997. http://dx.doi.org/10.1523/JNEUROSCI.17-18-07053.1997
- C.A. Saura, S. Choi, V. Beglopoulos, S. Malkani, D. Zhang, B. Rao, S. Chattarji, R.J. Kelleher, E.R. Kandel, K. Duff, A. Kirkwood, and J. Shen, "Loss of Presenilin Function Causes Impairments of Memory and Synaptic Plasticity Followed by Age-Dependent Neurodegeneration", Neuron, vol. 42, pp. 23-36, 2004. http://dx.doi.org/10.1016/S0896-6273(04)00182-5
- J. Shen, and R.J. Kelleher, "The presenilin hypothesis of Alzheimer's disease: Evidence for a loss-of-function pathogenic mechanism", Proceedings of the National Academy of Sciences, vol. 104, pp. 403-409, 2007. http://dx.doi.org/10.1073/pnas.0608332104
- E.A. Heilig, W. Xia, J. Shen, and R.J. Kelleher, "A Presenilin-1 Mutation Identified in Familial Alzheimer Disease with Cotton Wool Plaques Causes a Nearly Complete Loss of γ-Secretase Activity", Journal of Biological Chemistry, vol. 285, pp. 22350-22359, 2010. http://dx.doi.org/10.1074/jbc.M110.116962
- T. Saito, T. Suemoto, N. Brouwers, K. Sleegers, S. Funamoto, N. Mihira, Y. Matsuba, K. Yamada, P. Nilsson, J. Takano, M. Nishimura, N. Iwata, C. Van Broeckhoven, Y. Ihara, and T.C. Saido, "Potent amyloidogenicity and pathogenicity of Aβ43", Nature Neuroscience, vol. 14, pp. 1023-1032, 2011. http://dx.doi.org/10.1038/nn.2858
- B. Kretner, J. Trambauer, A. Fukumori, J. Mielke, P. Kuhn, E. Kremmer, A. Giese, S.F. Lichtenthaler, C. Haass, T. Arzberger, and H. Steiner, "Generation and deposition of Aβ43 by the virtually inactive presenilin‐1 L435F mutant contradicts the presenilin loss‐of‐function hypothesis of Alzheimer's disease", EMBO Molecular Medicine, vol. 8, pp. 458-465, 2016. http://dx.doi.org/10.15252/emmm.201505952
- S. Veugelen, T. Saito, T. Saido, L. Chávez-Gutiérrez, and B. De Strooper, "Familial Alzheimer’s Disease Mutations in Presenilin Generate Amyloidogenic Aβ Peptide Seeds", Neuron, vol. 90, pp. 410-416, 2016. http://dx.doi.org/10.1016/j.neuron.2016.03.010
- E.A. Heilig, U. Gutti, T. Tai, J. Shen, and R.J. Kelleher, "Trans-Dominant Negative Effects of Pathogenic PSEN1 Mutations on -Secretase Activity and A Production", Journal of Neuroscience, vol. 33, pp. 11606-11617, 2013. http://dx.doi.org/10.1523/JNEUROSCI.0954-13.2013
- R. Zhou, G. Yang, and Y. Shi, "Dominant negative effect of the loss-of-function γ-secretase mutants on the wild-type enzyme through heterooligomerization", Proceedings of the National Academy of Sciences, vol. 114, pp. 12731-12736, 2017. http://dx.doi.org/10.1073/pnas.1713605114
- L. Sun, R. Zhou, G. Yang, and Y. Shi, "Analysis of 138 pathogenic mutations in presenilin-1 on the in vitro production of Aβ42 and Aβ40 peptides by γ-secretase", Proceedings of the National Academy of Sciences, vol. 114, 2016. http://dx.doi.org/10.1073/pnas.1618657114
- M. Cacquevel, L. Aeschbach, J. Houacine, and P.C. Fraering, "Alzheimer's Disease-Linked Mutations in Presenilin-1 Result in a Drastic Loss of Activity in Purified γ-Secretase Complexes", PLoS ONE, vol. 7, pp. e35133, 2012. http://dx.doi.org/10.1371/journal.pone.0035133
- B. Wang, W. Yang, W. Wen, J. Sun, B. Su, B. Liu, D. Ma, D. Lv, Y. Wen, T. Qu, M. Chen, M. Sun, Y. Shen, and X. Zhang, "γ-Secretase Gene Mutations in Familial Acne Inversa", Science, vol. 330, pp. 1065-1065, 2010. http://dx.doi.org/10.1126/science.1196284
- M.S. Wolfe, "γ‐Secretase inhibitors and modulators for Alzheimer’s disease", Journal of Neurochemistry, vol. 120, pp. 89-98, 2011. http://dx.doi.org/10.1111/j.1471-4159.2011.07501.x
- T.E. Golde, E.H. Koo, K.M. Felsenstein, B.A. Osborne, and L. Miele, "γ-Secretase inhibitors and modulators", Biochimica et Biophysica Acta (BBA) - Biomembranes, vol. 1828, pp. 2898-2907, 2013. http://dx.doi.org/10.1016/j.bbamem.2013.06.005
- M.S. Shearman, D. Beher, E.E. Clarke, H.D. Lewis, T. Harrison, P. Hunt, A. Nadin, A.L. Smith, G. Stevenson, and J.L. Castro, "L-685,458, an Aspartyl Protease Transition State Mimic, Is a Potent Inhibitor of Amyloid β-Protein Precursor γ-Secretase Activity", Biochemistry, vol. 39, pp. 8698-8704, 2000. http://dx.doi.org/10.1021/bi0005456
- H.F. Dovey, V. John, J.P. Anderson, L.Z. Chen, P. De Saint Andrieu, L.Y. Fang, S.B. Freedman, B. Folmer, E. Goldbach, E.J. Holsztynska, K.L. Hu, K.L. Johnson‐Wood, S.L. Kennedy, D. Kholodenko, J.E. Knops, L.H. Latimer, M. Lee, Z. Liao, I.M. Lieberburg, R.N. Motter, L.C. Mutter, J. Nietz, K.P. Quinn, K.L. Sacchi, P.A. Seubert, G.M. Shopp, E.D. Thorsett, J.S. Tung, J. Wu, S. Yang, C.T. Yin, D.B. Schenk, P.C. May, L.D. Altstiel, M.H. Bender, L.N. Boggs, T.C. Britton, J.C. Clemens, D.L. Czilli, D.K. Dieckman‐McGinty, J.J. Droste, K.S. Fuson, B.D. Gitter, P.A. Hyslop, E.M. Johnstone, W. Li, S.P. Little, T.E. Mabry, F.D. Miller, B. Ni, J.S. Nissen, W.J. Porter, B.D. Potts, J.K. Reel, D. Stephenson, Y. Su, L.A. Shipley, C.A. Whitesitt, T. Yin, and J.E. Audia, "Functional gamma‐secretase inhibitors reduce beta‐amyloid peptide levels in brain", Journal of Neurochemistry, vol. 76, pp. 173-181, 2001. http://dx.doi.org/10.1046/j.1471-4159.2001.00012.x
- E. Siemers, M. Skinner, R.A. Dean, C. Gonzales, J. Satterwhite, M. Farlow, D. Ness, and P.C. May, "Safety, Tolerability, and Changes in Amyloid β Concentrations After Administration of a γ-Secretase Inhibitor in Volunteers", Clinical Neuropharmacology, vol. 28, pp. 126-132, 2005. http://dx.doi.org/10.1097/01.wnf.0000167360.27670.29
- B.K. Hadland, N.R. Manley, D. Su, G.D. Longmore, C.L. Moore, M.S. Wolfe, E.H. Schroeter, and R. Kopan, "γ-Secretase inhibitors repress thymocyte development", Proceedings of the National Academy of Sciences, vol. 98, pp. 7487-7491, 2001. http://dx.doi.org/10.1073/pnas.131202798
- P. Doerfler, M.S. Shearman, and R.M. Perlmutter, "Presenilin-dependent γ-secretase activity modulates thymocyte development", Proceedings of the National Academy of Sciences, vol. 98, pp. 9312-9317, 2001. http://dx.doi.org/10.1073/pnas.161102498
- A. Geling, H. Steiner, M. Willem, L. Bally‐Cuif, and C. Haass, "A γ‐secretase inhibitor blocks Notch signaling in vivo and causes a severe neurogenic phenotype in zebrafish", EMBO reports, vol. 3, pp. 688-694, 2002. http://dx.doi.org/10.1093/embo-reports/kvf124
- G.T. Wong, D. Manfra, F.M. Poulet, Q. Zhang, H. Josien, T. Bara, L. Engstrom, M. Pinzon-Ortiz, J.S. Fine, H.J. Lee, L. Zhang, G.A. Higgins, and E.M. Parker, "Chronic Treatment with the γ-Secretase Inhibitor LY-411,575 Inhibits β-Amyloid Peptide Production and Alters Lymphopoiesis and Intestinal Cell Differentiation", Journal of Biological Chemistry, vol. 279, pp. 12876-12882, 2004. http://dx.doi.org/10.1074/jbc.M311652200
- E.R. Andersson, and U. Lendahl, "Therapeutic modulation of Notch signalling — are we there yet?", Nature Reviews Drug Discovery, vol. 13, pp. 357-378, 2014. http://dx.doi.org/10.1038/nrd4252
- K.W. Gillman, J.E. Starrett, M.F. Parker, K. Xie, J.J. Bronson, L.R. Marcin, K.E. McElhone, C.P. Bergstrom, R.A. Mate, R. Williams, J.E. Meredith, C.R. Burton, D.M. Barten, J.H. Toyn, S.B. Roberts, K.A. Lentz, J.G. Houston, R. Zaczek, C.F. Albright, C.P. Decicco, J.E. Macor, and R.E. Olson, "Discovery and Evaluation of BMS-708163, a Potent, Selective and Orally Bioavailable γ-Secretase Inhibitor", ACS Medicinal Chemistry Letters, vol. 1, pp. 120-124, 2010. http://dx.doi.org/10.1021/ml1000239
- R.S. Doody, R. Raman, M. Farlow, T. Iwatsubo, B. Vellas, S. Joffe, K. Kieburtz, F. He, X. Sun, R.G. Thomas, P.S. Aisen, E. Siemers, G. Sethuraman, and R. Mohs, "A Phase 3 Trial of Semagacestat for Treatment of Alzheimer's Disease", New England Journal of Medicine, vol. 369, pp. 341-350, 2013. http://dx.doi.org/10.1056/NEJMoa1210951
- V. Coric, S. Salloway, C.H. van Dyck, B. Dubois, N. Andreasen, M. Brody, C. Curtis, H. Soininen, S. Thein, T. Shiovitz, G. Pilcher, S. Ferris, S. Colby, W. Kerselaers, R. Dockens, H. Soares, S. Kaplita, F. Luo, C. Pachai, L. Bracoud, M. Mintun, J.D. Grill, K. Marek, J. Seibyl, J.M. Cedarbaum, C. Albright, H.H. Feldman, and R.M. Berman, "Targeting Prodromal Alzheimer Disease With Avagacestat", JAMA Neurology, vol. 72, pp. 1324, 2015. http://dx.doi.org/10.1001/jamaneurol.2015.0607
- C.J. Crump, S.V. Castro, F. Wang, N. Pozdnyakov, T.E. Ballard, S.S. Sisodia, K.R. Bales, D.S. Johnson, and Y. Li, "BMS-708,163 Targets Presenilin and Lacks Notch-Sparing Activity", Biochemistry, vol. 51, pp. 7209-7211, 2012. http://dx.doi.org/10.1021/bi301137h
- N. Kakuda, S. Funamoto, S. Yagishita, M. Takami, S. Osawa, N. Dohmae, and Y. Ihara, "Equimolar Production of Amyloid β-Protein and Amyloid Precursor Protein Intracellular Domain from β-Carboxyl-terminal Fragment by γ-Secretase", Journal of Biological Chemistry, vol. 281, pp. 14776-14786, 2006. http://dx.doi.org/10.1074/jbc.M513453200
- C. McLendon, T. Xin, C. Ziani-Cherif, M.P. Murphy, K.A. Findlay, P.A. Lewis, I. Pinnix, K. Sambamurti, R. Wang, A. Fauq, and T.E. Golde, "Cell‐free assays for γ‐secretase activity", The FASEB Journal, vol. 14, pp. 2383-2386, 2000. http://dx.doi.org/10.1096/fj.00-0286fje
- I. Pinnix, U. Musunuru, H. Tun, A. Sridharan, T. Golde, C. Eckman, C. Ziani-Cherif, L. Onstead, and K. Sambamurti, "A Novel γ-Secretase Assay Based on Detection of the Putative C-terminal Fragment-γ of Amyloid β Protein Precursor", Journal of Biological Chemistry, vol. 276, pp. 481-487, 2001. http://dx.doi.org/10.1074/jbc.M005968200
- B. De Strooper, and L. Chávez Gutiérrez, "Learning by Failing: Ideas and Concepts to Tackle γ-Secretases in Alzheimer's Disease and Beyond", Annual Review of Pharmacology and Toxicology, vol. 55, pp. 419-437, 2015. http://dx.doi.org/10.1146/annurev-pharmtox-010814-124309
- S. Weggen, J.L. Eriksen, P. Das, S.A. Sagi, R. Wang, C.U. Pietrzik, K.A. Findlay, T.E. Smith, M.P. Murphy, T. Bulter, D.E. Kang, N. Marquez-Sterling, T.E. Golde, and E.H. Koo, "A subset of NSAIDs lower amyloidogenic Aβ42 independently of cyclooxygenase activity", Nature, vol. 414, pp. 212-216, 2001. http://dx.doi.org/10.1038/35102591
- S. Weggen, J.L. Eriksen, S.A. Sagi, C.U. Pietrzik, T. Golde, and E.H. Koo, "Aβ42-lowering Nonsteroidal Anti-inflammatory Drugs Preserve Intramembrane Cleavage of the Amyloid Precursor Protein (APP) and ErbB-4 Receptor and Signaling through the APP Intracellular Domain", Journal of Biological Chemistry, vol. 278, pp. 30748-30754, 2003. http://dx.doi.org/10.1074/jbc.M304824200
- S. Weggen, J.L. Eriksen, S.A. Sagi, C.U. Pietrzik, V. Ozols, A. Fauq, T. Golde, and E.H. Koo, "Evidence That Nonsteroidal Anti-inflammatory Drugs Decrease Amyloid β42 Production by Direct Modulation of γ-Secretase Activity", Journal of Biological Chemistry, vol. 278, pp. 31831-31837, 2003. http://dx.doi.org/10.1074/jbc.M303592200
- B.D. Moore, J. Martin, L. de Mena, J. Sanchez, P.E. Cruz, C. Ceballos-Diaz, T.B. Ladd, Y. Ran, Y. Levites, T.L. Kukar, J.J. Kurian, R. McKenna, E.H. Koo, D.R. Borchelt, C. Janus, D. Rincon-Limas, P. Fernandez-Funez, and T.E. Golde, "Short Aβ peptides attenuate Aβ42 toxicity in vivo", Journal of Experimental Medicine, vol. 215, pp. 283-301, 2017. http://dx.doi.org/10.1084/jem.20170600
- A. Lleó, O. Berezovska, L. Herl, S. Raju, A. Deng, B.J. Bacskai, M.P. Frosch, M. Irizarry, and B.T. Hyman, "Nonsteroidal anti-inflammatory drugs lower Aβ42 and change presenilin 1 conformation", Nature Medicine, vol. 10, pp. 1065-1066, 2004. http://dx.doi.org/10.1038/nm1112
- K. Takeo, S. Tanimura, T. Shinoda, S. Osawa, I.K. Zahariev, N. Takegami, Y. Ishizuka-Katsura, N. Shinya, S. Takagi-Niidome, A. Tominaga, N. Ohsawa, T. Kimura-Someya, M. Shirouzu, S. Yokoshima, S. Yokoyama, T. Fukuyama, T. Tomita, and T. Iwatsubo, "Allosteric regulation of γ-secretase activity by a phenylimidazole-type γ-secretase modulator", Proceedings of the National Academy of Sciences, vol. 111, pp. 10544-10549, 2014. http://dx.doi.org/10.1073/pnas.1402171111
- M.G. Bursavich, B.A. Harrison, and J. Blain, "Gamma Secretase Modulators: New Alzheimer’s Drugs on the Horizon?", Journal of Medicinal Chemistry, vol. 59, pp. 7389-7409, 2016. http://dx.doi.org/10.1021/acs.jmedchem.5b01960
- M.Z. Kounnas, A.M. Danks, S. Cheng, C. Tyree, E. Ackerman, X. Zhang, K. Ahn, P. Nguyen, D. Comer, L. Mao, C. Yu, D. Pleynet, P.J. Digregorio, G. Velicelebi, K.A. Stauderman, W.T. Comer, W.C. Mobley, Y. Li, S.S. Sisodia, R.E. Tanzi, and S.L. Wagner, "Modulation of γ-Secretase Reduces β-Amyloid Deposition in a Transgenic Mouse Model of Alzheimer's Disease", Neuron, vol. 67, pp. 769-780, 2010. http://dx.doi.org/10.1016/j.neuron.2010.08.018
- G. Siegel, H. Gerber, P. Koch, O. Bruestle, P.C. Fraering, and L. Rajendran, "The Alzheimer’s Disease γ-Secretase Generates Higher 42:40 Ratios for β-Amyloid Than for p3 Peptides", Cell Reports, vol. 19, pp. 1967-1976, 2017. http://dx.doi.org/10.1016/j.celrep.2017.05.034
- J. Sevigny, P. Chiao, T. Bussière, P.H. Weinreb, L. Williams, M. Maier, R. Dunstan, S. Salloway, T. Chen, Y. Ling, J. O’Gorman, F. Qian, M. Arastu, M. Li, S. Chollate, M.S. Brennan, O. Quintero-Monzon, R.H. Scannevin, H.M. Arnold, T. Engber, K. Rhodes, J. Ferrero, Y. Hang, A. Mikulskis, J. Grimm, C. Hock, R.M. Nitsch, and A. Sandrock, "The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease", Nature, vol. 537, pp. 50-56, 2016. http://dx.doi.org/10.1038/nature19323
ACKNOWLEDGMENTS
This work was supported by the DFG (FOR2290; H.S.) and in part by AMED (JP16dm0107056h0001; M.O., S.T.), the Collaborative Research Project (2017-2808) of the Brain Research Institute, Niigata University (M.O.), Grants-in-Aid for Scientific Research (T254617680; M.O.), JSPS KAKENHI (JP17K10304; A.F.), the Research Foundation for Dementia of Osaka (A.F.), the SENSHIN Medical Research Foundation (A.F., S.T.), and the Takeda Science Foundation (A.F.).
COPYRIGHT
© 2018

Making the final cut: pathogenic amyloid-β peptide generation by γ-secretase by Steiner et al. is licensed under a Creative Commons Attribution 4.0 International License.


