Reviews:
Cell Stress, Vol. 2, No. 6, pp. 125 - 140; doi: 10.15698/cst2018.06.139
Nucleolar Stress: hallmarks, sensing mechanism and diseases

1 Shanghai Key Laboratory for Tumor Microenvironment and Inflammation, Department of Biochemistry and Molecular Cell Biology, Shanghai Jiao Tong University School of Medicine, 280 South Chongqing Road, Shanghai, 200025, China.
2 Shanghai Key Laboratory for Prevention and Treatment of Bone and Joint Diseases with Integrated Chinese-Western Medicine, Shanghai Institute of Traumatology and Orthopedics, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, 197 Ruijin 2nd Road, Shanghai, 200025, China.
Keywords: nucleolar stress, ribosome biogenesis, NPM1, translocation, p53, MDM2, ribosomal proteins.
Abbreviations:
Act.D – Actinomycin D,
AD – Alzheimer’s disease,
AML – acute myeloid leukemia,
ARF – alternative reading frame,
CBS – cystathionine-β-synthase,
DFC – dense FC,
FC – fibrillar center,
GC – granular component,
KO – knockout,
PD – Parkinson’s disease,
Pol I – RNA Polymerase I
rDNA – ribosomal DNA,
RP – ribosomal protein,
rRNA – ribosomal RNA,
TF – transcription factor.
Received originally: 03/10/2017 Received in revised form: 02/04/2018
Accepted: 11/04/2018
Published: 10/05/2018
Correspondence:
Jing Yi, Shanghai Key Laboratory for Tumor Microenvironment and Inflammation, Department of Biochemistry and Molecular Cell Biology, Shanghai Jiao Tong University School of Medicine, 280 South Chongqing Road, Shanghai, 200025, China yijing@shsmu.edu.cn
Conflict of interest statement: There is no conflict of interest.
Please cite this article as: Kai Yang, Jie Yang and Jing Yi (2018). Nucleolar Stress: hallmarks, sensing mechanism and diseases. Cell Stress 2(6): 125-140. doi: 10.15698/cst2018.06.139
Abstract
The nucleolus is a prominent subnuclear compartment, where ribosome biosynthesis takes place. Recently, the nucleolus has gained attention for its novel role in the regulation of cellular stress. Nucleolar stress is emerging as a new concept, which is characterized by diverse cellular insult-induced abnormalities in nucleolar structure and function, ultimately leading to activation of p53 or other stress signaling pathways and alterations in cell behavior. Despite a number of comprehensive reviews on this concept, straightforward and clear-cut way criteria for a nucleolar stress state, regarding the factors that elicit this state, the morphological and functional alterations as well as the rationale for p53 activation are still missing. Based on literature of the past two decades, we herein summarize the evolution of the concept and provide hallmarks of nucleolar stress. Along with updated information and thorough discussion of existing confusions in the field, we pay particular attention to the current understanding of the sensing mechanisms, i.e., how stress is integrated by p53. In addition, we propose our own emphasis regarding the role of nucleolar protein NPM1 in the hallmarks of nucleolar stress and sensing mechanisms. Finally, the links of nucleolar stress to human diseases are briefly and selectively introduced.
INTRODUCTION
The nucleolus is a subnuclear compartment, which is primarily known for its role in ribosome biosynthesis. Within nucleoli, genes for ribosomal RNA (rDNA) are arranged in arrays of tandem repeats, precursors of ribosomal RNA (rRNA) are transcribed by the RNA polymerase I (Pol I) and processed, before the ribosomal proteins are incorporated and ribosomal subunits are assembled. However, during the past two decades, researchers have demonstrated that this is in fact an organelle having multiple complex functions. Several lines of evidence have revealed the most intriguing novel role of the nucleolus as a sensor for various cellular stresses, eventually leading to the concept of ‘nucleolar stress’. Numerous studies have listed triggers for nucleolar stress, characterized morphological and functional alterations, and dissected the molecules that induce activation of p53 signaling or other stress-responsive pathways. While these studies enriched our understanding of the general features of nucleolar stress, many questions, especially those regarding the sensing mechanism, remain unanswered. In this review we summarize the literature describing the evolution of the concept, focusing on the hallmarks and the sensing mechanisms for nucleolar stress. We will also discuss some key ambiguities in this field.
–
THE NUCLEOLUS: A MULTIFUNCTIONAL ORGANELLE AND ITS ROLE IN CELLULAR STRESS
Starting in the 1990s, evidence has gradually accumulated that transcripts other than rRNAs can be produced and processed in the nucleolus [1]. Indeed, a world of small nucleolar RNAs (snoRNAs) was discovered; these small RNAs are important for modifications of rRNA, tRNA and many small nuclear RNAs [2]. In addition, the nucleolus was also found to functionally interact with Cajal bodies, other nuclear sub-compartments, to promote non-ribosomal RNA species maturation [3]. Around the new millennium proteomics approaches were massively applied, which led to the identification of over 4500 nucleolar proteins in the nucleolus, of which only 30% have a clear relationship with ribosome biogenesis [4]. This intriguingly showed that the nucleolus is involved in such diverse cellular events as signal recognition particle assembly, cell cycle regulation, DNA replication and repair, control of aging, response to viral infection, modulation of telomerase, and others [5][6]. Overall, these clues indicate that the nucleolus is a multifunctional organelle.
–
In parallel, many researchers noticed that some nucleolus-enriched proteins are frequently shuttled between the nucleolus and nucleoplasm. Remarkably, a number of nucleolar proteins translocate to the nucleoplasm in response to various stress conditions [7][8][9][10][11][12][13]. This phenomenon was initially observed when ribosome biogenesis was blocked by Actinomycin D (Act.D), a Pol I inhibitor [14], and soon thereafter was also found in cells exposed to cytotoxic agents [9][15], viral proteins [16], ultraviolet radiation [10][17], heat shock [15] and agents inducing DNA damage [18][19], apoptosis or senescence [20][21][22].
–
The links between the nucleolus and cellular stress were eventually proposed based on the findings that the nucleolus participates in regulating the abundance of the stress responsive protein p53 [7][23][24][25][26][27][28]. In summary, the notion that the nucleolus plays a role in regulating cellular stress states represents at least two aspects of the same idea: p53 activation by nucleolar proteins. One hypothesis emphasizes that the nucleolus is a sensor for cellular stresses, in which stress-induced nucleoplasmic translocation of nucleolar proteins, such as NPM1 [29][30][31] and GLTSCR2 [32] initiates p53 activation. The other hypothesis proposes the engagement of ribosomal proteins (RPs) mainly RPL5 [33], RPL11 [34] and RPL23 [35], or nucleolus-resident proteins (e.g. ARF [36] among others) in p53 interaction with its negative regulator MDM2, but placing less emphasis on their translocation.
THE DEFINITION AND EVOLUTION OF THE CONCEPT OF NUCLEOLAR STRESS
Although the term ‘nucleolar stress’ is increasingly used, the exact description varies and (still) evolves, thus a precise definition has not yet been universally approved. The term was originally referred to the stressful events that impair the homeostasis of ribosome biogenesis and activate the cellular stress response. Therefore, nucleolar stress has also been referred to as ‘ribosomal stress’ or ‘ribotoxic stress’ [37][38][39][40], as typical inducers are the Pol I inhibitor Act.D [14], or aberrant expression of nucleolar proteins [25], which also impair ribosomal function. In general, nucleolar stress is now used to describe various stressor-induced impairments in nucleolar morphology and function that ultimately lead to disturbances in cell homeostasis through activation of p53 or other stress signaling (Figure 1).
–
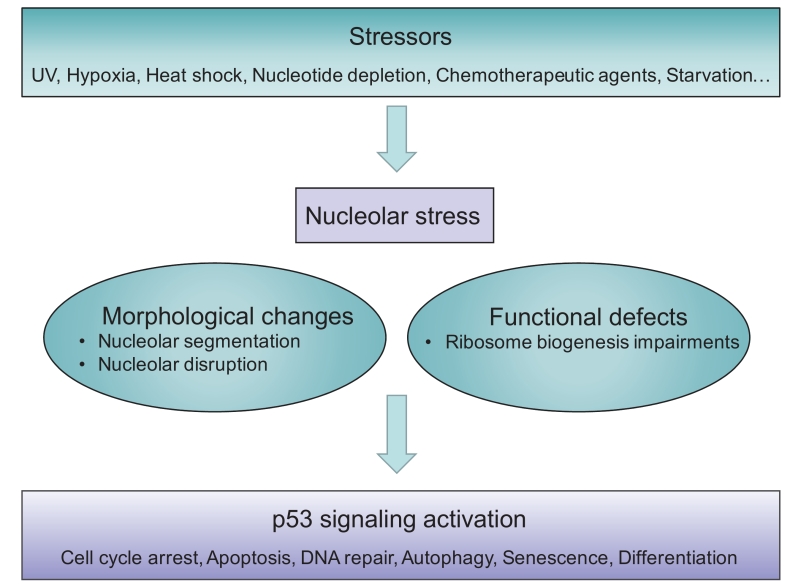 | FIGURE 1: Schematic illustration of nucleolar stress. Various stressors induce nucleolar stress, accompanied by morphological changes and functional defects, ultimately resulting in activation of p53 signaling pathway and altered cell behavior. |
–
The idea that cell cycle progression may depend on some aspect of ribosome biogenesis was first implied in early studies on the cell cycle [41]. In higher eukaryotes, Volarevic proved that deletion of RPS6 in the liver of adult mice abolished 40S ribosome biogenesis and inhibited cell proliferation following partial hepatectomy [42]. Identifying the correlation of interference of the nucleolar protein Bop1 and p53-dependent cell cycle arrest, Pestov et al. proposed that perturbation in ribosome biogenesis may cause nucleolar stress, leading to cell cycle arrest in a p53-dependent manner [25]. Indeed, this model of nucleolar stress, probably the first of its kind, is consistent with many observations under diverse p53-activating stressors [28][29][43].
–
The pioneering work conducted by Rubbi and Milner significantly solidified the notion of nucleolar stress [29]. They aimed at resolving the puzzle of how signals in a large variety of cellular stress situations can be integrated by a single molecule, namely p53. A common phenomenon in all p53-inducing stresses is nucleolar disruption. Based on a comparative meta-analysis of diverse stimuli that activate p53 signaling and induce nucleolar alteration, they hypothesized that the impairment of nucleolar function might stabilize p53. In fact, activation of p53 is induced by a wide range of cellular stresses, aside from the Pol I inhibitor Act.D, which all cause disruption of nucleolar organization. The translocation of nucleophosmin (NPM1, or B23), an abundant nucleolar protein that is the most frequently reported to move to the nucleoplasm and cytoplasm upon various cellular insults was set as the criterion for nucleolus disruption. Rubbi and Milner demonstrated that NPM1 translocation, or nucleolus disruption following micropore UV irradiation over the nucleoli occurs prior to and independent of p53 induction. Alternatively, p53 response can be induced by interfering with nucleolar function using an antibody against the nucleolar protein UBF (upstream binding factor) in the absence of any genotoxic insult. Therefore, the model they proposed was the only one that could provide a unifying and coherent explanation for the action of all known p53-stabilizing agents.
THE HALLMARKS OF NUCLEOLAR STRESS
Following the principle of cellular events in response to stress conditions, we describe the following elements as the hallmarks of nucleolar stress.
–
Ribosome biogenesis insults and a wide range of stimuli as stressors
Ribosome biogenesis comprises multiple steps accomplished in three distinct subnucleolar components, from Pol I transcription initiation to pre-rRNA processing and ribosomal assembly. Any error that causes disturbance in ribosome biogenesis will lead to nucleolar stress.
–
In fact, deletion or aberrant expression of a number of ribosomal proteins induce p53 stabilization and activation via disruption of ribosome biogenesis: Pestov et al. found that perturbation of the nucleolar protein Bop1 activity could induce ribosome biogenesis impairment, followed by a p53-dependent cell cycle arrest [25]. Genetic inactivation of TIF-1A, a basal transcription initiation factor for Pol I, leads to nucleolar disruption, cell cycle arrest and p53-mediated apoptosis [44]. Depletion of importin 7 (IPO7) or exportin 1 (XPO1) proteins impairs ribosome biogenesis and also initiates p53-dependent cell cycle arrest [45]. Microinjection of specific monoclonal antibodies against transcription factor UBF inhibits rRNA transcription and leads to p53 stabilization [29]. Overall, systematic screening analysis revealed an extensive connection of p53 stabilization with nucleolar disruption induced by ribosomal protein depletion [46].
–
The chemotherapeutic agent Act.D is the mostly used nucleolar stress inducer. It may inhibit three individual RNA polymerases at different concentrations [47]. It is believed that Act.D can induce DNA damage and inhibit general transcription at high concentrations, such as 430 nM, but selectively inhibits Pol I and induces ribosomal stress at low dose like 5 nM [34].
–
Strikingly, as summarized by Rubbi and Milner, stressful conditions that can induce p53 activation can all induce nucleolar stress: these include UV light, hypoxia, heat shock, nucleotide depletion and various chemotherapeutic agents [29]. These stimuli were confirmed to simultaneously induce nucleolar stress and p53 activation by subsequent studies [48][49]. The rationale that stressors of diverse nature can all induce nucleolar stress has not been adequately discussed, thus has remained unknown for a long time. Recently we found that common cellular insults that are able to induce p53 activation can also induce the translocation of NPM1, a hallmark of nucleolar stress, in a reactive oxygen species (ROS)-dependent manner. Moreover, our study added nutrient starvation and direct exposure to hydrogen peroxide (H2O2) to the growing list of nucleolar stress inducers [31].
–
In summary, the reported factors that induce nucleolar stress can be classified into two categories: canonical and non-canonical. The former points to those affecting homeostasis of ribosome biogenesis, whereas the latter includes a wide range of general cellular insults (Figure 2).
–
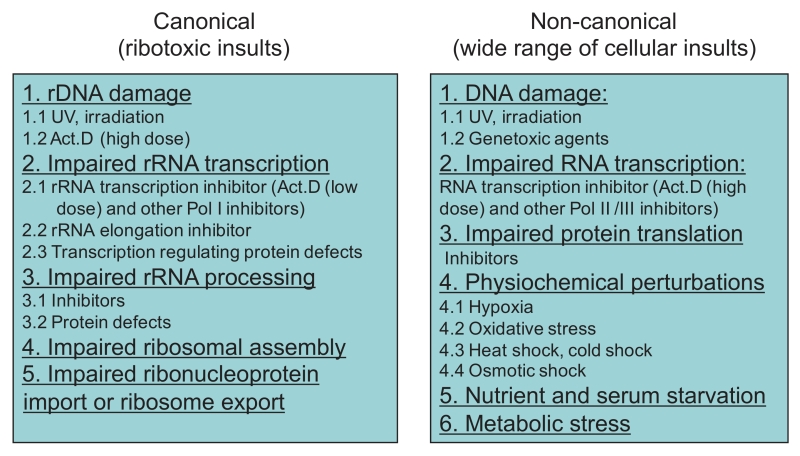 | FIGURE 2: Stressors eliciting nucleolar stress. Two categories of nucleolar stress inducers are direct ribotoxic insults and a wide range of cellular insults. |
–
Nucleoplasmic translocation of nucleolar proteins
Unlike membrane-limited organelles, there is no structural barrier between the nucleolus and the surrounding nucleoplasm. As a consequence, any soluble molecule can potentially traffic in and out of the nucleolus in a relatively free manner. This shuttling might occur at basal levels under non-stressful ‘resting’ conditions, but is significantly increased under various stress conditions. Nucleolar stress causes a lot of nucleolar molecules to redistribute in the nucleus, or in other words, to be released from the nucleolus to the nucleoplasm. This translocation or redistribution is thus considered as an indicator of nucleolar stress.
–
NPM1
NPM1 (also known as B23, nucleophosmin, numatrin or NO38) is the most abundant protein in the nucleolus [50][51] and under diverse scenarios can dynamically shuttle both within nucleoli and between the nucleolus and the nucleoplasm or the cytoplasm [52][53][54]. The known functions of this protein include the interaction with a plethora of macromolecules, for instance, Rb in the nucleus [55] and BAX in the cytoplasm [56], and chaperoning activity protecting proteins from aggregation in the crowded nucleolar environment [57][58]. At exit of mitosis, NPM1, among other ribosomal processing proteins, undergoes bidirectional traffic between incipient nucleoli and perinucleolar bodies, which may contribute to nucleolar assembly in early G1 phase [59]. NPM1 is also responsible for the nuclear export of ribosomal protein L5 [53].
–
Studies on NPM1 nucleoplasmic translocation are mostly based on contributions made by Busch, Chan and Yung [52]. Although the concept of nucleolar stress had not been proposed by then, the conditions under which they found NPM1 nucleoplasmic translocation belonged to general cellular stress or typical ribosomal stress. With immunofluorescence technology, they first found that upon 48 hours serum-free medium starvation, NPM1 was diminished in nucleoli and appeared in the nucleoplasm, whereas refeeding of serum-containing medium relocated NPM1 protein to nucleolus [60], indicating a reversible nucleoplasmic translocation capability of NPM1. They also noticed that ribosomal transcription inhibitors, such as Act.D, were all able to induce NPM1 nucleoplasmic translocation [52].
–
Furthermore, a wide range of anticancer agents aside from specific inhibitors also induce NPM1 nucleoplasmic translocation, including the inosine-5′-monophosphate (IMP) dehydrogenase inhibitor tiazofurin [61], DNA topoisomerase II (topo II) inhibitors doxorubicin and daunomycin [8][62], topo I inhibitors mitomycin C and camptothecin [63][64], phosphatidylinositol kinase inhibitor toyocamycin [65] and JAK/STAT3 inhibitor cucurbitacin B [21]. Even an iron chelator deferoxamine which showed anti-proliferation effects [66], UV radiation [30], viral infection [30], hypoxia and oxidative stress (H2O2) [31] all lead to nucleoplasmic translocation of NPM1.
–
Among the observations of nucleoplasmic translocation of NPM1, a great part described the association of this event with p53 signaling activation. Using the anti-cancer drug daunomycin, Chan et al. found a relationship between NPM1-translocation and apoptosis [62]. Then, Rubbi et al. elegantly proved a relationship between NPM1 translocation and p53 activation using different doses of UV irradiation in nucleolar areas and different anti-cancer drugs [29]. Furthermore, Kurki et al. found that UV irradiation-induced p53 activation was dependent on NPM1 interaction with HDM2, suggesting that NPM1 activates p53 in a regulated fashion [30]. Recently, we uncovered a redox mechanism of NPM1 for sensing nucleolar stress that causes p53 accumulation and activation [31].
–
Therefore, as a most frequent event, NPM1 translocation should be regarded as a conspicuous hallmark of nucleolar stress.
–
Other nucleolar proteins
The following nucleolar proteins exhibit nucleoplasmic translocation under particular types of nucleolar stresses. However, their translocations are not yet explored as universally under many stress conditions as NPM1.
–
Nucleolin, alias C23, a DNA and RNA binding protein, is essential for pre-RNA transcription, folding, processing and assembly [67]. Cyclin-dependent kinase inhibitor roscovitine induced both nucleolin translocation and nuclear accumulation of p53 [7]. Nucleostemin that functions in pre-RNA processing was also translocated to the nucleoplasm under doxorubicin and Act.D treatments in neonatal rat cardiomyocytes, which occurred concurrently with p53 accumulation [20].
–
PICT1, also called glioma tumor-suppressor candidate region gene 2 (GLTSCR2), a candidate tumor suppressor, is translocated to the nucleoplasm in response to hypoxia or Act.D treatment and enhanced p53 stability through ARF-independent direct physical interaction with p53 [32].
–
Morphological descriptions for nucleolar stress
According to the classical ‘tripartite’ model, the three main events for ribosome biogenesis, i.e., pre-rRNA transcription, processing, and ribosomal subunit assembly, are reflected in three distinct subnucleolar compartments named the fibrillar center (FC), the dense fibrillar component (DFC), and the granular component (GC). It is generally accepted that pre-rRNA is transcribed from rDNA in the FC or at the border between the FC and DFC. FCs are enriched in components of the RNA Pol I machinery, such as UBF, whereas the DFC harbors pre-rRNA processing factors, such as the snoRNAs and snoRNP proteins, fibrillarin and Nop58. Both the FC and the DFC are surrounded by the GC, where pre-ribosome subunit assembly takes place (reviewed in [5][68]). The morphology and size of nucleoli are linked to nucleolar activity, which are inevitably altered under stress conditions, showing a variety of reorganization.
–
The widely used descriptions of morphological alterations in nucleolar stress are based on immunostaining using fluorescence-labeled antibodies against known markers of the nucleolus, such as NPM1, fibrillarin, and UBF, that visualizes their redistribution under nucleolar stress conditions. Typically, under Act.D treatment, these nucleolar marker proteins aggregate in different regions, migrate towards the nucleolar periphery, or even distribute to the nucleoplasm, finally forming distinct staining structures, the nucleolar caps, and spots or foci that spread in the nucleoplasm. Nucleolar caps are shaped around the nucleolar remnant; they can be also formed by nucleoplasmic proteins (mostly RNA-binding proteins) or the Cajal body marker, coilin [69].
–
These kinds of morphological alterations have been designated as ‘nucleolar segregation’ or ‘nucleolar disintegration’ to reflect a state of loss of nucleolar integrity. As reviewed by Boulon et al., segregation is characterized by the condensation and subsequent separation of the FC and GC, together with the formation of nucleolar caps [49]. Nucleolar segregation is thought to be different from nucleolar fragmentation which occurs following inhibition of either RNA Pol II (but not I) or protein kinases [7][70] and leads to unraveling of the FC. Viral infections can also cause specific changes in nucleolar morphology, such as an increase in nucleolar size [71].
–
Nucleolar segregation has emerged as an indicator of nucleolar stress induced by, in particular, agents that cause rDNA damage and rRNA transcription impairment [72][73][74]. For instance, chemotherapeutic agents that inhibit rRNA transcription and early processing steps, but not late processing steps, lead to the loss of nucleolar integrity, which is marked by NPM1 translocation to the nucleoplasm [75]. Meanwhile, after chemotherapeutic agent treatment that mostly inhibit early and late rRNA processing steps, fibrillarin and the ribosomal biogenesis factor pescadillo are translocated into distinct morphological subnuclear structures, namely nuclear spots and nucleolar caps structures or even form ‘necklace’ structures (especially for fibrillarin) [75]. Interestingly, it was noticed that the nucleolar integrity was maintained for drugs without inhibitory activity on ribosome biogenesis [76].
–
A more frequently used term for morphological alteration is ‘nucleolar disruption‘ that was initiated by Rubbi and Milner, meaning the dispersion of the nucleolar structure [29]. Notably, this morphological description is characterized by the release of nucleolar proteins, in particular, NPM1, to the nucleoplasm, being seen as a homogenous staining in nucleoplasmic area or even whole nuclear area [29][31][52][60] (Figure 3). This characteristic alteration may simultaneously occur with the formation of nucleolar caps and foci, but is frequently used to independently indicate nucleolar stress.
–
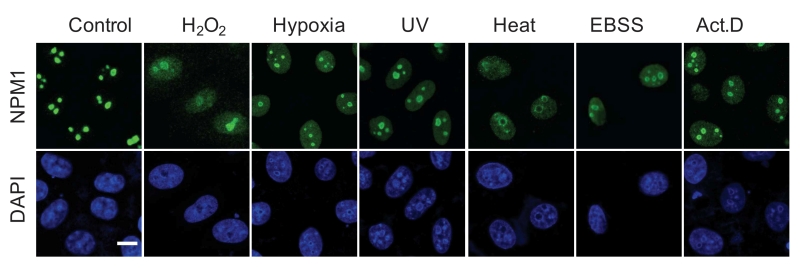 | FIGURE 3: NPM1 translocation under nucleolar stress. Representative images of NPM1 translocation in HeLa cells under various nucleolar stresses, including H2O2 (500 mM, 30 min), hypoxia (1% O2, 1 h), UV irradiation (100 J m-2), heat-shock (42°C, 30 min), EBSS starvation (6 h) and Act.D (8 nM, 1 h), examined by immunofluorescence with anti-NPM1 antibody. Bar, 5 mm. Image from ref [31]. |
–
Electron microscopic observations showed a segregation of nucleolar components when cells were exposed to the antitumoral drug VM26, a specific inhibitor of topoisomerase II [77]. Drug treatment caused double-strand breaks in the tandem repeat rDNA genes, leading to rDNA fragmentation, which might explain the morphology with a segregation of nucleolar components. Double immuno-gold labeling demonstrated redistribution of the nucleolar or nuclear proteins during nucleolar stress [78]. However, the immuno-electron microscopic characteristics of the nucleoli are technically challengeable and less quantitative compared to immunofluorescence under light microscope, thus are not widely applicable.
–
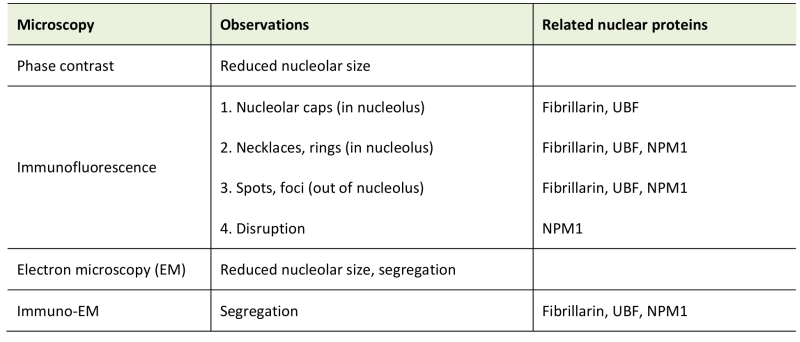
Interestingly, nucleolar atrophy is observed in neurons of patient brain autopsies [79] and pharmacological mouse models for some neurodegenerative disorders like Parkinson’s disease and Alzheimer’s disease [80][81], indicating that the size of the nucleolus can change under long-term stress. Of note, changes in nucleolar size and shape in cultured cells can be briefly observed by simple phase contract microscope [82]. We list the general morphological characteristics of nucleolar stress in Table 1.
–
TABLE 1. Morphology upon nucleolar stress. |
 |
–
Impaired rRNA transcription and processing
In mammalian cells, precursor ribosomal RNA (47S pre-rRNA) is transcribed by Pol I, then processed to 45S, 41S, 36S, and 32S rRNA intermediate precursors and finally matured 18S, 5.8S and 28S rRNA [83]. To curtail the role of rRNA maturation in nucleolar stress and to identify specific processing steps, which might be impaired under the respective stress condition, a number of methodological approaches are available: Inhibition of rRNA transcription can easily be confirmed by reduced amount of 47S pre-rRNA precursor. This could happen under Pol I-targeted chemotherapeutic drugs treatment [75], depletion of essential factors in Pol I complex (like TIF-IA and UBF), or specific Pol I component inhibitors (like Act.D) [47]. Inhibition of rRNA processing usually results in accumulated precursor levels, reduced amounts of products or both. If an early processing stage is impaired, it will result in reduced 41S, 36S and 32S rRNA products [75]; if a late stage is impaired, there will be accumulated 32S rRNA or reduced 28S rRNA [25][84]. The relative ratio changes of precursor and product (like 32S/28S) is widely accepted as a more accurate measurement of the processing [85][86]. Eventually, by analyzing the variation of the amount of precursors and products after treatments, one can figure out which steps in ribosome biogenesis are impaired.
–
Ethidium bromide (EtBr) staining and reversed-transcriptional PCR (RT-PCR) can be used as classical nucleic acid detecting methods for analyzing individual rRNA products. However, to get a more accurate result, radioactive probes for specific rRNAs and Northern hybridization analysis of precursors are widely applied [87]. For instance, with 32P or 3H labeling in culture medium, the major rRNA precursors can be visualized [75], and 32S precursor can be detected using radioactive ITS2-specific probes [88].
–
Activation of p53 signaling
The p53 tumor suppressor protein is considered as an integration point in response to various cellular stresses [89][90]. The activation of p53 can promote transcription of p21 leading to G1/S growth arrest [91], of 14-3-3 sigma inducing G2/M arrest [92], or of Bax inducing apoptosis [93]. It can also induce other factors involved in autophagy, DNA repair and metabolism [94].
–
The major negative regulator of p53 is the E3 ubiquitin ligase MDM2 (murine double minute 2, HDM2 in human). Mechanistically, MDM2 interacts with p53 via its C-terminal RING finger domain, promoting p53 ubiquitination and degradation by the 26S proteasome [95][96]. Therefore, p53 stabilization and activation in response to various stresses rely on a disruption this interaction between p53 and MDM2/HDM2. Simple readouts for p53 activation under nucleolar stress conditions are an increased p53 protein levels (stabilization or accumulation following blockage of ubiquitin-proteasomal degradation), reduced p53 binding to MDM2/HDM2, increased p53 mRNA levels under a long-lasting stress, elevated mRNA levels of p53 target genes, typically CDKN1A (p21) and BAX, and corresponding cell phenotypes such as cell cycle arrest, autophagy, DNA repair, senescence, or apoptosis [89].
–
Involvement of p53-independent stress signaling
In p53-/- or p53 inactivated cell lines, nucleolar stress can usually still invoke cell cycle arrest or apoptosis, implying that there is a stress response that is mediated by signaling pathways other than p53 [97].
–
Ribosomal proteins (RPs) regulating transcription factors (TFs)
The major non-p53 TFs that respond to ribosomal stress are c-Myc, E2Fs and SP1 [97][98][99][100]. Their downregulation or decreased transcriptional activities by RPs mediate cellular stress responses via altered transcription of target genes. Measurement of mRNA and/or protein levels of these TFs and their target genes, and analysis of TF binding with the target DNA, may indicate the involvement of these signaling pathways.
–
The oncoprotein c-Myc positively controls cell growth and proliferation [101] and serves as a direct regulator of ribosome biogenesis; many products of its transcriptional target genes are involved in ribosome biogenesis [102]. As a feedback mechanism, RPL5 and RPL11 are two critical negative regulators of c-Myc expression during ribosomal biogenesis; they form a complex with c-Myc mRNA and recruit microRNAs to repress c-Myc expression thus inhibiting the transcriptional activity of c-Myc [100]. RPS14 may also function as a negative regulator of c-Myc [103]. Consistently, upon nucleolar stress, as ribosome-free RPs, these proteins can lead to inhibition of cell proliferation through suppression of c-Myc and its target gene expression [102].
–
E2F-1 is a member of the E2Fs family of transcription factors; the expression of their target genes are important both for cell proliferation and apoptosis [104]. Independent of its regulatory control of p53, MDM2 prolongs the half-life of E2F-1 [99]. Under impaired rRNA biosynthesis, free RPL11 binds to MDM2 causing E2F-1 degradation, which is associated with the inhibition of cell proliferation [105].
–
Recently, RPL3 has been found as a pro-apoptotic factor under nucleolar stress induced by 5-fluorouracil in colon cancer cells devoid of p53. RPL3 in ribosome-free form, negatively regulates cystathionine-β-synthase (CBS) expression at the transcriptional level through inhibition of Sp1 binding to the CBS gene [98]. In addition, RPL3 can mediate p53-independent p21 upregulation, which requires the specific interaction between RPL3 and Sp1. Depending on its intracellular levels, p21 can either induce G1/S arrest of the cell cycle or mitochondria-mediated apoptosis [106].
–
RPs regulating non-TF proteins
RPL3 can not only negatively regulate CBS expression at the transcriptional level, but also trigger CBS translocation into mitochondria. Consequently, apoptosis is induced through the mitochondrial apoptotic cell death pathway [98].
–
Nucleolar proteins regulating TFs and non-TF proteins
There are several nucleolar proteins that bypass p53 and directly promote cell cycle arrest or apoptosis. These p53-independent regulators of apoptosis mainly include NPM1, PPAN, ARF and NuMA [107][108]. Both NPM1 and ARF are well-known for their roles in p53 signaling, however, several reports have demonstrated their involvement in p53-independent signaling [109]. In these cases, translocation of the nucleolar proteins and their interactions with the corresponding proteins may be analyzed. Interestingly, many RP or other nucleolar protein-mediated p53-independent stress responses require NPM1. In fact, NPM1 alone also interacts with apoptotic proteins. In conditions of nucleolar stress, NPM1 is transcriptionally induced and relocalizes from the nucleolus to the cytoplasm where it complexes with BAX, a crucial effector of the mitochondrial apoptosis pathway. Of note, cytosolic NPM1-BAX interaction has also been associated with cell resistance to death stimuli [109], therefore, the cellular response this direct interaction of NPM1 with apoptosis regulators does not necessarily result in cell death.
–
The Wnt target Peter Pan (PPAN) localizes to mitochondria in addition to its nucleolar localization and inhibits the mitochondrial apoptosis pathway in a p53-independent manner. Its role as an anti-apoptotic factor is indicated by the fact that knockdown of PPAN induces BAX stabilization, mitochondrial membrane depolarization and cytochrome c release. Staurosporine or Act.D-induced nucleolar stress and apoptosis disrupt nucleolar PPAN localization and induce its accumulation in the cytoplasm, which might be associated with impairment in its anti-apoptotic function [110].
–
Recently, the nuclear mitotic apparatus protein NuMA that locates in nucleoli in the interphase, has been demonstrated to be redistributed upon Act.D or doxorubicin- induced nucleolar stress. NuMA co-immunoprecipitates with Pol I, with RPL26 and RPL24, and with components of an ATP-dependent chromatin remodeling complex associated with rDNA transcription. Downregulation of NuMA expression triggers nucleolar stress, as shown by decreased nascent pre-rRNA synthesis, fibrillarin perinucleolar cap formation and upregulation of p27kip1, but not p53 [108].
–
Several studies reported that ARF binds and antagonizes the transcriptional activities of c-Myc and E2F-1, halting cell cycle progression in absence of p53 [111]. In addition to the regulation of the TFs, ARF controls proliferation by limiting nucleolar localization of the RNA helicase DDX5, which ultimately increases ribosome output [112].
THE MECHANISMS UNDERLYING NUCLEOLAR STRESS SENSING AND INTEGRATION TO p53 SIGNALING
Cumulative findings that any impairment in ribosome biogenesis by various insults can lead to p53 stabilization and activation, has led to the hypothesis that a low p53 level under non-stress condition relies on normal homeostasis of ribosome biogenesis in the nucleolus. This default state is ensured by the intactness of the ribosome biogenesis procedures and nucleolar structure. The evidence supporting this default model is robust, as p53 activation in nucleolar stress can be induced by the aberrant expression of those nucleolar proteins that are indispensable for ribosome biogenesis, or by various stimuli.
–
This notion then brought up an outstanding question how the errors or hurdles within the nucleolus signal to p53. Of note, since p53 is mainly controlled by its negative regulator MDM2/HDM2, one should specifically ask, how nucleolar stress interrupts MDM2-p53 association. We here summarize several aspects of studies addressing this question [30][48][97][113], including some frequent confusions or ambiguities.
–
From nucleolar stress to MDM2/HDM2-p53
The role of ribosomal proteins
In response to nucleolar stress, several RPs bind to MDM2 and block MDM2-mediated p53 ubiquitination and degradation, resulting in p53 stabilization and activation. After cells are exposed to low doses of Act.D, serum starvation or other insults, there is an increased binding of RPL5, RPL11 and RPL23 to MDM2 [33][34][114][115]. RPL5, RPL11, and RPL23 all bind to the central acidic region of MDM2, but importantly, each of them requires specific sequences to interact [43][116][117]. Therefore, the RP-MDM2-p53 signaling pathway has been proposed and believed to constitute a surveillance network monitoring the integrity of ribosomal biogenesis [113].
–
If an increased binding of free RPs with MDM2 is a prerequisite for this monitoring or sensing, the questions arises where these increased free RPs come from under nucleolar stress conditions, and in which subnuclear compartment they interact with MDM2, given that RPs mostly reside in the nucleolus and the cytoplasm, whereas MDM2 often stays in the nucleoplasm. However, these critical points have not been paid adequate attention to, and thus remain unclear. To our knowledge, first of all, few publications addressed the issue of subnuclear localization or fractionation of RPs and MDM2/HDM2 before and after stressor exposure. And secondly, the reported findings were controversial and highly context-dependent:
–
The early work by Lohrum et al. [114] showed that in unstressed U2OS cells under ectopic expression, (RP)L11 and HDM2 displayed complex localization. When expressed alone, L11 was predominantly nucleolar and HDM2 confined to the nucleoplasm, but 24 hours after co-transfection, L11 and HDM2 were both localized to the nucleoplasm when the L11:HDM2 ratio was low (2:1); or both localized to the nucleolus when L11 levels were higher (4:1). However, again under ectopic expression, in response to nucleolar stress induced by low levels of Act.D, both L11 and HDM2 co-localized with endogenous NPM1 to discrete subnuclear bodies, and, after longer treatment, both were co-localized with NPM1 in the nucleoplasm. Apparently, one is hardly able to draw conclusions from these observations that L11 is released from the nucleolus to the nucleoplasm upon stress.
–
Bursac et al. reported differential events in 2012 [74]. Using cell fractionation to purify the nucleolus extract, they found that endogenous nucleolar L5 and L11 were not reduced upon Act.D treatment, and using YFP-L11 transfection and fibrillarin immunostaining, they found that L11 was not translocated to the nucleoplasm upon Act.D treatment. The authors claimed that whereas several other newly synthesized ribosomal proteins are degraded by the proteasome upon Act.D treatment, L5 and L11 accumulate in the ribosome-free fraction where they bind to MDM2. Furthermore, the endogenous, newly synthesized L5 and L11 continued to be imported into nucleoli even after nucleolar disruption and co-localized with MDM2, p53, and PML. Therefore, in contrast to findings by others, their results suggest that the disrupted nucleoli may provide a platform for L5- and L11-dependent p53 activation.
–
Watkins and Thomas labs both pointed out that 5S RNA complexes with L5 and L11, and functions either in pre-60S assembly, or inhibiting MDM2 induced p53 degradation [118][119].
Recently, using immunostaining, Kayama et al. demonstrated a nucleoplasmic translocation of FLAG-tagged RPL11 in HCT 116 cells upon low dosed Act.D treatment [120].
–
In our experiments, immunostaining of RPL11 in U2OS cells showed a predominant location in the cytoplasm and no redistribution to the nucleoli or nucleoplasm was detectable upon Act.D-induced nucleolar stress (unpublished data).
–
A number of review articles stated or implied that the increased levels of ribosome-free RPs may originate from disrupted nucleoli [97][113]. To our knowledge, this notion lacks direct evidence, and thus remains speculative, unless free endogenous RP translocation from the nucleolus is detected under specific or general stress conditions.
–
An increased RPs-MDM2 interaction could follow diverse cellular disturbances, such as global translation inhibition [121], or the breakdown of ribosomal polysomes in the cytoplasm [33][43][115]. One can predict that there would be an accumulation of free RPs in the cells under these circumstances. However, in order to validate the postulation that excessive and ‘wandering’ free RPs find their way toward the nucleoplasm and there interact with MDM2, more detailed analysis based on subcellular fractionation and localization approaches are required. Taken together, it is worth further investigating how free RPs sense the nucleolar stress and then transmit signals to p53.
–
The role of ARF
ARF (alternative reading frame protein, p19 in mouse, p14 in humans) induces p53 activation in response to certain types of DNA damage [122] or several ontogenetic stresses [123][124][125], and is therefore categorized as a tumor suppressor. ARF is considered to localize to nucleoli of non-stressed cells [126]. As a basic nucleolar protein rich in arginine residues, it binds to multiple ribosomal proteins, and participates in 47S rRNA transcription and 32S processing events [127]. Although several studies revealed that ARF was released to nucleoplasm under nucleolar stress and targeted the central acidic domain of MDM2 to inhibit p53 degradation [128][129], some other reports implied that a resident nucleoplasmic fraction of total ARF is involved in the interaction with MDM2 under stress conditions [128][130]. Moreover, some studies indicated that ARF is dispensable for nucleolar stress-induced p53 accumulation [131][132].
–
In our study [31], we found that endogenous or exogenous ARF was similarly distributed both in the nucleolus and nucleoplasm of U2OS cells; the nucleolar ARF did not move out following Act.D treatment and, in contrast, the nucleolus/nucleoplasm ratio of ARF was slightly increased, while NPM1 was redistributed to the nucleoplasm under the same condition.
–
Therefore, the role of ARF in nucleolar stress-induced p53 activation may be cell type-dependent and/or context-dependent. In addition, the subnuclear compartment where ARF interacts with MDM2 still needs to be determined.
–
The role of translocated nucleolar proteins
As outlined above, NPM1 can interact directly with p53 via its C-terminal domain [17] and also binds to HDM2, competing with p53 and blocking HDM2-mediated p53 ubiquitination [30]. The wide range of stress conditions which provoke NPM1 translocation [16][31][133][134] might explain how p53 can integrate various stimuli. In our own work, we aimed at answering the question, if the translocated, nucleoplasmic fraction of NPM1 was the sole trigger for p53 activation. Therefore, we constructed an NPM1 mutant, unable to move upon Act.D treatment. In contrast to wild-type NPM1, this mutant was unable to disrupt the p53-HDM2 interaction, and thus greatly compromised the activation of p53 [31]. Interestingly, compared with the mock DNA, overexpression of the p53 activator ARF in U2OS cells with either normal or silenced NPM1 led to an increased accumulation of p53 under non-stress condition. However, after cells were exposed to Act.D, overexpression of ARF alone did not produce further accumulation of p53 in NPM1 knockdown cells. An enhanced p53 accumulation was observed when wild-type NPM1 was restored, but reintroducing the unmovable mutant NPM1 could not cause an increase of p53. A co-immunoprecipitation assay showed that the disruption of the HDM2-p53 interaction occurred in the cells bearing the wild-type but not mutant NPM1, whereas amounts of ARF bond to HDM2 appeared similar in both cell groups. These data suggest that the nucleoplasmic fraction of ARF alone is able to induce p53 accumulation under basal conditions. However, further p53 accumulation under stress conditions is determined by the presence of the nucleoplasmic NPM1, independent of ARF.
–
We also found that the p53-HDM2 interaction was prevented only by the wild-type but not the mutant NPM1 that lost nucleoplasmic translocation, whereas RPL23 remained bound to HDM2 in both samples. This means that, although RPL23 is required for p53 stabilization under stress, only NPM1 translocation to the nucleoplasm dictates the final outcome of p53 accumulation through NPM1’s binding with HDM2 and thus the disruption of the HDM2-p53 interaction. Collectively, the binding of ribosomal proteins or ARF with HDM2, which had been thought to be sufficient for p53 stabilization [33][115][135][136], is actually insufficient when NPM1 stays within the nucleoli. These results highlight that nucleoplasmic translocation of NPM1 is a prerequisite for stress-induced activation of p53.
–
From nucleolar stress to NPM1 translocation
As discussed above, nucleoplasmic translocation of NPM1 is the most prominent hallmark of nucleolar stress. However, the upstream causes of this translocation remained unclear. In other words, how various cellular insults trigger NPM1 translocation had not been ever asked.
–
Using single live-cell imaging and the redox biosensors, we demonstrated that nucleolar oxidation is a general response to various cellular stresses and a trigger for NPM1 translocation. This conclusion was supported by that antioxidant N-acetyl-cystein pretreatment was able to prevent the NPM1 translocation to a great extent, while treatment with a protein reducing agent completely inhibited NPM1 translocation. We showed that during nucleolar oxidation, NPM1 undergoes S-glutathionylation on cysteine 275, which triggers the dissociation of NPM1 from nucleolar nucleic acids. Accordingly, the NPM1 C275S mutant, unable to be glutathionylated, remained in the nucleolus under nucleolar stress, and greatly compromised the activation of p53. In sum, our findings provide a redox mechanism underlying the nucleolar stress sensing by NPM1 [31] (Figure 4).
–
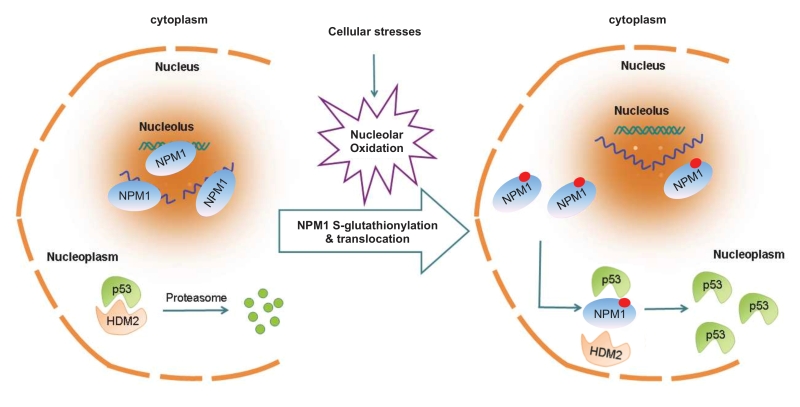 | FIGURE 4: NPM1 sensing for nucleolar stress. Nucleolar oxidation is a general response to nucleolar stress. S-glutathionylation and nucleoplasmic translocation of NPM1 are indispensable for p53 activation in nucleolar stress [31]. |
NUCLEOLAR STRESS AND HUMAN DISEASES
Natural mutations in genes that encode ribosomal proteins or proteins regulating ribosome biogenesis within the nucleolus result in a class of genetic disorders entitled ‘ribosomopathies’. These diseases usually have dramatic systemic phenotypes and severe outcomes [137][138][139][140][141][142][143][144]. Of note, although ribosomopathies display nucleolar stress, there are no therapeutic options directly targeting nucleolar stress to delay disease progression.
–
Nucleolar stress is a common event in neurodegenerative disorders such as Parkinson’s disease (PD) [137], Alzheimer’s disease (AD) [80] and Huntington’s disease (HD) [141]. Despite the fact that these disorders are polycausal, nucleolar stress may be one of the significant mediators in the degeneration or loss of neurons [138]. Besides morphological and functional manifestations in tissues and cell culture, the direct causal relation between these diseases and nucleolar stress has been established using the TIF-IA mouse model (see below) [79][145]. A related application, aiming to alleviate nucleolar stress for the prevention and treatment of these disorders will likely ensue.
–
Large and abnormal nucleoli are commonly observed in cancer cells [146]. The hyperactivation of ribosome biogenesis likely contributes to increased cancer cell survival and proliferation. In addition, cancer treatment faces challenges in chemo-radio-resistance cancers and those insensitive to other killing approaches. Potentiating nucleolar stress in these cancer cells may be a novel therapeutic strategy. Indeed, some typical nucleolar stress-inducing agents are under clinical investigation for remedy of leukemia, which has shown promising outcomes [147][148].
–
Hereafter, we present a brief overview on these two types of nucleolar stress-related diseases. We recommend several comprehensive and in-depth reviews [137][147][149][150], and try to provide some updated information.
–
Neurodegenerative disorders
Neurodegenerative disorders are chronic diseases, characterized by the progressive loss of specific neurons in the central or peripheral nervous system. These diseases are characterized by degeneration or loss of a specific subpopulation of neurons. Nucleolar stress is an emerging element of the degenerative process, caused by impaired rRNA transcription and altered nucleolar integrity [151].
–
PD is associated with the loss of dopaminergic (DA) neurons. Reduced nucleolar volume usually reflects reduced rRNA synthesis, while reduced rRNA synthesis has been reported in neurodegenerative disorders [152]. Early data show that nucleolar volume in DA neurons is decreased in PD patients and this is inversely correlated with disease duration [153][154]. Decreased nucleolar volume has been reported in the partial unilateral intrastriatal 6-hydroxydopamine oxidative stress rat model of PD [81]. Along these lines, a pharmacological mouse model of PD displays disruption of nucleolar integrity [79], while there is also a significant atrophy of the nucleoli in AD [80]. Interestingly, DNA damage in neurons can cause NPM1 translocation to the nucleoplasm [155], thus eliciting another hallmark of nucleolar stress.
–
Mouse models for conditional knockout (KO) of the transcription factor TIF-IA have been generated, in which nucleolar stress is induced in specific neuronal populations at a defined time-point [44]. Therefore, mice with conditional KO of TIF-IA not only confirmed a causal correlation of nucleolar stress with neuronal degeneration, but also served as an efficient model to study nucleolar stress itself. Cell-specific TIF-IA KO in distinct postmitotic neurons resulted in their slow, progressive degeneration, showing that neurons can survive for several months under nucleolar stress [79][145][156][157]. This slow progression allows the analysis of the sequence of events triggered by nucleolar stress in distinct neuronal populations. Interestingly, DA-specific TIF-IA KO mice show behavioral and cellular features of PD [79]. Notably, nucleolar impairment at the age of two months in DA neurons leads to mitochondrial dysfunction and increased oxidative damage, characteristics shared by various neurodegenerative diseases, such as PD, AD and HD. Interestingly, DA-specific TIF-IA KO in a double KO background of the PD-related genes DJ-1/PINK1 displayed an early phenotype similar to those mice lacking TIF-IA alone [158]. This conditional TIF-IA KO mouse model has also revealed a dual role of nucleolar stress. It could trigger a neuroprotective defense response at early stages, probably through inducing autophagy [145] and p53-dependent antioxidant response [159], but, on the long run, lead to impaired mitochondrial function and increased oxidative stress, and ultimately neuronal death [79][145].
–
Further dissection of the regulatory factors in nucleolar stress at a temporal resolution may help to better understand the pathophysiology of neurodegenerative disorders and create novel interfering strategies for prevention and treatment.
–
Malignancies
Given the pivotal role of NPM1 in nucleolar stress transmission to p53, one of the main tumorsuppressors, there are multiple NPM1-based therapeutic strategies for cancer treatment. Indeed, a vast part of anticancer drugs triggers apoptotic cell death through p53-activation pathway. Because of a causal relationship between anticancer chemical drug induced cell apoptosis and NPM1 translocation [63][134][160], NPM1 translocation could be regarded as a effective drug screening marker for novel antitumor agents selection [8]. As a safe and tolerable drug in clinical phase II trial, CIGB-300 exerts a broad original and synergistic antiproliferative effect on different cell lines [161][162][163][164]. Perera et al. revealed that CIGB-300 directly binds to NPM1, induces a rapid nucleoplasmic translocation of NPM1, and leads to a nucleolar disassembly-dependent apoptosis [165].
–
Additionally, there are also some selective Pol I inhibitors that reveal promising clinical effects [148][166]. For example, CX-3543 exhibits broad anti-proliferative and apoptotic effects on cancer cells and demonstrated impressive anti-tumor growth properties in xenograft models of breast and pancreatic cancer [167]. Intriguingly, the next generation of CX-3543, CX-5461 showed effectiveness in human cancer cells that experience overloaded ribosomal biogenesis compared to normal cells [168][169].
–
The cytotoxic marine natural product Avrainvillamide specifically binds the C-terminus of NPM1 and leads to its disassociation from nucleolar nucleic acid [170][171]. Furthermore, a G-quadruplex ligand TmPyP4 could perfectly compete and inhibit nucleolar nucleic acid binding activity of NPM1 [172]. Both of them caused NPM1 nucleoplasmic translocation. Because the NPM1 C-terminal domain surface is responsible for NPM1 nucleolar localization [173], it is likely that these two C-terminus targeting compounds are also related with nucleolar stress.
–
Additionally, there are some compounds targeting other NPM1 functional domains. NSC348884 and YTR107 interact with the N-terminus of NPM1 and inhibit its normal oligomerization function [174]. REV-NLS targets the N-terminal NPM1 surface and inhibits its protein-protein interaction functions [175]. A small synthetic RNA, named 1A1, identified in a screen for binding to full-length NPM1, binds to the central region of NPM1 [176]. These NPM1-targeting compounds both showed excellent anticancer effects. Importantly, both of them induced increased p53 levels and trancriptional activity, although their effects on NPM1 localization has not been investigated so far.
–
The NPM1 gene was identified to harbor the most frequent genetic (30%) lesions in adult acute myeloid leukemia (AML) [177][178]. Immunohistochemical staining in bone marrow specimens reveals a constant cytoplasmic localization for NPM1 [179]. Due to a frame shift mutation in one allele of the last exon of NPM1 gene [180], this mutant, termed as NPMc+ (cytoplasmic positive), holds a nuclear export signal (NES) and a misfolded structure in its C-terminus, resulting in disassociation from nucleic acid and cytoplasmic export [173][181][182]. In addition, this mutant also causes a cytoplasmic retention effect of wild-type NPM1, leaving only trace amounts of wild-type NPM1 in the nucleoli [183]. Fortunately, an intact functional p53 response pathway is still preserved in NPM1c+ AML cells [178], thus NPM1c+ AML cells remain sensitive to nucleolar stress-induced p53 activation [184][185]. Thus, Falini et al. focused on Act.D induced nucleolar stress and utilized the clinical recommended dose of Act.D to treat seven refractory or relapsed NPM1c+ mutant only (without FLT3 mutations) AML patients. Three of them showed hematologic complete remission within six weeks of therapy, and one of them even manifested molecular complete remission lasting for 14 months [186].
–
Considering our recently uncovered sensing mechanism of NPM1 in response to nucleolar stress [31], we strongly expect that the chemical compounds that enable NPM1 glutathionylation or specifically target the NPM1 nucleic acid binding site would exert better efficacy in therapies for malignancies like NPM1c+ AML.
References
- S. Gerbi, "The nucleolus: a site of ribonucleoprotein maturation", Current Opinion in Cell Biology, vol. 15, pp. 318-325, 2003. http://dx.doi.org/10.1016/s0955-0674(03)00049-8
- T. Kiss, "Small nucleolar RNAs: an abundant group of noncoding RNAs with diverse cellular functions.", Cell, 2002. http://www.ncbi.nlm.nih.gov/pubmed/12007400
- U. Scheer, M. Thiry, and G. Goessens, "Structure, function and assembly of the nucleolus.", Trends in cell biology, 1993. http://www.ncbi.nlm.nih.gov/pubmed/14731759
- Y. Ahmad, F. Boisvert, P. Gregor, A. Cobley, and A.I. Lamond, "NOPdb: Nucleolar Proteome Database--2008 update", Nucleic Acids Research, vol. 37, pp. D181-D184, 2009. http://dx.doi.org/10.1093/nar/gkn804
- F. Boisvert, S. van Koningsbruggen, J. Navascués, and A.I. Lamond, "The multifunctional nucleolus", Nature Reviews Molecular Cell Biology, vol. 8, pp. 574-585, 2007. http://dx.doi.org/10.1038/nrm2184
- J.S. Andersen, Y.W. Lam, A.K.L. Leung, S. Ong, C.E. Lyon, A.I. Lamond, and M. Mann, "Nucleolar proteome dynamics", Nature, vol. 433, pp. 77-83, 2005. http://dx.doi.org/10.1038/nature03207
- T. David-Pfeuty, "Potent inhibitors of cyclin-dependent kinase 2 induce nuclear accumulation of wild-type p53 and nucleolar fragmentation in human untransformed and tumor-derived cells", Oncogene, vol. 18, pp. 7409-7422, 1999. http://dx.doi.org/10.1038/sj.onc.1203103
- P.K. Chan, M.B. Aldrich, and B.Y. Yung, "Nucleolar protein B23 translocation after doxorubicin treatment in murine tumor cells.", Cancer research, 1987. http://www.ncbi.nlm.nih.gov/pubmed/2439199
- S. Desnoyers, S.H. Kaufmann, and G.G. Poirier, "Alteration of the Nucleolar Localization of Poly(ADP-ribose) Polymerase upon Treatment with Transcription Inhibitors", Experimental Cell Research, vol. 227, pp. 146-153, 1996. http://dx.doi.org/10.1006/excr.1996.0259
- H.W. Thielmann, O. Popanda, and H.J. Staab, "Subnuclear distribution of DNA topoisomerase I and Bax protein in normal and xeroderma pigmentosum fibroblasts after irradiation with UV light and gamma rays or treatment with topotecan.", Journal of cancer research and clinical oncology, 1999. http://www.ncbi.nlm.nih.gov/pubmed/10235474
- C. Mayer, and I. Grummt, "Cellular Stress and Nucleolar Function", Cell Cycle, vol. 4, pp. 1036-1038, 2005. http://dx.doi.org/10.4161/cc.4.8.1925
- L. Perlaky, B.C. Valdez, and H. Busch, "Effects of Cytotoxic Drugs on Translocation of Nucleolar RNA Helicase RH-II/Gu", Experimental Cell Research, vol. 235, pp. 413-420, 1997. http://dx.doi.org/10.1006/excr.1997.3686
- B.C. Valdez, L. Perlaky, Z.J. Cai, D. Henning, and H. Busch, "Green fluorescent protein tag for studies of drug-induced translocation of nucleolar protein RH-II/Gu.", BioTechniques, 1998. http://www.ncbi.nlm.nih.gov/pubmed/9631199
- B.Y. Yung, R.K. Busch, H. Busch, A.B. Mauger, and P.K. Chan, "Effects of actinomycin D analogs on nucleolar phosphoprotein B23 (37,000 daltons/pI 5.1).", Biochemical pharmacology, 1985. http://www.ncbi.nlm.nih.gov/pubmed/2415133
- P.K. Chan, D.A. Bloom, and T.T. Hoang, "The N-Terminal Half of NPM Dissociates from Nucleoli of HeLa Cells after Anticancer Drug Treatments", Biochemical and Biophysical Research Communications, vol. 264, pp. 305-309, 1999. http://dx.doi.org/10.1006/bbrc.1999.1255
- D.A. Matthews, "Adenovirus Protein V Induces Redistribution of Nucleolin and B23 from Nucleolus to Cytoplasm", Journal of Virology, vol. 75, pp. 1031-1038, 2001. http://dx.doi.org/10.1128/JVI.75.2.1031-1038.2001
- E. Colombo, J. Marine, D. Danovi, B. Falini, and P.G. Pelicci, "Nucleophosmin regulates the stability and transcriptional activity of p53", Nature Cell Biology, vol. 4, pp. 529-533, 2002. http://dx.doi.org/10.1038/ncb814
- J. Kim, K. Seok, Y. Kim, W.K. Bae, S. Lee, and J. Park, "Involvement of GLTSCR2 in the DNA Damage Response", The American Journal of Pathology, vol. 179, pp. 1257-1264, 2011. http://dx.doi.org/10.1016/j.ajpath.2011.05.041
- O. Yogev, K. Saadon, S. Anzi, K. Inoue, and E. Shaulian, "DNA Damage–Dependent Translocation of B23 and p19ARF Is Regulated by the Jun N-Terminal Kinase Pathway", Cancer Research, vol. 68, pp. 1398-1406, 2008. http://dx.doi.org/10.1158/0008-5472.CAN-07-2865
- D. Avitabile, B. Bailey, C.T. Cottage, B. Sundararaman, A. Joyo, M. McGregor, N. Gude, S. Truffa, A. Zarrabi, M. Konstandin, M. Khan, S. Mohsin, M. Völkers, H. Toko, M. Mason, Z. Cheng, S. Din, R. Alvarez, K. Fischer, and M.A. Sussman, "Nucleolar stress is an early response to myocardial damage involving nucleolar proteins nucleostemin and nucleophosmin", Proceedings of the National Academy of Sciences, vol. 108, pp. 6145-6150, 2011. http://dx.doi.org/10.1073/pnas.1017935108
- S. Duangmano, P. Sae-lim, A. Suksamrarn, F.E. Domann, and P. Patmasiriwat, "Cucurbitacin B inhibits human breast cancer cell proliferation through disruption of microtubule polymerization and nucleophosmin/B23 translocation", BMC Complementary and Alternative Medicine, vol. 12, 2012. http://dx.doi.org/10.1186/1472-6882-12-185
- B. kar, B. Liu, Z. Zhou, and Y.W. Lam, "Quantitative nucleolar proteomics reveals nuclear re-organization during stress- induced senescence in mouse fibroblast", BMC Cell Biology, vol. 12, 2011. http://dx.doi.org/10.1186/1471-2121-12-33
- M. Ljungman, F. Zhang, F. Chen, A.J. Rainbow, and B.C. McKay, "Inhibition of RNA polymerase II as a trigger for the p53 response", Oncogene, vol. 18, pp. 583-592, 1999. http://dx.doi.org/10.1038/sj.onc.1202356
- C.J. Sherr, and J.D. Weber, "The ARF/p53 pathway.", Current opinion in genetics & development, 2000. http://www.ncbi.nlm.nih.gov/pubmed/10679383
- D.G. Pestov, �. Strezoska, and L.F. Lau, "Evidence of p53-Dependent Cross-Talk between Ribosome Biogenesis and the Cell Cycle: Effects of Nucleolar Protein Bop1 on G1/S Transition", Molecular and Cellular Biology, vol. 21, pp. 4246-4255, 2001. http://dx.doi.org/10.1128/MCB.21.13.4246-4255.2001
- J. Klein, and I. Grummt, "Cell cycle-dependent regulation of RNA polymerase I transcription: the nucleolar transcription factor UBF is inactive in mitosis and early G1.", Proceedings of the National Academy of Sciences of the United States of America, 1999. http://www.ncbi.nlm.nih.gov/pubmed/10339547
- A. Budde, and I. Grummt, "p53 represses ribosomal gene transcription", Oncogene, vol. 18, pp. 1119-1124, 1999. http://dx.doi.org/10.1038/sj.onc.1202402
- T. David-Pfeuty, Y. Nouvian-Dooghe, V. Sirri, P. Roussel, and D. Hernandez-Verdun, "Common and reversible regulation of wild-type p53 function and of ribosomal biogenesis by protein kinases in human cells", Oncogene, vol. 20, pp. 5951-5963, 2001. http://dx.doi.org/10.1038/sj.onc.1204741
- C.P. Rubbi, "Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stresses", The EMBO Journal, vol. 22, pp. 6068-6077, 2003. http://dx.doi.org/10.1093/emboj/cdg579
- S. Kurki, K. Peltonen, L. Latonen, T.M. Kiviharju, P.M. Ojala, D. Meek, and M. Laiho, "Nucleolar protein NPM interacts with HDM2 and protects tumor suppressor protein p53 from HDM2-mediated degradation", Cancer Cell, vol. 5, pp. 465-475, 2004. http://dx.doi.org/10.1016/s1535-6108(04)00110-2
- K. Yang, M. Wang, Y. Zhao, X. Sun, Y. Yang, X. Li, A. Zhou, H. Chu, H. Zhou, J. Xu, M. Wu, J. Yang, and J. Yi, "A redox mechanism underlying nucleolar stress sensing by nucleophosmin", Nature Communications, vol. 7, 2016. http://dx.doi.org/10.1038/ncomms13599
- S. Lee, J. Kim, Y. Kim, K. Seok, J. Kim, Y. Chang, H. Kang, and J. Park, "Nucleolar protein GLTSCR2 stabilizes p53 in response to ribosomal stresses", Cell Death & Differentiation, vol. 19, pp. 1613-1622, 2012. http://dx.doi.org/10.1038/cdd.2012.40
- M. Dai, and H. Lu, "Inhibition of MDM2-mediated p53 Ubiquitination and Degradation by Ribosomal Protein L5", Journal of Biological Chemistry, vol. 279, pp. 44475-44482, 2004. http://dx.doi.org/10.1074/jbc.M403722200
- Y. Zhang, G.W. Wolf, K. Bhat, A. Jin, T. Allio, W.A. Burkhart, and Y. Xiong, "Ribosomal Protein L11 Negatively Regulates Oncoprotein MDM2 and Mediates a p53-Dependent Ribosomal-Stress Checkpoint Pathway", Molecular and Cellular Biology, vol. 23, pp. 8902-8912, 2003. http://dx.doi.org/10.1128/mcb.23.23.8902-8912.2003
- M. Dai, S.X. Zeng, Y. Jin, X. Sun, L. David, and H. Lu, "Ribosomal Protein L23 Activates p53 by Inhibiting MDM2 Function in Response to Ribosomal Perturbation but Not to Translation Inhibition", Molecular and Cellular Biology, vol. 24, pp. 7654-7668, 2004. http://dx.doi.org/10.1128/MCB.24.17.7654-7668.2004
- Y. Zhang, "The ARF-B23 Connection: Implications for Growth Control and Cancer Treatment", Cell Cycle, vol. 3, pp. 257-260, 2004. http://dx.doi.org/10.4161/cc.3.3.719
- M.S. Iordanov, D. Pribnow, J.L. Magun, T.H. Dinh, J.A. Pearson, S.L. Chen, and B.E. Magun, "Ribotoxic stress response: activation of the stress-activated protein kinase JNK1 by inhibitors of the peptidyl transferase reaction and by sequence-specific RNA damage to the alpha-sarcin/ricin loop in the 28S rRNA.", Molecular and cellular biology, 1997. http://www.ncbi.nlm.nih.gov/pubmed/9154836
- V.I. Shifrin, and P. Anderson, "Trichothecene mycotoxins trigger a ribotoxic stress response that activates c-Jun N-terminal kinase and p38 mitogen-activated protein kinase and induces apoptosis.", The Journal of biological chemistry, 1999. http://www.ncbi.nlm.nih.gov/pubmed/10318810
- M.S. Iordanov, and B.E. Magun, "Loss of cellular K+ mimics ribotoxic stress. Inhibition of protein synthesis and activation of the stress kinases SEK1/MKK4, stress-activated protein kinase/c-Jun NH2-terminal kinase 1, and p38/HOG1 by palytoxin.", The Journal of biological chemistry, 1998. http://www.ncbi.nlm.nih.gov/pubmed/9452478
- J.D. Laskin, "The Ribotoxic Stress Response as a Potential Mechanism for MAP Kinase Activation in Xenobiotic Toxicity", Toxicological Sciences, vol. 69, pp. 289-291, 2002. http://dx.doi.org/10.1093/toxsci/69.2.289
- R. Baserga, R.D. Estensen, and R.O. Petersen, "Inhibition of DNA synthesis in Ehrlich ascites cells by actinomycin D. II. The presynthetic block in the cell cycle.", Proceedings of the National Academy of Sciences of the United States of America, 1965. http://www.ncbi.nlm.nih.gov/pubmed/5220060
- S. Volarević, M.J. Stewart, B. Ledermann, F. Zilberman, L. Terracciano, E. Montini, M. Grompe, S.C. Kozma, and G. Thomas, "Proliferation, But Not Growth, Blocked by Conditional Deletion of 40 S Ribosomal Protein S6", Science, vol. 288, pp. 2045-2047, 2000. http://dx.doi.org/10.1126/science.288.5473.2045
- D.M. Gilkes, L. Chen, and J. Chen, "MDMX regulation of p53 response to ribosomal stress", The EMBO Journal, vol. 25, pp. 5614-5625, 2006. http://dx.doi.org/10.1038/sj.emboj.7601424
- X. Yuan, Y. Zhou, E. Casanova, M. Chai, E. Kiss, H. Gröne, G. Schütz, and I. Grummt, "Genetic Inactivation of the Transcription Factor TIF-IA Leads to Nucleolar Disruption, Cell Cycle Arrest, and p53-Mediated Apoptosis", Molecular Cell, vol. 19, pp. 77-87, 2005. http://dx.doi.org/10.1016/j.molcel.2005.05.023
- L. Golomb, D. Bublik, S. Wilder, R. Nevo, V. Kiss, K. Grabusic, S. Volarevic, and M. Oren, "Importin 7 and Exportin 1 Link c-Myc and p53 to Regulation of Ribosomal Biogenesis", Molecular Cell, vol. 45, pp. 222-232, 2012. http://dx.doi.org/10.1016/j.molcel.2011.11.022
- E. Nicolas, P. Parisot, C. Pinto-Monteiro, R. de Walque, C. De Vleeschouwer, and D.L.J. Lafontaine, "Involvement of human ribosomal proteins in nucleolar structure and p53-dependent nucleolar stress", Nature Communications, vol. 7, 2016. http://dx.doi.org/10.1038/ncomms11390
- R.P. Perry, and D.E. Kelley, "Persistent synthesis of 5S RNA when production of 28S and 18S ribosomal RNA is inhibited by low doses of actinomycin D", Journal of Cellular Physiology, vol. 72, pp. 235-245, 1968. http://dx.doi.org/10.1002/jcp.1040720311
-
A. Suzuki, R. Kogo, K. Kawahara, M. Sasaki, M. Nishio, T. Maehama, T. Sasaki, K. Mimori, and M. Mori, "A new
PICT ure of nucleolar stress", Cancer Science, vol. 103, pp. 632-637, 2012. http://dx.doi.org/10.1111/j.1349-7006.2012.02219.x - S. Boulon, B.J. Westman, S. Hutten, F. Boisvert, and A.I. Lamond, "The Nucleolus under Stress", Molecular Cell, vol. 40, pp. 216-227, 2010. http://dx.doi.org/10.1016/j.molcel.2010.09.024
- B.Y. Yung, E.K. Hui, and P.K. Chan, "Protein B23 (M.W./pI = 37 kD/5.1) is the only major protein extracted from HeLa nucleoli with 3M urea.", Life sciences, 1992. http://www.ncbi.nlm.nih.gov/pubmed/1518368
- D.L. Spector, R.L. Ochs, and H. Busch, "Silver staining, immunofluorescence, and immunoelectron microscopic localization of nucleolar phosphoproteins B23 and C23.", Chromosoma, 1984. http://www.ncbi.nlm.nih.gov/pubmed/6206987
- B.Y. Yung, H. Busch, and P.K. Chan, "Translocation of nucleolar phosphoprotein B23 (37 kDa/pI 5.1) induced by selective inhibitors of ribosome synthesis.", Biochimica et biophysica acta, 1985. http://www.ncbi.nlm.nih.gov/pubmed/3907710
- Y. Yu, L.B. Maggi, S.N. Brady, A.J. Apicelli, M. Dai, H. Lu, and J.D. Weber, "Nucleophosmin Is Essential for Ribosomal Protein L5 Nuclear Export", Molecular and Cellular Biology, vol. 26, pp. 3798-3809, 2006. http://dx.doi.org/10.1128/MCB.26.10.3798-3809.2006
- E. Muro, T.Q. Hoang, A. Jobart‐Malfait, and D. Hernandez‐Verdun, "In nucleoli, the steady state of nucleolar proteins is leptomycin B‐sensitive", Biology of the Cell, vol. 100, pp. 303-313, 2008. http://dx.doi.org/10.1042/BC20070117
- M. Takemura, K. Sato, M. Nishio, T. Akiyama, H. Umekawa, and S. Yoshida, "Nucleolar Protein B23.1 Binds to Retinoblastoma Protein and Synergistically Stimulates DNA Polymerase Activity", Journal of Biochemistry, vol. 125, pp. 904-909, 1999. http://dx.doi.org/10.1093/oxfordjournals.jbchem.a022367
- L.E. Kerr, J. Birse-Archbold, D.M. Short, A.L. McGregor, I. Heron, D.C. MacDonald, J. Thompson, G.J. Carlson, J.S. Kelly, J. McCulloch, and J. Sharkey, "Nucleophosmin is a novel Bax chaperone that regulates apoptotic cell death", Oncogene, vol. 26, pp. 2554-2562, 2006. http://dx.doi.org/10.1038/sj.onc.1210044
- M.S. Lindström, "NPM1/B23: A Multifunctional Chaperone in Ribosome Biogenesis and Chromatin Remodeling", Biochemistry Research International, vol. 2011, pp. 1-16, 2011. http://dx.doi.org/10.1155/2011/195209
- A. Szebeni, and M.O. Olson, "Nucleolar protein B23 has molecular chaperone activities", Protein Science, vol. 8, pp. 905-912, 1999. http://dx.doi.org/10.1110/ps.8.4.905
- E. Muro, J. Gébrane-Younès, A. Jobart-Malfait, E. Louvet, P. Roussel, and D. Hernandez-Verdun, "The traffic of proteins between nucleolar organizer regions and prenucleolar bodies governs the assembly of the nucleolus at exit of mitosis", Nucleus, vol. 1, pp. 202-211, 2010. http://dx.doi.org/10.4161/nucl.1.2.11334
- P.K. Chan, M. Aldrich, and H. Busch, "Alterations in immunolocalization of the phosphoprotein B23 in HeLa cells during serum starvation.", Experimental cell research, 1985. http://www.ncbi.nlm.nih.gov/pubmed/2414117
- R.A. Finch, G.R. Revankar, and P.K. Chan, "Nucleolar localization of nucleophosmin/B23 requires GTP.", The Journal of biological chemistry, 1993. http://www.ncbi.nlm.nih.gov/pubmed/8095498
- P.K. Chan, and F.Y. Chan, "A study of correlation between NPM-translocation and apoptosis in cells induced by daunomycin.", Biochemical pharmacology, 1999. http://www.ncbi.nlm.nih.gov/pubmed/10230770
- P.K. Chan, M.B. Aldrich, and S. Chakrabarty, "Assessment of tumor cell sensitivity to mitomycin C by "B23 translocation" assay.", Cancer letters, 1988. http://www.ncbi.nlm.nih.gov/pubmed/3133107
- P.K. Chan, "Characterization and cellular localization of nucleophosmin/B23 in HeLa cells treated with selected cytotoxic agents (studies of B23-translocation mechanism).", Experimental cell research, 1992. http://www.ncbi.nlm.nih.gov/pubmed/1426041
- R.A. Finch, G.R. Revankar, and P.K. Chan, "Structural and functional relationships of toyocamycin on NPM-translocation.", Anti-cancer drug design, 1997. http://www.ncbi.nlm.nih.gov/pubmed/9154111
- B.Y. Yung, Y.H. Yang, and A.M. Bor, "Nucleolar protein B23 translocation after deferoxamine treatment in a human leukemia cell line.", International journal of cancer, 1991. http://www.ncbi.nlm.nih.gov/pubmed/2071236
- H. Ginisty, "Nucleolin functions in the first step of ribosomal RNA processing", The EMBO Journal, vol. 17, pp. 1476-1486, 1998. http://dx.doi.org/10.1093/emboj/17.5.1476
- V. Sirri, S. Urcuqui-Inchima, P. Roussel, and D. Hernandez-Verdun, "Nucleolus: the fascinating nuclear body", Histochemistry and Cell Biology, vol. 129, pp. 13-31, 2007. http://dx.doi.org/10.1007/s00418-007-0359-6
- Y. Shav-Tal, J. Blechman, X. Darzacq, C. Montagna, B.T. Dye, J.G. Patton, R.H. Singer, and D. Zipori, "Dynamic Sorting of Nuclear Components into Distinct Nucleolar Caps during Transcriptional Inhibition", Molecular Biology of the Cell, vol. 16, pp. 2395-2413, 2005. http://dx.doi.org/10.1091/mbc.E04-11-0992
- T. Haaf, and D.C. Ward, "Inhibition of RNA Polymerase II Transcription Causes Chromatin Decondensation, Loss of Nucleolar Structure, and Dispersion of Chromosomal Domains", Experimental Cell Research, vol. 224, pp. 163-173, 1996. http://dx.doi.org/10.1006/excr.1996.0124
- M. Arcangeletti, F. De Conto, F. Ferraglia, F. Pinardi, R. Gatti, G. Orlandini, A. Calderaro, F. Motta, M. Medici, M. Martinelli, P. Valcavi, S. Razin, C. Chezzi, and G. Dettori, "Human cytomegalovirus proteins PP65 and IEP72 are targeted to distinct compartments in nuclei and nuclear matrices of infected human embryo fibroblasts", Journal of Cellular Biochemistry, vol. 90, pp. 1056-1067, 2003. http://dx.doi.org/10.1002/jcb.10655
- G.I. SCHOEFL, "THE EFFECT OF ACTINOMYCIN D ON THE FINE STRUCTURE OF THE NUCLEOLUS.", Journal of ultrastructure research, 1964. http://www.ncbi.nlm.nih.gov/pubmed/14166291
- K.V. Hadjiolova, A.A. Hadjiolov, and J.P. Bachellerie, "Actinomycin D stimulates the transcription of rRNA minigenes transfected into mouse cells. Implications for the in vivo hypersensitivity of rRNA gene transcription.", European journal of biochemistry, 1995. http://www.ncbi.nlm.nih.gov/pubmed/7737154
- S. Bursać, M.C. Brdovčak, M. Pfannkuchen, I. Orsolić, L. Golomb, Y. Zhu, C. Katz, L. Daftuar, K. Grabušić, I. Vukelić, V. Filić, M. Oren, C. Prives, and S. Volarević, "Mutual protection of ribosomal proteins L5 and L11 from degradation is essential for p53 activation upon ribosomal biogenesis stress", Proceedings of the National Academy of Sciences, vol. 109, pp. 20467-20472, 2012. http://dx.doi.org/10.1073/pnas.1218535109
- K. Burger, B. Mühl, T. Harasim, M. Rohrmoser, A. Malamoussi, M. Orban, M. Kellner, A. Gruber-Eber, E. Kremmer, M. Hölzel, and D. Eick, "Chemotherapeutic Drugs Inhibit Ribosome Biogenesis at Various Levels", Journal of Biological Chemistry, vol. 285, pp. 12416-12425, 2010. http://dx.doi.org/10.1074/jbc.M109.074211
- M. Franek, A. Kovaříková, E. Bártová, and S. Kozubek, "Nucleolar Reorganization Upon Site-Specific Double-Strand Break Induction", Journal of Histochemistry & Cytochemistry, vol. 64, pp. 669-686, 2016. http://dx.doi.org/10.1369/0022155416668505
- M. Govoni, F. Farabegoli, A. Pession, and F. Novello, "Inhibition of Topoisomerase II Activity and Its Effect on Nucleolar Structure and Function", Experimental Cell Research, vol. 211, pp. 36-41, 1994. http://dx.doi.org/10.1006/excr.1994.1055
- M. Sokka, K. Rilla, I. Miinalainen, H. Pospiech, and J.E. Syväoja, "High levels of TopBP1 induce ATR-dependent shut-down of rRNA transcription and nucleolar segregation", Nucleic Acids Research, vol. 43, pp. 4975-4989, 2015. http://dx.doi.org/10.1093/nar/gkv371
- C. Rieker, D. Engblom, G. Kreiner, A. Domanskyi, A. Schober, S. Stotz, M. Neumann, X. Yuan, I. Grummt, G. Schütz, and R. Parlato, "Nucleolar Disruption in Dopaminergic Neurons Leads to Oxidative Damage and Parkinsonism through Repression of Mammalian Target of Rapamycin Signaling", The Journal of Neuroscience, vol. 31, pp. 453-460, 2011. http://dx.doi.org/10.1523/JNEUROSCI.0590-10.2011
- D. Iacono, R. O'Brien, S.M. Resnick, A.B. Zonderman, O. Pletnikova, G. Rudow, Y. An, M.J. West, B. Crain, and J.C. Troncoso, "Neuronal Hypertrophy in Asymptomatic Alzheimer Disease", Journal of Neuropathology & Experimental Neurology, vol. 67, pp. 578-589, 2008. http://dx.doi.org/10.1097/NEN.0b013e3181772794
- M. Healy-Stoffel, S.O. Ahmad, J.A. Stanford, and B. Levant, "Altered nucleolar morphology in substantia nigra dopamine neurons following 6-hydroxydopamine lesion in rats", Neuroscience Letters, vol. 546, pp. 26-30, 2013. http://dx.doi.org/10.1016/j.neulet.2013.04.033
- K. Holmberg Olausson, M. Nistér, and M.S. Lindström, "p53 -Dependent and -Independent Nucleolar Stress Responses", Cells, vol. 1, pp. 774-798, 2012. http://dx.doi.org/10.3390/cells1040774
-
A.K. Henras, C. Plisson‐Chastang, M. O'Donohue, A. Chakraborty, and P. Gleizes, "An overview of pre‐ribosomal
RNA processing in eukaryotes", WIREs RNA, vol. 6, pp. 225-242, 2014. http://dx.doi.org/10.1002/wrna.1269 - J.C.R. Politz, I. Polena, I. Trask, D.P. Bazett-Jones, and T. Pederson, "A Nonribosomal Landscape in the Nucleolus Revealed by the Stem Cell Protein Nucleostemin", Molecular Biology of the Cell, vol. 16, pp. 3401-3410, 2005. http://dx.doi.org/10.1091/mbc.E05-02-0106
- E. Finkbeiner, M. Haindl, and S. Muller, "The SUMO system controls nucleolar partitioning of a novel mammalian ribosome biogenesis complex", The EMBO Journal, vol. 30, pp. 1067-1078, 2011. http://dx.doi.org/10.1038/emboj.2011.33
- E. Finkbeiner, M. Haindl, N. Raman, and S. Muller, "SUMO routes ribosome maturation", Nucleus, vol. 2, pp. 527-532, 2011. http://dx.doi.org/10.4161/nucl.2.6.17604
- D.G. Pestov, Y.R. Lapik, and L.F. Lau, "Assays for Ribosomal RNA Processing and Ribosome Assembly", Current Protocols in Cell Biology, vol. 39, 2008. http://dx.doi.org/10.1002/0471143030.cb2211s39
- C.D. Castle, E.K. Cassimere, and C. Denicourt, "LAS1L interacts with the mammalian Rix1 complex to regulate ribosome biogenesis", Molecular Biology of the Cell, vol. 23, pp. 716-728, 2012. http://dx.doi.org/10.1091/mbc.E11-06-0530
- K.T. Bieging, S.S. Mello, and L.D. Attardi, "Unravelling mechanisms of p53-mediated tumour suppression", Nature Reviews Cancer, vol. 14, pp. 359-370, 2014. http://dx.doi.org/10.1038/nrc3711
- A.C. Joerger, and A.R. Fersht, "The p53 Pathway: Origins, Inactivation in Cancer, and Emerging Therapeutic Approaches", Annual Review of Biochemistry, vol. 85, pp. 375-404, 2016. http://dx.doi.org/10.1146/annurev-biochem-060815-014710
- W.S. el-Deiry, J.W. Harper, P.M. O'Connor, V.E. Velculescu, C.E. Canman, J. Jackman, J.A. Pietenpol, M. Burrell, D.E. Hill, and Y. Wang, "WAF1/CIP1 is induced in p53-mediated G1 arrest and apoptosis.", Cancer research, 1994. http://www.ncbi.nlm.nih.gov/pubmed/8118801
- H. Hermeking, C. Lengauer, K. Polyak, T.C. He, L. Zhang, S. Thiagalingam, K.W. Kinzler, and B. Vogelstein, "14-3-3sigma is a p53-regulated inhibitor of G2/M progression.", Molecular cell, 1997. http://www.ncbi.nlm.nih.gov/pubmed/9659898
- T. Miyashita, and J.C. Reed, "Tumor suppressor p53 is a direct transcriptional activator of the human bax gene.", Cell, 1995. http://www.ncbi.nlm.nih.gov/pubmed/7834749
- M. Dai, H. Arnold, X. Sun, R. Sears, and H. Lu, "Inhibition of c-Myc activity by ribosomal protein L11", The EMBO Journal, vol. 26, pp. 3332-3345, 2007. http://dx.doi.org/10.1038/sj.emboj.7601776
- Y. Haupt, R. Maya, A. Kazaz, and M. Oren, "Mdm2 promotes the rapid degradation of p53", Nature, vol. 387, pp. 296-299, 1997. http://dx.doi.org/10.1038/387296a0
- M.H.G. Kubbutat, S.N. Jones, and K.H. Vousden, "Regulation of p53 stability by Mdm2", Nature, vol. 387, pp. 299-303, 1997. http://dx.doi.org/10.1038/387299a0
- A. Russo, and G. Russo, "Ribosomal Proteins Control or Bypass p53 during Nucleolar Stress", International Journal of Molecular Sciences, vol. 18, pp. 140, 2017. http://dx.doi.org/10.3390/ijms18010140
- V. Pagliara, A. Saide, E. Mitidieri, R. d’Emmanuele di Villa Bianca, R. Sorrentino, G. Russo, and A. Russo, "5-FU targets rpL3 to induce mitochondrial apoptosis via cystathionine-β-synthase in colon cancer cells lacking p53", Oncotarget, vol. 7, pp. 50333-50348, 2016. http://dx.doi.org/10.18632/oncotarget.10385
- Z. Zhang, H. Wang, M. Li, E.R. Rayburn, S. Agrawal, and R. Zhang, "Stabilization of E2F1 protein by MDM2 through the E2F1 ubiquitination pathway", Oncogene, vol. 24, pp. 7238-7247, 2005. http://dx.doi.org/10.1038/sj.onc.1208814
- M. Dai, R. Sears, and H. Lu, "Feedback Regulation of c-Myc by Ribosomal Protein L11", Cell Cycle, vol. 6, pp. 2735-2741, 2007. http://dx.doi.org/10.4161/cc.6.22.4895
- C.V. Dang, "c-Myc target genes involved in cell growth, apoptosis, and metabolism.", Molecular and cellular biology, 1999. http://www.ncbi.nlm.nih.gov/pubmed/9858526
- J. van Riggelen, A. Yetil, and D.W. Felsher, "MYC as a regulator of ribosome biogenesis and protein synthesis", Nature Reviews Cancer, vol. 10, pp. 301-309, 2010. http://dx.doi.org/10.1038/nrc2819
- X. Zhou, Q. Hao, J. Liao, P. Liao, and H. Lu, "Ribosomal Protein S14 Negatively Regulates c-Myc Activity", Journal of Biological Chemistry, vol. 288, pp. 21793-21801, 2013. http://dx.doi.org/10.1074/jbc.M112.445122
- D.K. Dimova, and N.J. Dyson, "The E2F transcriptional network: old acquaintances with new faces", Oncogene, vol. 24, pp. 2810-2826, 2005. http://dx.doi.org/10.1038/sj.onc.1208612
- H. Wang, T. Chen, C. Weng, C. Yang, and M. Tang, "Acrolein preferentially damages nucleolus eliciting ribosomal stress and apoptosis in human cancer cells", Oncotarget, vol. 7, pp. 80450-80464, 2016. http://dx.doi.org/10.18632/oncotarget.12608
- A. Russo, D. Esposito, M. Catillo, C. Pietropaolo, E. Crescenzi, and G. Russo, "Human rpL3 induces G₁/S arrest or apoptosis by modulating p21waf1/cip1 levels in a p53-independent manner", Cell Cycle, vol. 12, pp. 76-87, 2012. http://dx.doi.org/10.4161/cc.22963
- A. James, Y. Wang, H. Raje, R. Rosby, and P. DiMario, "Nucleolar stress with and without p53", Nucleus, vol. 5, pp. 402-426, 2014. http://dx.doi.org/10.4161/nucl.32235
- S. Jayaraman, S. Chittiboyina, Y. Bai, P.C. Abad, P. Vidi, C.V. Stauffacher, and S.A. Lelièvre, "The nuclear mitotic apparatus protein NuMA controls rDNA transcription and mediates the nucleolar stress response in a p53-independent manner", Nucleic Acids Research, vol. 45, pp. 11725-11742, 2017. http://dx.doi.org/10.1093/nar/gkx782
- S. Lo, L. Fan, Y. Tsai, K. Lin, H. Huang, T. Wang, H. Liu, T. Chen, S. Huang, C. Chang, Y. Lin, B.Y. Yung, and S. Hsieh, "A novel interaction of nucleophosmin with BCL2-associated X protein regulating death evasion and drug sensitivity in human hepatoma cells", Hepatology, vol. 57, pp. 1893-1905, 2013. http://dx.doi.org/10.1002/hep.26209
- A.S. Pfister, M. Keil, and M. Kühl, "The Wnt Target Protein Peter Pan Defines a Novel p53-independent Nucleolar Stress-Response Pathway", Journal of Biological Chemistry, vol. 290, pp. 10905-10918, 2015. http://dx.doi.org/10.1074/jbc.M114.634246
- B. Eymin, P. Claverie, C. Salon, C. Leduc, E. Col, E. Brambilla, S. Khochbin, and S. Gazzeri, "p14ARF Activates a Tip60-Dependent and p53-Independent ATM/ATR/CHK Pathway in Response to Genotoxic Stress", Molecular and Cellular Biology, vol. 26, pp. 4339-4350, 2006. http://dx.doi.org/10.1128/MCB.02240-05
- A.J. Saporita, H. Chang, C.L. Winkeler, A.J. Apicelli, R.D. Kladney, J. Wang, R.R. Townsend, L.S. Michel, and J.D. Weber, "RNA Helicase DDX5 Is a p53-Independent Target of ARF That Participates in Ribosome Biogenesis", Cancer Research, vol. 71, pp. 6708-6717, 2011. http://dx.doi.org/10.1158/0008-5472.CAN-11-1472
- Y. Zhang, and H. Lu, "Signaling to p53: Ribosomal Proteins Find Their Way", Cancer Cell, vol. 16, pp. 369-377, 2009. http://dx.doi.org/10.1016/j.ccr.2009.09.024
- M.A. Lohrum, R.L. Ludwig, M.H. Kubbutat, M. Hanlon, and K.H. Vousden, "Regulation of HDM2 activity by the ribosomal protein L11", Cancer Cell, vol. 3, pp. 577-587, 2003. http://dx.doi.org/10.1016/s1535-6108(03)00134-x
- A. Jin, K. Itahana, K. O'Keefe, and Y. Zhang, "Inhibition of HDM2 and Activation of p53 by Ribosomal Protein L23", Molecular and Cellular Biology, vol. 24, pp. 7669-7680, 2004. http://dx.doi.org/10.1128/MCB.24.17.7669-7680.2004
- M. Dai, D. Shi, Y. Jin, X. Sun, Y. Zhang, S.R. Grossman, and H. Lu, "Regulation of the MDM2-p53 Pathway by Ribosomal Protein L11 Involves a Post-ubiquitination Mechanism", Journal of Biological Chemistry, vol. 281, pp. 24304-24313, 2006. http://dx.doi.org/10.1074/jbc.M602596200
- M.S. Lindstroöm, A. Jin, C. Deisenroth, G. White Wolf, and Y. Zhang, "Cancer-Associated Mutations in the MDM2 Zinc Finger Domain Disrupt Ribosomal Protein Interaction and Attenuate MDM2-Induced p53 Degradation", Molecular and Cellular Biology, vol. 27, pp. 1056-1068, 2007. http://dx.doi.org/10.1128/MCB.01307-06
- G. Donati, S. Peddigari, C. Mercer, and G. Thomas, "5S Ribosomal RNA Is an Essential Component of a Nascent Ribosomal Precursor Complex that Regulates the Hdm2-p53 Checkpoint", Cell Reports, vol. 4, pp. 87-98, 2013. http://dx.doi.org/10.1016/j.celrep.2013.05.045
- K. Sloan, M. Bohnsack, and N. Watkins, "The 5S RNP Couples p53 Homeostasis to Ribosome Biogenesis and Nucleolar Stress", Cell Reports, vol. 5, pp. 237-247, 2013. http://dx.doi.org/10.1016/j.celrep.2013.08.049
-
K. Kayama, S. Watanabe, T. Takafuji, T. Tsuji, K. Hironaka, M. Matsumoto, K.I. Nakayama, M. Enari, T. Kohno, K. Shiraishi, T. Kiyono, K. Yoshida, N. Sugimoto, and M. Fujita, "
GRWD 1 negatively regulates p53 via theRPL 11–MDM 2 pathway and promotes tumorigenesis", EMBO reports, vol. 18, pp. 123-137, 2016. http://dx.doi.org/10.15252/embr.201642444 - E.V. Schmidt, "The role of c-myc in regulation of translation initiation", Oncogene, vol. 23, pp. 3217-3221, 2004. http://dx.doi.org/10.1038/sj.onc.1207548
- F. Zindy, C.M. Eischen, D.H. Randle, T. Kamijo, J.L. Cleveland, C.J. Sherr, and M.F. Roussel, "Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization.", Genes & development, 1998. http://www.ncbi.nlm.nih.gov/pubmed/9694806
- I. Palmero, C. Pantoja, and M. Serrano, "p19ARF links the tumour suppressor p53 to Ras", Nature, vol. 395, pp. 125-126, 1998. http://dx.doi.org/10.1038/25870
- E. de Stanchina, M.E. McCurrach, F. Zindy, S.Y. Shieh, G. Ferbeyre, A.V. Samuelson, C. Prives, M.F. Roussel, C.J. Sherr, and S.W. Lowe, "E1A signaling to p53 involves the p19(ARF) tumor suppressor.", Genes & development, 1998. http://www.ncbi.nlm.nih.gov/pubmed/9694807
- A. Radfar, I. Unnikrishnan, H.W. Lee, R.A. DePinho, and N. Rosenberg, "p19(Arf) induces p53-dependent apoptosis during abelson virus-mediated pre-B cell transformation.", Proceedings of the National Academy of Sciences of the United States of America, 1998. http://www.ncbi.nlm.nih.gov/pubmed/9789064
- M.S. Lindström, U. Klangby, R. Inoue, P. Pisa, K.G. Wiman, and C.E. Asker, "Immunolocalization of Human p14ARF to the Granular Component of the Interphase Nucleolus", Experimental Cell Research, vol. 256, pp. 400-410, 2000. http://dx.doi.org/10.1006/excr.2000.4854
- D. Bertwistle, M. Sugimoto, and C.J. Sherr, "Physical and Functional Interactions of the Arf Tumor Suppressor Protein with Nucleophosmin/B23", Molecular and Cellular Biology, vol. 24, pp. 985-996, 2004. http://dx.doi.org/10.1128/mcb.24.3.985-996.2004
- S. Llanos, P.A. Clark, J. Rowe, and G. Peters, "Stabilization of p53 by p14ARF without relocation of MDM2 to the nucleolus", Nature Cell Biology, vol. 3, pp. 445-452, 2001. http://dx.doi.org/10.1038/35074506
- C.J. Sherr, "Divorcing ARF and p53: an unsettled case", Nature Reviews Cancer, vol. 6, pp. 663-673, 2006. http://dx.doi.org/10.1038/nrc1954
- S. Moulin, S. Llanos, S. Kim, and G. Peters, "Binding to nucleophosmin determines the localization of human and chicken ARF but not its impact on p53", Oncogene, vol. 27, pp. 2382-2389, 2007. http://dx.doi.org/10.1038/sj.onc.1210887
- F.J. Stott, "The alternative product from the human CDKN2A locus, p14ARF, participates in a regulatory feedback loop with p53 and MDM2", The EMBO Journal, vol. 17, pp. 5001-5014, 1998. http://dx.doi.org/10.1093/emboj/17.17.5001
- E. Macias, A. Jin, C. Deisenroth, K. Bhat, H. Mao, M.S. Lindström, and Y. Zhang, "An ARF-Independent c-MYC-Activated Tumor Suppression Pathway Mediated by Ribosomal Protein-Mdm2 Interaction", Cancer Cell, vol. 18, pp. 231-243, 2010. http://dx.doi.org/10.1016/j.ccr.2010.08.007
- B.Y. Yung, A.M. Bor, and P.K. Chan, "Short exposure to actinomycin D induces "reversible" translocation of protein B23 as well as "reversible" inhibition of cell growth and RNA synthesis in HeLa cells.", Cancer research, 1990. http://www.ncbi.nlm.nih.gov/pubmed/1697505
- P.K. Chan, Y. Qi, J. Amley, and C.A. Koller, "Quantitation of the nucleophosmin/B23-translocation using imaging analysis.", Cancer letters, 1996. http://www.ncbi.nlm.nih.gov/pubmed/8620441
- M. Sasaki, K. Kawahara, M. Nishio, K. Mimori, R. Kogo, K. Hamada, B. Itoh, J. Wang, Y. Komatsu, Y.R. Yang, H. Hikasa, Y. Horie, T. Yamashita, T. Kamijo, Y. Zhang, Y. Zhu, C. Prives, T. Nakano, T.W. Mak, T. Sasaki, T. Maehama, M. Mori, and A. Suzuki, "Regulation of the MDM2-P53 pathway and tumor growth by PICT1 via nucleolar RPL11", Nature Medicine, vol. 17, pp. 944-951, 2011. http://dx.doi.org/10.1038/nm.2392
- Y. Zhang, Y. Xiong, and W.G. Yarbrough, "ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways.", Cell, 1998. http://www.ncbi.nlm.nih.gov/pubmed/9529249
- R. Parlato, and B. Liss, "How Parkinson's disease meets nucleolar stress", Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease, vol. 1842, pp. 791-797, 2014. http://dx.doi.org/10.1016/j.bbadis.2013.12.014
- J. Hallgren, M. Pietrzak, G. Rempala, P.T. Nelson, and M. Hetman, "Neurodegeneration-associated instability of ribosomal DNA", Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease, vol. 1842, pp. 860-868, 2014. http://dx.doi.org/10.1016/j.bbadis.2013.12.012
- S.R. Ellis, "Nucleolar stress in Diamond Blackfan anemia pathophysiology", Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease, vol. 1842, pp. 765-768, 2014. http://dx.doi.org/10.1016/j.bbadis.2013.12.013
- P.A. Trainor, and A.E. Merrill, "Ribosome biogenesis in skeletal development and the pathogenesis of skeletal disorders", Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease, vol. 1842, pp. 769-778, 2014. http://dx.doi.org/10.1016/j.bbadis.2013.11.010
- J. Lee, Y.J. Hwang, H. Ryu, N.W. Kowall, and H. Ryu, "Nucleolar dysfunction in Huntington's disease", Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease, vol. 1842, pp. 785-790, 2014. http://dx.doi.org/10.1016/j.bbadis.2013.09.017
- N. Hariharan, and M.A. Sussman, "Stressing on the nucleolus in cardiovascular disease", Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease, vol. 1842, pp. 798-801, 2014. http://dx.doi.org/10.1016/j.bbadis.2013.09.016
- J.E. Quin, J.R. Devlin, D. Cameron, K.M. Hannan, R.B. Pearson, and R.D. Hannan, "Targeting the nucleolus for cancer intervention", Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease, vol. 1842, pp. 802-816, 2014. http://dx.doi.org/10.1016/j.bbadis.2013.12.009
- A. Salvetti, and A. Greco, "Viruses and the nucleolus: The fatal attraction", Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease, vol. 1842, pp. 840-847, 2014. http://dx.doi.org/10.1016/j.bbadis.2013.12.010
- G. Kreiner, H. Bierhoff, M. Armentano, J. Rodriguez-Parkitna, K. Sowodniok, J.R. Naranjo, L. Bonfanti, B. Liss, G. Schütz, I. Grummt, and R. Parlato, "A neuroprotective phase precedes striatal degeneration upon nucleolar stress", Cell Death & Differentiation, vol. 20, pp. 1455-1464, 2013. http://dx.doi.org/10.1038/cdd.2013.66
- V. Moskvina, "Analysis of Genome-Wide Association Studies of Alzheimer Disease and of Parkinson Disease to Determine If These 2 Diseases Share a Common Genetic Risk", JAMA Neurology, 2013. http://dx.doi.org/10.1001/jamaneurol.2013.448
- A. Di Matteo, M. Franceschini, S. Chiarella, S. Rocchio, C. Travaglini-Allocatelli, and L. Federici, "Molecules that target nucleophosmin for cancer treatment: an update", Oncotarget, vol. 7, pp. 44821-44840, 2016. http://dx.doi.org/10.18632/oncotarget.8599
- D. Drygin, S.E. O’Brien, R.D. Hannan, G.A. McArthur, and D.D. Von Hoff, "Targeting the nucleolus for cancer-specific activation of p53", Drug Discovery Today, vol. 19, pp. 259-265, 2014. http://dx.doi.org/10.1016/j.drudis.2013.08.012
- S. Grisendi, C. Mecucci, B. Falini, and P.P. Pandolfi, "Nucleophosmin and cancer", Nature Reviews Cancer, vol. 6, pp. 493-505, 2006. http://dx.doi.org/10.1038/nrc1885
- E. Colombo, M. Alcalay, and P.G. Pelicci, "Nucleophosmin and its complex network: a possible therapeutic target in hematological diseases", Oncogene, vol. 30, pp. 2595-2609, 2011. http://dx.doi.org/10.1038/onc.2010.646
- M. Hetman, and M. Pietrzak, "Emerging roles of the neuronal nucleolus", Trends in Neurosciences, vol. 35, pp. 305-314, 2012. http://dx.doi.org/10.1016/j.tins.2012.01.002
- R. Parlato, and G. Kreiner, "Nucleolar activity in neurodegenerative diseases: a missing piece of the puzzle?", Journal of Molecular Medicine, vol. 91, pp. 541-547, 2012. http://dx.doi.org/10.1007/s00109-012-0981-1
- D.M. Mann, and P.O. Yates, "Pathogenesis of Parkinson's disease.", Archives of neurology, 1982. http://www.ncbi.nlm.nih.gov/pubmed/7115143
- H.J. Gertz, A. Siegers, and J. Kuchinke, "Stability of cell size and nucleolar size in Lewy body containing neurons of substantia nigra in Parkinson's disease.", Brain research, 1994. http://www.ncbi.nlm.nih.gov/pubmed/8180816
- K. Kalita, D. Makonchuk, C. Gomes, J. Zheng, and M. Hetman, "Inhibition of nucleolar transcription as a trigger for neuronal apoptosis", Journal of Neurochemistry, vol. 105, pp. 2286-2299, 2008. http://dx.doi.org/10.1111/j.1471-4159.2008.05316.x
- R. Parlato, G. Kreiner, G. Erdmann, C. Rieker, S. Stotz, E. Savenkova, S. Berger, I. Grummt, and G. Schütz, "Activation of an Endogenous Suicide Response after Perturbation of rRNA Synthesis Leads to Neurodegeneration in Mice", The Journal of Neuroscience, vol. 28, pp. 12759-12764, 2008. http://dx.doi.org/10.1523/JNEUROSCI.2439-08.2008
- A. Kiryk, K. Sowodniok, G. Kreiner, J. Rodriguez-Parkitna, A. Sönmez, T. Górkiewicz, H. Bierhoff, M. Wawrzyniak, A.K. Janusz, B. Liss, W. Konopka, G. Schütz, L. Kaczmarek, and R. Parlato, "Impaired rRNA synthesis triggers homeostatic responses in hippocampal neurons", Frontiers in Cellular Neuroscience, vol. 7, 2013. http://dx.doi.org/10.3389/fncel.2013.00207
- V. Evsyukov, A. Domanskyi, H. Bierhoff, S. Gispert, R. Mustafa, F. Schlaudraff, B. Liss, and R. Parlato, "Genetic mutations linked to Parkinson's disease differentially control nucleolar activity in pre-symptomatic mouse models", Disease Models & Mechanisms, 2017. http://dx.doi.org/10.1242/dmm.028092
- C. Zhang, M. Lin, R. Wu, X. Wang, B. Yang, A.J. Levine, W. Hu, and Z. Feng, "Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect", Proceedings of the National Academy of Sciences, vol. 108, pp. 16259-16264, 2011. http://dx.doi.org/10.1073/pnas.1113884108
- B.Y. Yung, F.J. Chang, and K.J. Luo, "Dipyridamole enhancement of doxorubicin-induced translocation of nucleophosmin and inhibition of cell growth in HL-60 cells.", International journal of cancer, 1991. http://www.ncbi.nlm.nih.gov/pubmed/1917161
- S.E. Perea, O. Reyes, I. Baladron, Y. Perera, H. Farina, J. Gil, A. Rodriguez, D. Bacardi, J.L. Marcelo, K. Cosme, M. Cruz, C. Valenzuela, P.A. López-Saura, Y. Puchades, J.M. Serrano, O. Mendoza, L. Castellanos, A. Sanchez, L. Betancourt, V. Besada, R. Silva, E. López, V. Falcón, I. Hernández, M. Solares, A. Santana, A. Díaz, T. Ramos, C. López, J. Ariosa, L.J. González, H. Garay, D. Gómez, R. Gómez, D.F. Alonso, H. Sigman, L. Herrera, and B. Acevedo, "CIGB-300, a novel proapoptotic peptide that impairs the CK2 phosphorylation and exhibits anticancer properties both in vitro and in vivo", Molecular and Cellular Biochemistry, vol. 316, pp. 163-167, 2008. http://dx.doi.org/10.1007/s11010-008-9814-5
- Y. Perera, H.C. Costales, Y. Diaz, O. Reyes, H.G. Farina, L. Mendez, R.E. Gómez, B.E. Acevedo, D.E. Gomez, D.F. Alonso, and S.E. Perea, "Sensitivity of tumor cells towards CIGB‐300 anticancer peptide relies on its nucleolar localization", Journal of Peptide Science, vol. 18, pp. 215-223, 2012. http://dx.doi.org/10.1002/psc.1432
- Y. PERERA, N.D. TORO, L. GOROVAYA, J. FERNANDEZ-DE-COSSIO, H.G. FARINA, and S.E. PEREA, "Synergistic interactions of the anti-casein kinase 2 CIGB-300 peptide and chemotherapeutic agents in lung and cervical preclinical cancer models", Molecular and Clinical Oncology, vol. 2, pp. 935-944, 2014. http://dx.doi.org/10.3892/mco.2014.338
- D. Destouches, N. Page, Y. Hamma-Kourbali, V. Machi, O. Chaloin, S. Frechault, C. Birmpas, P. Katsoris, J. Beyrath, P. Albanese, M. Maurer, G. Carpentier, J. Strub, A. Van Dorsselaer, S. Muller, D. Bagnard, J.P. Briand, and J. Courty, "A Simple Approach to Cancer Therapy Afforded by Multivalent Pseudopeptides That Target Cell-Surface Nucleoproteins", Cancer Research, vol. 71, pp. 3296-3305, 2011. http://dx.doi.org/10.1158/0008-5472.CAN-10-3459
- Y. Perera, H.G. Farina, J. Gil, A. Rodriguez, F. Benavent, L. Castellanos, R.E. Gómez, B.E. Acevedo, D.F. Alonso, and S.E. Perea, "Anticancer peptide CIGB-300 binds to nucleophosmin/B23, impairs its CK2-mediated phosphorylation, and leads to apoptosis through its nucleolar disassembly activity", Molecular Cancer Therapeutics, vol. 8, pp. 1189-1196, 2009. http://dx.doi.org/10.1158/1535-7163.MCT-08-1056
- S.J. Woods, K.M. Hannan, R.B. Pearson, and R.D. Hannan, "The nucleolus as a fundamental regulator of the p53 response and a new target for cancer therapy", Biochimica et Biophysica Acta (BBA) - Gene Regulatory Mechanisms, vol. 1849, pp. 821-829, 2015. http://dx.doi.org/10.1016/j.bbagrm.2014.10.007
- D. Drygin, A. Siddiqui-Jain, S. O'Brien, M. Schwaebe, A. Lin, J. Bliesath, C.B. Ho, C. Proffitt, K. Trent, J.P. Whitten, J.K.C. Lim, D. Von Hoff, K. Anderes, and W.G. Rice, "Anticancer Activity of CX-3543: A Direct Inhibitor of rRNA Biogenesis", Cancer Research, vol. 69, pp. 7653-7661, 2009. http://dx.doi.org/10.1158/0008-5472.CAN-09-1304
- D. Drygin, A. Lin, J. Bliesath, C.B. Ho, S.E. O'Brien, C. Proffitt, M. Omori, M. Haddach, M.K. Schwaebe, A. Siddiqui-Jain, N. Streiner, J.E. Quin, E. Sanij, M.J. Bywater, R.D. Hannan, D. Ryckman, K. Anderes, and W.G. Rice, "Targeting RNA Polymerase I with an Oral Small Molecule CX-5461 Inhibits Ribosomal RNA Synthesis and Solid Tumor Growth", Cancer Research, vol. 71, pp. 1418-1430, 2011. http://dx.doi.org/10.1158/0008-5472.CAN-10-1728
- M. Bywater, G. Poortinga, E. Sanij, N. Hein, A. Peck, C. Cullinane, M. Wall, L. Cluse, D. Drygin, K. Anderes, N. Huser, C. Proffitt, J. Bliesath, M. Haddach, M. Schwaebe, D. Ryckman, W. Rice, C. Schmitt, S. Lowe, R. Johnstone, R. Pearson, G. McArthur, and R. Hannan, "Inhibition of RNA Polymerase I as a Therapeutic Strategy to Promote Cancer-Specific Activation of p53", Cancer Cell, vol. 22, pp. 51-65, 2012. http://dx.doi.org/10.1016/j.ccr.2012.05.019
- J.E. Wulff, R. Siegrist, and A.G. Myers, "The Natural Product Avrainvillamide Binds to the Oncoprotein Nucleophosmin", Journal of the American Chemical Society, vol. 129, pp. 14444-14451, 2007. http://dx.doi.org/10.1021/ja075327f
- H. Mukherjee, K. Chan, V. Andresen, M.L. Hanley, B.T. Gjertsen, and A.G. Myers, "Interactions of the Natural Product (+)-Avrainvillamide with Nucleophosmin and Exportin-1 Mediate the Cellular Localization of Nucleophosmin and its AML-Associated Mutants", ACS Chemical Biology, vol. 10, pp. 855-863, 2015. http://dx.doi.org/10.1021/cb500872g
- S. Chiarella, A. De Cola, G.L. Scaglione, E. Carletti, V. Graziano, D. Barcaroli, C. Lo Sterzo, A. Di Matteo, C. Di Ilio, B. Falini, A. Arcovito, V. De Laurenzi, and L. Federici, "Nucleophosmin mutations alter its nucleolar localization by impairing G-quadruplex binding at ribosomal DNA", Nucleic Acids Research, vol. 41, pp. 3228-3239, 2013. http://dx.doi.org/10.1093/nar/gkt001
- C.G. Grummitt, F.M. Townsley, C.M. Johnson, A.J. Warren, and M. Bycroft, "Structural Consequences of Nucleophosmin Mutations in Acute Myeloid Leukemia", Journal of Biological Chemistry, vol. 283, pp. 23326-23332, 2008. http://dx.doi.org/10.1074/jbc.M801706200
- W. Qi, K. Shakalya, A. Stejskal, A. Goldman, S. Beeck, L. Cooke, and D. Mahadevan, "NSC348884, a nucleophosmin inhibitor disrupts oligomer formation and induces apoptosis in human cancer cells", Oncogene, vol. 27, pp. 4210-4220, 2008. http://dx.doi.org/10.1038/onc.2008.54
- H.J. Chan, J.J. Weng, and B.Y.M. Yung, "Nucleophosmin/B23-binding peptide inhibits tumor growth and up-regulates transcriptional activity of p53", Biochemical and Biophysical Research Communications, vol. 333, pp. 396-403, 2005. http://dx.doi.org/10.1016/j.bbrc.2005.04.176
- S.M. Nimjee, C.P. Rusconi, and B.A. Sullenger, "Aptamers: An Emerging Class of Therapeutics", Annual Review of Medicine, vol. 56, pp. 555-583, 2005. http://dx.doi.org/10.1146/annurev.med.56.062904.144915
- B. Falini, C. Mecucci, E. Tiacci, M. Alcalay, R. Rosati, L. Pasqualucci, R. La Starza, D. Diverio, E. Colombo, A. Santucci, B. Bigerna, R. Pacini, A. Pucciarini, A. Liso, M. Vignetti, P. Fazi, N. Meani, V. Pettirossi, G. Saglio, F. Mandelli, F. Lo-Coco, P. Pelicci, and M.F. Martelli, "Cytoplasmic Nucleophosmin in Acute Myelogenous Leukemia with a Normal Karyotype", New England Journal of Medicine, vol. 352, pp. 254-266, 2005. http://dx.doi.org/10.1056/NEJMoa041974
- . , "Genomic and Epigenomic Landscapes of Adult De Novo Acute Myeloid Leukemia", New England Journal of Medicine, vol. 368, pp. 2059-2074, 2013. http://dx.doi.org/10.1056/NEJMoa1301689
- B. Falini, "Immunohistochemistry predicts nucleophosmin (NPM) mutations in acute myeloid leukemia", Blood, vol. 108, pp. 1999-2005, 2006. http://dx.doi.org/10.1182/blood-2006-03-007013
- N. Bolli, I. Nicoletti, M.F. De Marco, B. Bigerna, A. Pucciarini, R. Mannucci, M.P. Martelli, A. Liso, C. Mecucci, F. Fabbiano, M.F. Martelli, B.R. Henderson, and B. Falini, "Born to Be Exported: COOH-Terminal Nuclear Export Signals of Different Strength Ensure Cytoplasmic Accumulation of Nucleophosmin Leukemic Mutants", Cancer Research, vol. 67, pp. 6230-6237, 2007. http://dx.doi.org/10.1158/0008-5472.CAN-07-0273
- F. Scaloni, L. Federici, M. Brunori, and S. Gianni, "Deciphering the folding transition state structure and denatured state properties of Nucleophosmin C-terminal domain", Proceedings of the National Academy of Sciences, vol. 107, pp. 5447-5452, 2010. http://dx.doi.org/10.1073/pnas.0910516107
- A. De Cola, L. Pietrangelo, F. Forlì, D. Barcaroli, M.C. Budani, V. Graziano, F. Protasi, C. Di Ilio, V. De Laurenzi, and L. Federici, "AML cells carrying NPM1 mutation are resistant to nucleophosmin displacement from nucleoli caused by the G-quadruplex ligand TmPyP4", Cell Death & Disease, vol. 5, pp. e1427-e1427, 2014. http://dx.doi.org/10.1038/cddis.2014.402
- B. Falini, "Both carboxy-terminus NES motif and mutated tryptophan(s) are crucial for aberrant nuclear export of nucleophosmin leukemic mutants in NPMc+ AML", Blood, vol. 107, pp. 4514-4523, 2006. http://dx.doi.org/10.1182/blood-2005-11-4745
- B. Falini, and M.P. Martelli, "NPM1-mutated AML: targeting by disassembling", Blood, vol. 118, pp. 2936-2938, 2011. http://dx.doi.org/10.1182/blood-2011-07-366146
- B. Falini, I. Gionfriddo, F. Cecchetti, S. Ballanti, V. Pettirossi, and M.P. Martelli, "Acute myeloid leukemia with mutated nucleophosmin (NPM1): Any hope for a targeted therapy?", Blood Reviews, vol. 25, pp. 247-254, 2011. http://dx.doi.org/10.1016/j.blre.2011.06.001
- B. Falini, L. Brunetti, and M.P. Martelli, "Dactinomycin in NPM1-Mutated Acute Myeloid Leukemia", New England Journal of Medicine, vol. 373, pp. 1180-1182, 2015. http://dx.doi.org/10.1056/NEJMc1509584
ACKNOWLEDGMENTS
This work was supported by grant from the National Natural Science Foundation of China (31471263).
COPYRIGHT
© 2018

Nucleolar Stress: hallmarks, sensing mechanism and diseases by Yang et al. is licensed under a Creative Commons Attribution 4.0 International License.


