Reviews:
Cell Stress, Vol. 3, No. 1, pp. 9 - 18; doi: 10.15698/cst2019.01.171
Regulation of T cell antitumor immune response by tumor induced metabolic stress

1 Cancer Biology Research Platform, Centre Georges-François Leclerc, Dijon, France.
2 Université de Bourgogne-Franche Comté.
3 GIMI Genetic and Immunology Medical Institute, Dijon, France.
4 INSERM UMR1231, Dijon, France.
Keywords: antitumor immmunity, metabolic stress, acidosis, hypoxia, amino acids, fatty acid, T cells.
Abbreviations:
HIF – hypoxia inducible factor,
IDO - indoleamine-2,3-dioxygenase,
LDH – lactate dehydrogenase,
PD-1 – programmed cell death 1,
PD-L1 – programmed death-ligand 1,
ROR – RAR-related orphan receptor,
TME – tumor microrenvironment,
Treg – regulatory T cell.
Received originally: 29/09/2018 Received in revised form: 26/10/2018
Accepted: 30/10/2018
Published: 27/11/2018
Correspondence:
Prof. François Ghiringhelli, Centre Georges François Leclerc, Genetic and Immunology Medical Institute GIMI, 1 rue du professeur Marion, 21000 Dijon, France. Phone: (33) 3-80-39-33-53; Fax: (33) 3-80-39-34-34 fghiringhelli@cgfl.fr
Conflict of interest statement: Authors declare no conflict of interest relevant to this article.
Please cite this article as: Fanny Chalmin, Mélanie Bruchard, Frederique Vegran and Francois Ghiringhelli (2019). Regulation of T cell antitumor immune response by tumor induced metabolic stress. Cell Stress 3(1): 9-18. doi: 10.15698/cst2019.01.171
Abstract
Adaptive T cell immune response is essential for tumor growth control. The efficacy of immune checkpoint inhibitors is regulated by intratumoral immune response. The tumor microenvironment has a major role in adaptive immune response tuning. Tumor cells generate a particular metabolic environment in comparison to other tissues. Tumors are characterized by glycolysis, hypoxia, acidosis, amino acid depletion and fatty acid metabolism modification. Such metabolic changes promote tumor growth, impair immune response and lead to resistance to therapies. This review will detail how these modifications strongly affect CD8 and CD4 T cell functions and impact immunotherapy efficacy.
INTRODUCTION
The tumor microenvironment (TME) plays an important role in tumor progression and response to therapy. A growing number of publications show that CD8 T lymphocytes accumulation in tumor bed is a biomarker of a good clinical outcome in most cancer types [1]. Moreover, such an immune response is also a surrogate marker of chemotherapy efficacy in breast cancer setting and a biomarker of checkpoint inhibitors efficacy [2][3]. Antitumor immunotherapy and in particular immune-checkpoint-targeting inhibitors are revolutionizing cancer therapy [4]. Checkpoint inhibitors targeting PD-1 (programmed cell death protein-1)/PD-L1 (programmed death-ligand 1) lead to a response rate in many tumor types. However, in prevalent tumor types, such as colorectal cancer, lung cancer and breast cancer, substantial responses to checkpoint blockade have only been observed in specific subsets of patients, thus suggesting that both patient selection and therapy combination may be crucial [5]. Currently two concepts evolve in parallel to predict checkpoint efficacy: the presence of mutations in tumor cells and the presence of immune infiltrate at tumor site (the concept of cold vs hot tumor). The ability of a tumor to respond to immunotherapy depends on the presence of CD8 at the tumor site. However, a CD8 infiltrate does not perfectly correlate to the checkpoint response rate, thus suggesting that in addition to the number of immune cells, functional characteristics of intratumoral infiltrating T cells must be taken into account. In addition to CD8 T cell infiltrate, many other cells influence antitumor immune response. For example, CD4 T cells are essential and different subsets are defined. Regulatory T cells (Treg) and Th2 cells have immunosuppressive functions while Th1 cells have an antitumoral effect and sustain CD8 antitumoral effects [1]. Th17 cells can have different effects depending on the tumor type but frequently promote inflammation and neoangiogenesis [6]. The myeloid component of the immune system is also important to promote antitumoral T cell immune response or to drive immunosuppression. The presence of mature myeloid dendritic cells is essential for a good immune response. Myeloid derived suppressor cells (MDSC) are an essential component of the tumor induced tolerance and the ratio of Type 2/Type 1 Tumor Associated Macrophages is important to balance immune reaction from immunosuppression versus antitumoral response [6]. The recruitment and functions of immune cells in the TME markedly vary between patients even in the same tumor type for unknown reason.
–
While immune response is essential to control tumor growth and to promote checkpoint inhibitor efficacy, tumor environment physical conditions may influence T cell response. Tumors are characterized by low oxygen level and hypoxia, extracellular milieu acidification, oxidative stress and glucose deprivation. In this review we will resume how these physical modifications of TME affect T cell antitumoral immune response.
ACIDOSIS AND GLYCOLYSIS
HYPOXIA
The physiological oxygen fractions called normoxia largely vary between tissues and within the same tissue [7][8][9]. For example, the maximum value of oxygen found in the body reaches 14% in lung alveoli but only 1% in the skin. These values have to be put in balance with the atmospheric level of oxygen of 21%, frequently used for in vitro experiments.
–
Hypoxic areas can often be found within solid tumors. The oxygen level in tumors is frequently low, below 1%, and a high level of hypoxia is often associated with poor prognosis [10]. At the cellular level, hypoxia promotes tumor cell heterogeneity, epithelial to mesenchymal transition, tumor cell stemness, migration and metastatic process, and resistance to classical cytotoxic treatments such as radiotherapy and chemotherapy [11][12][13][14]. Molecular mechanisms underlying hypoxia mainly rely on the stabilization of hypoxia inducible factors (HIF1 and 2). These transcription factors mediate the cellular response to hypoxia by regulating the expression of different genes such as proangiogenic factors like VEGF (vascular endothelial growth factor) and glycolysis related genes. Indeed, low oxygen may impair energy production via oxidative phosphorylation and requires glycolysis which is less dependent on oxygen level.
–
It was recently shown that HIF-1 is able to regulate the balance between Treg and Th17 differentiation in CD4 T cells. Although TGF-? (transforming growth factor) is required for both Th17 and Treg differentiation, these cell types have opposing functions. While Th17 are pro-inflammatory cells, Tregs have an anti-inflammatory role [15][16][17][18][19][20]. Tregs master regulator is the transcriptional factor FoxP3 (forkhead box P3). In addition to TGF-?, Th17 cells require IL-6 for differentiation and expression of the transcriptional factor ROR?t (RAR-related orphan receptor), the master regulator of this cell type. Hypoxia promotes accumulation of Th17 cells and decreases the number of Tregs (Figure 1 top). Mechanistically, HIF-1? enhances Th17 development. HIF cooperates with STAT3 (Signal transducer and activator of transcription) to promote expression of ROR?t and then cooperates with ROR?t and p300 to transactivate IL-17 production. In contrast, HIF-1? blunts Treg differentiation by binding to FoxP3, promoting its ubiquitination and subsequent degradation by the proteasome [21]. While Tregs frequently promote tumor growth and mediate immunosuppression, we can hypothesize that such mechanism could promote antitumoral immune response by limiting Treg dependent immunosuppression and activating proinflammatory Th17 cells which could exert some antitumoral effects.
–
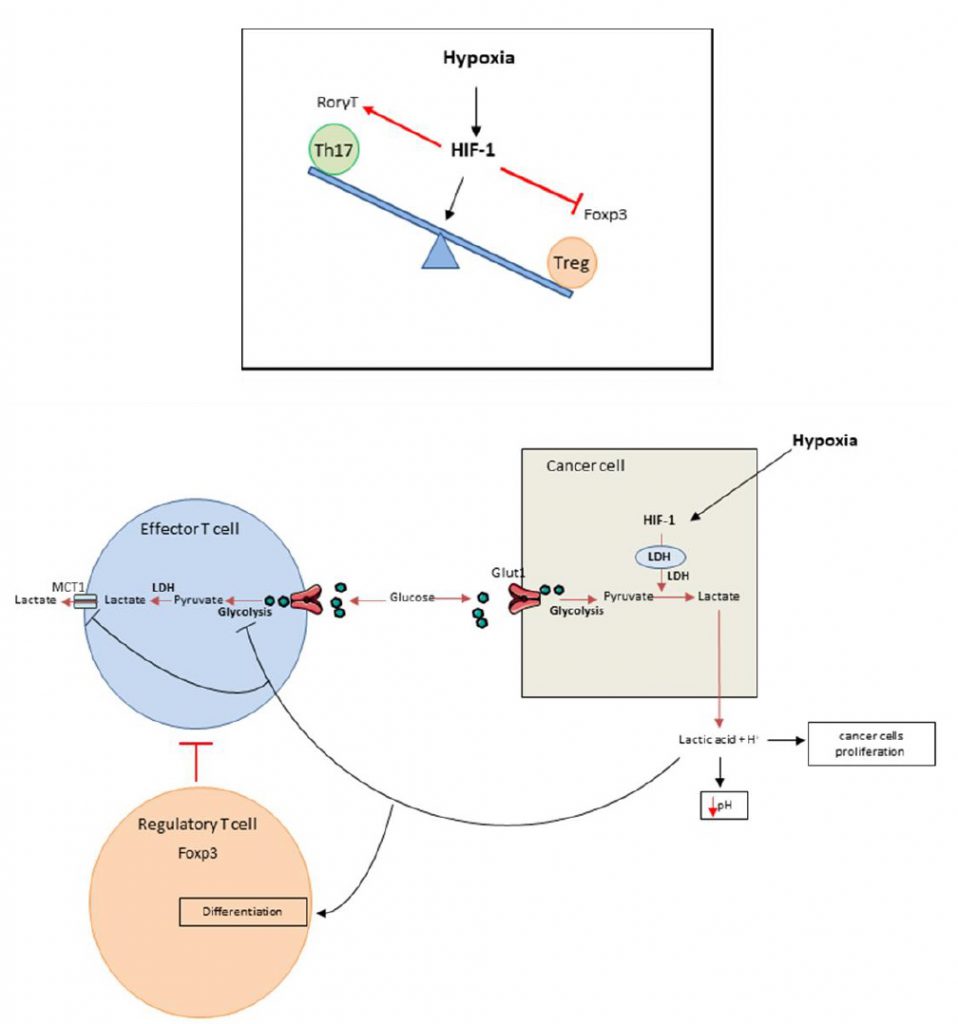 | FIGURE 1. Top: Role of hypoxia in Th17/Treg balance disregulation. Hypoxia within the tumors enhances the Th17 development over the Treg development. This is due to the induction of HIF-1 that will in one side promote the expression of ROR?t through its cooperation with STAT3 and in the other side bind to Foxp3 inducing its ubiquitination and degradation. Bottom: Hypoxia promotes immunosuppression. Cancer cells have a high glycolytic activity manifested by glucose intake through Glut1 transporter that is metabolized into pyruvate and then into lactic acid through LDH activation induced by HIF-1. The lactic acid is exported within the tumor microenvironment and induces its acidification. Effector T lymphocytes are also dependent of their glycolytic activity and must release lactate by SLC16A1; best known as MCT1. In this context of acidification, MCT1 is inhibited thus blocking the glycolysis and consequently the activation of the effector T cells. Moreover the high lactate concentration within the tumor microenvironment will promote Treg biology by inducing FoxP3 expression. In these conditions hypoxia leads to immunosuppression through the activation or inhibition of different immune cells populations. |
–
CD8 T cell priming under hypoxia can promote differentiation toward lytic effector cells, with increased expression of interferon gamma (IFN?), granzym B (GZMB) and Fas ligand (FASL), but might reduce cell expansion [22][23][24][25]. Hypoxia promotes a metabolic switch from an oxidative phosphorylation metabolism toward a glycolytic metabolism [26] which promotes effector and limits memory differentiation, largely dependent on oxidative phosphorylation and fatty acid oxidation [26][27]. Such data suggest that the use of hypoxia to generate ex vivo transgenic T cells or Chimeric Antigen Receptor-T cells for adoptive anticancer immunotherapy could be attractive. Moreover, it would be interesting to compare the efficacy of adoptive transfer of cells differentiated under hypoxia with increased cytotoxic effector functions and less stem cell memory properties, to the transfer of younger cells less cytotoxic with stemness capacity and better persistence and self-renewal [28][29][30].
–
Hypoxia can also affect activated memory CD8 T cells. This context is closer to tumor reality since memory cells migrate to tumor site and are then reactivated. Hypoxia prevents memory CD8 T cell expansion by decreasing both cell proliferation rate and viability, partly through apoptosis induction. Additionally, hypoxia promotes adenosine production by TME and adenosine could inhibit CD8 T cell functions. Hypoxia effect is also dependent on T cell receptor (TCR) engagement and no effect of hypoxia is observed on resting memory T cells [26]. Hypoxia could also have positive effects and enhance IL-10 production in CD8 T cells. Although IL-10 could have an immunosuppressive function, it could also sustain the development of memory CD8 T cells. In addition, hypoxia could promote CD25 and CD137 expression. CD137 is a checkpoint activator that can be targeted to reinvigorate CD8 T cells [31][32][33]. In some tumor models, hypoxia enhances PD-L1 expression on tumor cells and thus it might enhance the efficacy of checkpoint inhibitors targeting PD-1/PD-L1 [34].
ACIDOSIS AND GLYCOLYSIS
Median extracellular pH in human tumors ranges between 6.9 and 7.0 (compared to 7.4 in normal tissues) [35] while intracellular tumor pH remains unaltered in tumor bed [36]. Acidification of the TME has direct protumoral functions such as angiogenesis, prometastatic effect, and resistance to radiation or cytotoxic chemotherapies and is thus associated with poor prognosis [37][38][39][40][41][42][43][44][45][46][47][48][49][50][51][52][53][54][55][56][57][58][59][60][61][62]. Acidification of TME is due to local metabolism, which favors glycolysis and lactic acid production (Figure 1 bottom) [63][64][65]. Anaerobic glycolysis and production of lactic acid are strongly correlated with hypoxia but glycolysis could also arise in normoxic conditions. As glycolysis is energetically less efficient than oxidative phosphorylation, tumors must develop an important glycolytic flux to generate enough energy [66], this process is called the Warburg effect. Consequently lactic acid accumulates inside the TME [56], thus reducing pH. This acidification has a negative impact on T cell behavior and many studies demonstrated that low intra-tumoral pH leads to downregulation of anti-tumor immune responses [67]. In vitro experiments showed that at pH lower than 6.6 and similar to tumor pH [35], T cell proliferation, cytotoxicity and cytokine production are impaired [68]. This effect on T cells is rather dependent on pH than on the presence of lactate [25][69][70]. Interestingly, T cell function could be restored after pH neutralization [25][69][70][71][72]. Such data underline that acidosis mostly inhibits T cell function rather than inducing T cell death [70][71][72]. In vivo, tumor-derived lactic acid also impedes anti-tumor immunity [73]. The LDHA (lactate dehydrogenase) gene, which codes for the LDH-2 protein, converts pyruvate into lactate. LDHA gene deficient tumors grow slower than control tumors in immunocompetent mice, but not in immunodeficient mice, thus demonstrating that lactate impedes immune response in vivo. Effector T lymphocytes are also dependent on their glycolytic activity and release lactate by SLC16A1 (best known as monocarboxylate transporter 1 MCT1). In TME, in the context of acidification, MCT1 is inhibited, thus blocking glycolysis and consequently the activation of effector T cells. In this context, we have observed reduced IFN? and GZMB production by T cells. In humans, LDH expression in melanomas negatively correlates with T cell survival and activation [73]. On the other hand, Treg biology is promoted by high lactate concentration. FoxP3, Treg master regulator, shifts cellular metabolism from glycolysis toward oxidative phosphorylation [74]. Lactic acid inhibits T cell glycolysis leading to FoxP3 expression and promoting Treg differentiation [75][76][77]. Moreover, lactate uptake is required for Treg immunosuppressive effects [78]. At a mechanistic level, lactate is secreted by cancer cells via a monocarboxylate co-transporter, which induces acidification of the tumor with the release of lactate and H+. A high concentration of lactate and H+ blocks the monocarboxylate co-transporter of T cells. This blockade induces accumulation of these compounds in T cells, thus blunting glycolysis [79]. This leads to a reduction of the intracellular phosphoenolpyruvat level, a crucial glycolysis metabolite necessary for TCR mediated activation [73].
–
Recently it has been shown that tumor bed acidification blunts the efficacy of checkpoint inhibitors [80][81][82]. High LDH activity in blood is negatively correlated with the clinical outcome in melanoma patients treated with ipilimumab [83], pembrolizumab [82], or a combination of CTLA-4 and PD-1 blockade [84]. Similar results were observed with lung cancer treated with anti PD-1 [85], suggesting that combination of checkpoint inhibitors with drugs that lower tumor acidity could be interesting. Many drugs are currently tested such as glycolysis, lactate transporter and proton transporter inhibitors but also buffer therapies. Glycolysis and lactate are essential for T cell biology. Consequently, therapies targeting either glycolysis or lactate transporters are probably not ideal. In contrast, T cells are less dependent on proton transporters. Therefore, proton pump inhibitors and bicarbonate based therapies, which both can neutralize acidification, are probably better candidates to enhance immune response and to promote checkpoint inhibitors efficacy and adoptive T cell therapies.
AMINO ACIDS
In addition to glucose, amino acids are essential elements for energy generation in tumor cells and immune cells [86]. Cancer cells have the ability to consume a high level of amino acids, leading to T cell deprivation. Both arginine and tryptophan are essential for T cells and cannot be produced by T cell metabolism. Consequently, consumption of these amino acids by cancer cells controls the local immune response by inducing T cell metabolic stress (Figure 2).
–
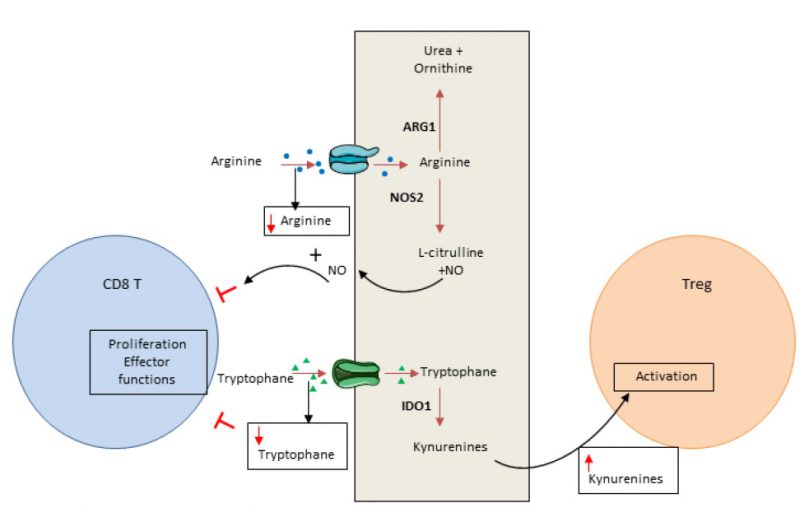 | FIGURE 2: Amino acid consumption by cancer cells promotes immunosuppression. Cancer cells deplete essential amino acids for T cell activity leading to a decrease of their antitumoral function. For instance cancer cells import arginine that will be metabolized in Urea by Arginase 1 (ARG1) or in L-citruline + Nitric oxide (NO) by Nictric oxide synthase 2 (NOS2). The diminution of Arginine within the tumoral microenvironment as well as the production of NO by the cancer cells are inhibitor of T cells. The diminution of tryptophan in the tumoral microenvironment will also inhibit T cell. Moreover, the increase in kynurenine (tryptophan metabolite produced by IDO) will promote Treg cell activity. |
–
Arginine can be used in oxidative phosphorylation and as a substrate for glycolysis in T cells [87]. Arginine availability favors memory T cell generation [88]. Arginine is converted by arginase or nitric oxide synthase, normally expressed in myeloid cells such as myeloid-derived suppressor cells, macrophages, dendritic cells and cancer cells. Arginase is highly expressed in many tumor types and induces T cell function inhibition via arginine deprivation [89]. Nitric oxide synthase, also frequently expressed in tumors, degrades arginine into nitric oxide. Nitric oxide could directly blunt T cell proliferation and secreting functions and promote T cell apoptosis [90].
–
Tryptophan is critical for several metabolic pathways and proliferation. Indoleamine-2,3-dioxygenase (IDO) 1 and 2 are key enzymes that transform tryptophan into its metabolite kynurenine. In tumors, IDO induces tryptophan deprivation and kynurenin accumulation. Tryptophan is essential for T cell biology and its depletion induces eukaryotic translation initiation factor 2 alpha kinase 4 (EIF2AK4; also known as GCN2, General Control Non-derepressible 2 kinase) activation and CD3 ?-chain downregulation. These events reduce T cell effector functions and limit their proliferation [91][92]. Similarly, kynurenine restrains T cell proliferation [93] and could activate arylhydrocarbon receptor, promoting the switch of CD4 T cells into Treg cells [94]. Recent data demonstrated that both cancer cells and tumor infiltrating myeloid cells could have a high level of IDO enzyme expression [95][96][97]. IDO is not constitutively expressed and its induction is dependent on inflammatory signal stimulation such as IFN? [95][96][97][98][99]. IDO acts as a negative feedback loop of Th1 response in cancer.
–
Inhibition of IDO and arginase could restore T cell functions and could improve the effector T cells/Treg ratio. Multiple IDO and arginase inhibitors are currently in development, associated to adoptive T cell therapy or checkpoint inhibitors [94]. However first reports are disappointing and phase III clinical trials evaluating efficacy of combination therapies involving IDO1 inhibitors and pembrolizumab in patients with melanoma are stopped (ECHO-301/KEYNOTE-252 study).
FATTY ACID METABOLISM
Alterations in lipid metabolism are frequently observed in cancer cells [100]. Tumor aggressiveness is linked to its capacity to store high levels of lipid and cholesterol [101][102][103].
–
Fatty acid metabolism has a role in T cell differentiation. Effector CD8 T cells use de novo fatty acid synthase and fatty acid uptake, whereas memory T cells degrade endogenous esterified fatty acids [104]. Endogenous fatty acid generation is essential to maintain energy level after PD-1 activation [105]. PD-1 activation impairs glucose and glutamine uptake but promotes fatty acid oxidation and utilization of endogenous lipids. Endogenous T cell lipid reserves provide energy and may be related to T cell exhaustion and T cell ability to be reactivated by checkpoint inhibitors [106]. Lipids produced by tumor cells could also have an impact on T cells by their transformation into prostaglandin by cyclooxygenase 2. Prostaglandin could then induce inflammation [107].
–
Concerning CD4 T cells, competition between de novo fatty acid synthase and exogenous uptake controls the decision between Th17 and Treg cells differentiation [108][109]. Inhibition of acetyl-CoA carboxylase 1 and the related de novo fatty acid synthase restrains Th17 differentiation and promotes Treg cells. Such data suggest that in tumor tissue where fatty acids are mostly directed to tumor cells, the deficit in exogenous fatty acids promotes de novo fatty acid synthase and Th17 response [108]. The molecule mTOR (mammalian target of rapamycin) is essential to control Treg differentiation, function, and survival notably by its ability to control many lipid metabolism genes [110][111].
–
Targeting fatty acid metabolism could be useful to improve antitumor immune response [109]. Fatty acid oxidase is required not only for memory CD8 T cell development but also for Treg cell differentiation [112], therefore its blockade limits Treg dependent immunosuppression. Similarly, fatty acid oxidase has a critical role in MDSC-mediated T cell suppressive function [113][114]. Thus, inhibiting fatty acid metabolism may affect multiple immune populations and could have unpredictable outcomes. In contrast, fibrate which enhances fatty acid oxidase activity and enhances endogenous production of fatty acids, may enhance functions of exhausted CD8 T cells and delay tumor growth when used together with PD-1-blocking immunotherapy [115].
CONCLUSION
TME is metabolically different from healthy tissues. Tumors are characterized by glycolysis, hypoxia, acidosis, amino acid depletion and fatty acid metabolism modifications. These modifications strongly affect CD8 T cell functions and T helper cell differentiation. Consequently, better understanding of tumor environment metabolic changes will provide key information for the development of novel therapies that improve T cell immune functions. A better knowledge of the metabolic pathways not shared between cancer and immune cells will allow the selection of drugs targeting specifically cancer or immune cells. The use of these novel drugs in combination with immunotherapies such as checkpoint inhibitors or adoptive cell transfer may open new opportunities to improve cancer treatment.
REFERENCES
- Fridman WH, Pages F, Sautes-Fridman C, Galon J (2012). The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer 12(4): 298-306. doi: 10.1038/nrc3245
- Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G (2017). Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol 17(2): 97-111. doi: 10.1038/nri.2016.107
- Pitt JM, Marabelle A, Eggermont A, Soria JC, Kroemer G, Zitvogel L (2016). Targeting the tumor microenvironment: removing obstruction to anticancer immune responses and immunotherapy. Ann Oncol 27(8): 1482-1492. doi: 10.1093/annonc/mdw168
- Zou W, Wolchok JD, Chen L (2016). PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci Transl Med 8(328): 328rv324. doi: 10.1126/scitranslmed.aad7118
- Gibney GT, Weiner LM, Atkins MB (2016). Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol 17(12): e542-e551. doi: 10.1016/S1470-2045(16)30406-5
- Martin F, Apetoh L, Ghiringhelli F (2012). Controversies on the role of Th17 in cancer: a TGF-beta-dependent immunosuppressive activity? Trends Mol Med 18(12): 742-749. doi: 10.1016/j.molmed.2012.09.007
- Ivanovic Z (2009). Hypoxia or in situ normoxia: The stem cell paradigm. J Cell Physiol 219(2): 271-275. doi: 10.1002/jcp.21690
- McNamee EN, Korns Johnson D, Homann D, Clambey ET (2013). Hypoxia and hypoxia-inducible factors as regulators of T cell development, differentiation, and function. Immunol Res 55(1-3): 58-70. doi: 10.1007/s12026-012-8349-8
- Carreau A, El Hafny-Rahbi B, Matejuk A, Grillon C, Kieda C (2011). Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J Cell Mol Med 15(6): 1239-1253. doi: 10.1111/j.1582-4934.2011.01258.x
- Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S, Shi Q, Cao Y, Lathia J, McLendon RE, Hjelmeland AB, Rich JN (2009). Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 15(6): 501-513. doi: 10.1016/j.ccr.2009.03.018
- Harris AL (2002). Hypoxia–a key regulatory factor in tumour growth. Nat Rev Cancer 2(1): 38-47. doi: 10.1038/nrc704
- Semenza GL (2010). Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 29(5): 625-634. doi: 10.1038/onc.2009.441
- Noman MZ, Hasmim M, Messai Y, Terry S, Kieda C, Janji B, Chouaib S (2015). Hypoxia: a key player in antitumor immune response. A Review in the Theme: Cellular Responses to Hypoxia. Am J Physiol Cell Physiol 309(9): C569-579. doi: 10.1152/ajpcell.00207.2015
- Chouaib S, Noman MZ, Kosmatopoulos K, Curran MA (2017). Hypoxic stress: obstacles and opportunities for innovative immunotherapy of cancer. Oncogene 36(4): 439-445. doi: 10.1038/onc.2016.225
- Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK (2006). Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441(7090): 235-238. doi: 10.1038/nature04753
- Dong C (2008). TH17 cells in development: an updated view of their molecular identity and genetic programming. Nat Rev Immunol 8(5): 337-348. doi: 10.1038/nri2295
- Littman DR, Rudensky AY (2010). Th17 and regulatory T cells in mediating and restraining inflammation. Cell 140(6): 845-858. doi: 10.1016/j.cell.2010.02.021
- Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT (2006). Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 441(7090): 231-234. doi: 10.1038/nature04754
- O’Quinn DB, Palmer MT, Lee YK, Weaver CT (2008). Emergence of the Th17 pathway and its role in host defense. Adv Immunol 99(115-163. doi: 10.1016/S0065-2776(08)00605-6
- Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B (2006). TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 24(2): 179-189. doi: 10.1016/j.immuni.2006.01.001
- Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, Bordman Z, Fu J, Kim Y, Yen HR, Luo W, Zeller K, Shimoda L, Topalian SL, Semenza GL, Dang CV, Pardoll DM, Pan F (2011). Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell 146(5): 772-784. doi: 10.1016/j.cell.2011.07.033
- Vuillefroy de Silly R, Ducimetiere L, Yacoub Maroun C, Dietrich PY, Derouazi M, Walker PR (2015). Phenotypic switch of CD8(+) T cells reactivated under hypoxia toward IL-10 secreting, poorly proliferative effector cells. Eur J Immunol 45(8): 2263-2275. doi: 10.1002/eji.201445284
- Caldwell CC, Kojima H, Lukashev D, Armstrong J, Farber M, Apasov SG, Sitkovsky MV (2001). Differential effects of physiologically relevant hypoxic conditions on T lymphocyte development and effector functions. J Immunol 167(11): 6140-6149. doi: 10.4049/jimmunol.167.11.6140
- Xu Y, Chaudhury A, Zhang M, Savoldo B, Metelitsa LS, Rodgers J, Yustein JT, Neilson JR, Dotti G (2016). Glycolysis determines dichotomous regulation of T cell subsets in hypoxia. J Clin Invest 126(7): 2678-2688. doi: 10.1172/JCI85834
- Nakagawa Y, Negishi Y, Shimizu M, Takahashi M, Ichikawa M, Takahashi H (2015). Effects of extracellular pH and hypoxia on the function and development of antigen-specific cytotoxic T lymphocytes. Immunol Lett 167(2): 72-86. doi: 10.1016/j.imlet.2015.07.003
- Larbi A, Zelba H, Goldeck D, Pawelec G (2010). Induction of HIF-1alpha and the glycolytic pathway alters apoptotic and differentiation profiles of activated human T cells. J Leukoc Biol 87(2): 265-273. doi: 10.1189/jlb.0509304
- Finlay DK, Rosenzweig E, Sinclair LV, Feijoo-Carnero C, Hukelmann JL, Rolf J, Panteleyev AA, Okkenhaug K, Cantrell DA (2012). PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J Exp Med 209(13): 2441-2453. doi: 10.1084/jem.20112607
- Gattinoni L, Klebanoff CA, Palmer DC, Wrzesinski C, Kerstann K, Yu Z, Finkelstein SE, Theoret MR, Rosenberg SA, Restifo NP (2005). Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J Clin Invest 115(6): 1616-1626. doi: 10.1172/JCI24480
- Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, Almeida JR, Gostick E, Yu Z, Carpenito C, Wang E, Douek DC, Price DA, June CH, Marincola FM, Roederer M, Restifo NP (2011). A human memory T cell subset with stem cell-like properties. Nat Med 17(10): 1290-1297. doi: 10.1038/nm.2446
- Sukumar M, Liu J, Mehta GU, Patel SJ, Roychoudhuri R, Crompton JG, Klebanoff CA, Ji Y, Li P, Yu Z, Whitehill GD, Clever D, Eil RL, Palmer DC, Mitra S, Rao M, Keyvanfar K, Schrump DS, Wang E, Marincola FM, Gattinoni L, Leonard WJ, Muranski P, Finkel T, Restifo NP (2016). Mitochondrial Membrane Potential Identifies Cells with Enhanced Stemness for Cellular Therapy. Cell Metab 23(1): 63-76. doi: 10.1016/j.cmet.2015.11.002
- Palazon A, Teijeira A, Martinez-Forero I, Hervas-Stubbs S, Roncal C, Penuelas I, Dubrot J, Morales-Kastresana A, Perez-Gracia JL, Ochoa MC, Ochoa-Callejero L, Martinez A, Luque A, Dinchuk J, Rouzaut A, Jure-Kunkel M, Melero I (2011). Agonist anti-CD137 mAb act on tumor endothelial cells to enhance recruitment of activated T lymphocytes. Cancer Res 71(3): 801-811. doi: 10.1158/0008-5472.CAN-10-1733
- Palazon A, Martinez-Forero I, Teijeira A, Morales-Kastresana A, Alfaro C, Sanmamed MF, Perez-Gracia JL, Penuelas I, Hervas-Stubbs S, Rouzaut A, de Landazuri MO, Jure-Kunkel M, Aragones J, Melero I (2012). The HIF-1alpha hypoxia response in tumor-infiltrating T lymphocytes induces functional CD137 (4-1BB) for immunotherapy. Cancer Discov 2(7): 608-623. doi: 10.1158/2159-8290.CD-11-0314
- Yonezawa A, Dutt S, Chester C, Kim J, Kohrt HE (2015). Boosting Cancer Immunotherapy with Anti-CD137 Antibody Therapy. Clin Cancer Res 21(14): 3113-3120. doi: 10.1158/1078-0432.CCR-15-0263
- Semaan A, Dietrich D, Bergheim D, Dietrich J, Kalff JC, Branchi V, Matthaei H, Kristiansen G, Fischer HP, Goltz D (2017). CXCL12 expression and PD-L1 expression serve as prognostic biomarkers in HCC and are induced by hypoxia. Virchows Arch 470(2): 185-196. doi: 10.1007/s00428-016-2051-5
- Wike-Hooley JL, Haveman J, Reinhold HS (1984). The relevance of tumour pH to the treatment of malignant disease. Radiother Oncol 2(4): 343-366. doi: 10.1016/s0167-8140(84)80077-8
- Gerweck LE, Seetharaman K (1996). Cellular pH gradient in tumor versus normal tissue: potential exploitation for the treatment of cancer. Cancer Res 56(6): 1194-1198. PMID: 8640796
- Hunt TK, Aslam RS, Beckert S, Wagner S, Ghani QP, Hussain MZ, Roy S, Sen CK (2007). Aerobically derived lactate stimulates revascularization and tissue repair via redox mechanisms. Antioxid Redox Signal 9(8): 1115-1124. doi: 10.1089/ars.2007.1674
- Vegran F, Boidot R, Michiels C, Sonveaux P, Feron O (2011). Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-kappaB/IL-8 pathway that drives tumor angiogenesis. Cancer Res 71(7): 2550-2560. doi: 10.1158/0008-5472.CAN-10-2828
- Sonveaux P, Copetti T, De Saedeleer CJ, Vegran F, Verrax J, Kennedy KM, Moon EJ, Dhup S, Danhier P, Frerart F, Gallez B, Ribeiro A, Michiels C, Dewhirst MW, Feron O (2012). Targeting the lactate transporter MCT1 in endothelial cells inhibits lactate-induced HIF-1 activation and tumor angiogenesis. PLoS One 7(3): e33418. doi: 10.1371/journal.pone.0033418
- Shi Q, Le X, Wang B, Abbruzzese JL, Xiong Q, He Y, Xie K (2001). Regulation of vascular endothelial growth factor expression by acidosis in human cancer cells. Oncogene 20(28): 3751-3756. doi: 10.1038/sj.onc.1204500
- Goetze K, Walenta S, Ksiazkiewicz M, Kunz-Schughart LA, Mueller-Klieser W (2011). Lactate enhances motility of tumor cells and inhibits monocyte migration and cytokine release. Int J Oncol 39(2): 453-463. doi: 10.3892/ijo.2011.1055
- Martinez-Zaguilan R, Seftor EA, Seftor RE, Chu YW, Gillies RJ, Hendrix MJ (1996). Acidic pH enhances the invasive behavior of human melanoma cells. Clin Exp Metastasis 14(2): 176-186. doi: 10.1007/bf00121214
- Kato Y, Nakayama Y, Umeda M, Miyazaki K (1992). Induction of 103-kDa gelatinase/type IV collagenase by acidic culture conditions in mouse metastatic melanoma cell lines. J Biol Chem 267(16): 11424-11430. PMID: 1317866
- Kato Y, Ozawa S, Tsukuda M, Kubota E, Miyazaki K, St-Pierre Y, Hata R (2007). Acidic extracellular pH increases calcium influx-triggered phospholipase D activity along with acidic sphingomyelinase activation to induce matrix metalloproteinase-9 expression in mouse metastatic melanoma. FEBS J 274(12): 3171-3183. doi: 10.1111/j.1742-4658.2007.05848.x
- Coman D, Huang Y, Rao JU, De Feyter HM, Rothman DL, Juchem C, Hyder F (2016). Imaging the intratumoral-peritumoral extracellular pH gradient of gliomas. NMR Biomed 29(3): 309-319. doi: 10.1002/nbm.3466
- Chen Y, Kung HN, Chen CH, Huang SH, Chen KH, Wang SM (2011). Acidic extracellular pH induces p120-catenin-mediated disruption of adherens junctions via the Src kinase-PKCdelta pathway. FEBS Lett 585(4): 705-710. doi: 10.1016/j.febslet.2011.01.022
- Robey IF, Baggett BK, Kirkpatrick ND, Roe DJ, Dosescu J, Sloane BF, Hashim AI, Morse DL, Raghunand N, Gatenby RA, Gillies RJ (2009). Bicarbonate increases tumor pH and inhibits spontaneous metastases. Cancer Res 69(6): 2260-2268. doi: 10.1158/0008-5472.CAN-07-5575
- Rizwan A, Serganova I, Khanin R, Karabeber H, Ni X, Thakur S, Zakian KL, Blasberg R, Koutcher JA (2013). Relationships between LDH-A, lactate, and metastases in 4T1 breast tumors. Clin Cancer Res 19(18): 5158-5169. doi: 10.1158/1078-0432.CCR-12-3300
- Brizel DM, Schroeder T, Scher RL, Walenta S, Clough RW, Dewhirst MW, Mueller-Klieser W (2001). Elevated tumor lactate concentrations predict for an increased risk of metastases in head-and-neck cancer. Int J Radiat Oncol Biol Phys 51(2): 349-353. doi: 10.1016/s0360-3016(00)80147-9
- Walenta S, Salameh A, Lyng H, Evensen JF, Mitze M, Rofstad EK, Mueller-Klieser W (1997). Correlation of high lactate levels in head and neck tumors with incidence of metastasis. Am J Pathol 150(2): 409-415. PMID: 9033256
- Liu R, Cao J, Gao X, Zhang J, Wang L, Wang B, Guo L, Hu X, Wang Z (2016). Overall survival of cancer patients with serum lactate dehydrogenase greater than 1000 IU/L. Tumour Biol 37(10): 14083-14088. doi: 10.1007/s13277-016-5228-2
- Li G, Wang Z, Xu J, Wu H, Cai S, He Y (2016). The prognostic value of lactate dehydrogenase levels in colorectal cancer: a meta-analysis. BMC cancer 16: 249. doi: 10.1186/s12885-016-2276-3
- Walenta S, Schroeder T, Mueller-Klieser W (2004). Lactate in solid malignant tumors: potential basis of a metabolic classification in clinical oncology. Curr Med Chem 11(16): 2195-2204. doi: 10.2174/0929867043364711
- Walenta S, Wetterling M, Lehrke M, Schwickert G, Sundfor K, Rofstad EK, Mueller-Klieser W (2000). High lactate levels predict likelihood of metastases, tumor recurrence, and restricted patient survival in human cervical cancers. Cancer Res 60(4): 916-921. PMID: 10706105
- Zhang J, Yao YH, Li BG, Yang Q, Zhang PY, Wang HT (2015). Prognostic value of pretreatment serum lactate dehydrogenase level in patients with solid tumors: a systematic review and meta-analysis. Sci Rep 5: 9800. doi: 10.1038/srep09800
- Walenta S, Mueller-Klieser WF (2004). Lactate: mirror and motor of tumor malignancy. Semin Radiat Oncol 14(3): 267-274. doi: 10.1016/j.semradonc.2004.04.004
- Wen Q, Meng X, Xie P, Wang S, Sun X, Yu J (2017). Evaluation of factors associated with platinum-sensitivity status and survival in limited-stage small cell lung cancer patients treated with chemoradiotherapy. Oncotarget 8(46): 81405-81418. doi: 10.18632/oncotarget.19073
- Sauvant C, Nowak M, Wirth C, Schneider B, Riemann A, Gekle M, Thews O (2008). Acidosis induces multi-drug resistance in rat prostate cancer cells (AT1) in vitro and in vivo by increasing the activity of the p-glycoprotein via activation of p38. Int J Cancer 123(11): 2532-2542. doi: 10.1002/ijc.23818
- Wachsberger PR, Landry J, Storck C, Davis K, O’Hara MD, Owen CS, Leeper DB, Coss RA (1997). Mammalian cells adapted to growth at pH 6.7 have elevated HSP27 levels and are resistant to cisplatin. Int J Hyperthermia 13(3): 251-255; discussion 257-259. doi: 10.3109/02656739709023533
- Phua LC, Goh S, Tai DWM, Leow WQ, Alkaff SMF, Chan CY, Kam JH, Lim TKH, Chan ECY (2018). Metabolomic prediction of treatment outcome in pancreatic ductal adenocarcinoma patients receiving gemcitabine. Cancer Chemother Pharmacol 81(2): 277-289. doi: 10.1007/s00280-017-3475-6
- Park HJ, Lee SH, Chung H, Rhee YH, Lim BU, Ha SW, Griffin RJ, Lee HS, Song CW, Choi EK (2003). Influence of environmental pH on G2-phase arrest caused by ionizing radiation. Radiat Res 159(1): 86-93. doi: 10.1667/0033-7587(2003)159[0086:ioepog]2.0.co;2
- Lee HS, Park HJ, Lyons JC, Griffin RJ, Auger EA, Song CW (1997). Radiation-induced apoptosis in different pH environments in vitro. Int J Radiat Oncol Biol Phys 38(5): 1079-1087. doi: 10.1016/s0959-8049(97)84471-1
- Volk T, Jahde E, Fortmeyer HP, Glusenkamp KH, Rajewsky MF (1993). pH in human tumour xenografts: effect of intravenous administration of glucose. Br J Cancer 68(3): 492-500. doi: 10.1038/bjc.1993.375
- Jain RK, Shah SA, Finney PL (1984). Continuous noninvasive monitoring of pH and temperature in rat Walker 256 carcinoma during normoglycemia and hyperglycemia. J Natl Cancer Inst 73(2): 429-436. doi: 10.1093/jnci/73.2.429
- Kallinowski F, Tyler G, Mueller-Klieser W, Vaupel P (1989). Growth-related changes of oxygen consumption rates of tumor cells grown in vitro and in vivo. J Cell Physiol 138(1): 183-191. doi: 10.1002/jcp.1041380124
- Holm E, Hagmuller E, Staedt U, Schlickeiser G, Gunther HJ, Leweling H, Tokus M, Kollmar HB (1995). Substrate balances across colonic carcinomas in humans. Cancer Res 55(6): 1373-1378. doi: 10.1016/0261-5614(88)90316-0
- Gillies RJ, Raghunand N, Karczmar GS, Bhujwalla ZM (2002). MRI of the tumor microenvironment. J Magn Reson Imaging 16(4): 430-450. doi: 10.1002/jmri.10181
- Bosticardo M, Ariotti S, Losana G, Bernabei P, Forni G, Novelli F (2001). Biased activation of human T lymphocytes due to low extracellular pH is antagonized by B7/CD28 costimulation. Eur J Immunol 31(9): 2829-2838. doi: 10.1002/1521-4141(200109)31:9<2829::AID-IMMU2829>3.0.CO;2-U
- Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, Gottfried E, Schwarz S, Rothe G, Hoves S, Renner K, Timischl B, Mackensen A, Kunz-Schughart L, Andreesen R, Krause SW, Kreutz M (2007). Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 109(9): 3812-3819. doi: 10.1182/blood-2006-07-035972
- Mendler AN, Hu B, Prinz PU, Kreutz M, Gottfried E, Noessner E (2012). Tumor lactic acidosis suppresses CTL function by inhibition of p38 and JNK/c-Jun activation. Int J Cancer 131(3): 633-640. doi: 10.1002/ijc.26410
- Calcinotto A, Filipazzi P, Grioni M, Iero M, De Milito A, Ricupito A, Cova A, Canese R, Jachetti E, Rossetti M, Huber V, Parmiani G, Generoso L, Santinami M, Borghi M, Fais S, Bellone M, Rivoltini L (2012). Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res 72(11): 2746-2756. doi: 10.1158/0008-5472.CAN-11-1272
- Pilon-Thomas S, Kodumudi KN, El-Kenawi AE, Russell S, Weber AM, Luddy K, Damaghi M, Wojtkowiak JW, Mule JJ, Ibrahim-Hashim A, Gillies RJ (2016). Neutralization of Tumor Acidity Improves Antitumor Responses to Immunotherapy. Cancer Res 76(6): 1381-1390. doi: 10.1158/0008-5472.CAN-15-1743
- Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, Matos C, Bruss C, Klobuch S, Peter K, Kastenberger M, Bogdan C, Schleicher U, Mackensen A, Ullrich E, Fichtner-Feigl S, Kesselring R, Mack M, Ritter U, Schmid M, Blank C, Dettmer K, Oefner PJ, Hoffmann P, Walenta S, Geissler EK, Pouyssegur J, Villunger A, Steven A, Seliger B, et al. (2016). LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab 24(5): 657-671. doi: 10.1016/j.cmet.2016.08.011
- Angelin A, Gil-de-Gomez L, Dahiya S, Jiao J, Guo L, Levine MH, Wang Z, Quinn WJ, 3rd, Kopinski PK, Wang L, Akimova T, Liu Y, Bhatti TR, Han R, Laskin BL, Baur JA, Blair IA, Wallace DC, Hancock WW, Beier UH (2017). Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab 25(6): 1282-1293 e1287. doi: 10.1016/j.cmet.2016.12.018
- Eleftheriadis T, Pissas G, Karioti A, Antoniadi G, Antoniadis N, Liakopoulos V, Stefanidis I (2013). Dichloroacetate at therapeutic concentration alters glucose metabolism and induces regulatory T-cell differentiation in alloreactive human lymphocytes. J Basic Clin Physiol Pharmacol 24(4): 271-276. doi: 10.1515/jbcpp-2013-0001
- Eleftheriadis T, Sounidaki M, Pissas G, Antoniadi G, Liakopoulos V, Stefanidis I (2016). In human alloreactive CD4(+) T-cells, dichloroacetate inhibits aerobic glycolysis, induces apoptosis and favors differentiation towards the regulatory T-cell subset instead of effector T-cell subsets. Mol Med Rep 13(4): 3370-3376. doi: 10.3892/mmr.2016.4912
- Ostroukhova M, Goplen N, Karim MZ, Michalec L, Guo L, Liang Q, Alam R (2012). The role of low-level lactate production in airway inflammation in asthma. Am J Physiol Lung Cell Mol Physiol 302(3): L300-307. doi: 10.1152/ajplung.00221.2011
- McLane W, Whetstone R, Deshpande R, Menk A, Scharping N, Morrison B, Gellhaus Wendell S, Delgoffe G (2017). Lactic acid as a mediator of metabolic symbiosis between regulatory T Cells and the tumor microenvironment. In: 32nd annual meeting and pre-conference programs of the society for immunotherapy of cancer (SITC 2017, Abstract). National harbor, Maryland.
- Huber V, Camisaschi C, Berzi A, Ferro S, Lugini L, Triulzi T, Tuccitto A, Tagliabue E, Castelli C, Rivoltini L (2017). Cancer acidity: An ultimate frontier of tumor immune escape and a novel target of immunomodulation. Semin Cancer Biol 43: 74-89. doi: 10.1016/j.semcancer.2017.03.001.
- Martens A, Wistuba-Hamprecht K, Geukes Foppen M, Yuan J, Postow MA, Wong P, Romano E, Khammari A, Dreno B, Capone M, Ascierto PA, Di Giacomo AM, Maio M, Schilling B, Sucker A, Schadendorf D, Hassel JC, Eigentler TK, Martus P, Wolchok JD, Blank C, Pawelec G, Garbe C, Weide B (2016). Baseline Peripheral Blood Biomarkers Associated with Clinical Outcome of Advanced Melanoma Patients Treated with Ipilimumab. Clin Cancer Res 22(12): 2908-2918. doi: 10.1158/1078-0432.CCR-15-2412
- Damuzzo V, Solito S, Pinton L, Carrozzo E, Valpione S, Pigozzo J, Arboretti Giancristofaro R, Chiarion-Sileni V, Mandruzzato S (2016). Clinical implication of tumor-associated and immunological parameters in melanoma patients treated with ipilimumab. Oncoimmunology 5(12): e1249559. doi: 10.1080/2162402X.2016.1249559
- Weide B, Martens A, Hassel JC, Berking C, Postow MA, Bisschop K, Simeone E, Mangana J, Schilling B, Di Giacomo AM, Brenner N, Kahler K, Heinzerling L, Gutzmer R, Bender A, Gebhardt C, Romano E, Meier F, Martus P, Maio M, Blank C, Schadendorf D, Dummer R, Ascierto PA, Hospers G, Garbe C, Wolchok JD (2016). Baseline Biomarkers for Outcome of Melanoma Patients Treated with Pembrolizumab. Clin Cancer Res 22(22): 5487-5496. doi: 10.1158/1078-0432.CCR-16-0127
- Kelderman S, Heemskerk B, van Tinteren H, van den Brom RR, Hospers GA, van den Eertwegh AJ, Kapiteijn EW, de Groot JW, Soetekouw P, Jansen RL, Fiets E, Furness AJ, Renn A, Krzystanek M, Szallasi Z, Lorigan P, Gore ME, Schumacher TN, Haanen JB, Larkin JM, Blank CU (2014). Lactate dehydrogenase as a selection criterion for ipilimumab treatment in metastatic melanoma. Cancer Immunol Immunother 63(5): 449-458. doi: 10.1007/s00262-014-1528-9
- Larkin J, Chiarion-Sileni V, Gonzalez R GJ, Cowey CL, Lao CD, Wagstaff J, Hogg D, Hill A, Carlino MS, Wolter P, Lebbé C, Schachter J, Thomas L, Hassel JC, Lorigan P, Walker D, Jiang J, Hodi FS, JD W (2015). Efficacy and safety in key patient subgroups of nivolumab (NIVO) alone or combined with ipilimumab (IPI) versus IPI alone in treatment-naïve patients with advanced melanoma (MEL) (CheckMate 067). The European Cancer Congress 2015 (abstract 3003). Vienna, Austria.
- Taniguchi Y, Tamiya A, Isa SI, Nakahama K, Okishio K, Shiroyama T, Suzuki H, Inoue T, Tamiya M, Hirashima T, Imamura F, Atagi S (2017). Predictive Factors for Poor Progression-free Survival in Patients with Non-small Cell Lung Cancer Treated with Nivolumab. Anticancer Res 37(10): 5857-5862. doi: 10.21873/anticanres.12030
- Munoz-Pinedo C, El Mjiyad N, Ricci JE (2012). Cancer metabolism: current perspectives and future directions. Cell Death Dis 3: e248. doi: 10.1038/cddis.2011.123
- Rodriguez PC, Quiceno DG, Ochoa AC (2007). L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood 109(4): 1568-1573. doi: 10.1182/blood-2006-06-031856
- Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, Kogadeeva M, Picotti P, Meissner F, Mann M, Zamboni N, Sallusto F, Lanzavecchia A (2016). L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 167(3): 829-842 e813. doi: 10.1016/j.cell.2016.09.031
- Ugel S, De Sanctis F, Mandruzzato S, Bronte V (2015). Tumor-induced myeloid deviation: when myeloid-derived suppressor cells meet tumor-associated macrophages. J Clin Invest 125(9): 3365-3376. doi: 10.1172/JCI80006
- Mazzoni A, Bronte V, Visintin A, Spitzer JH, Apolloni E, Serafini P, Zanovello P, Segal DM (2002). Myeloid suppressor lines inhibit T cell responses by an NO-dependent mechanism. J Immunol 168(2): 689-695. doi: 10.4049/jimmunol.168.2.689
- Fallarino F, Grohmann U, You S, McGrath BC, Cavener DR, Vacca C, Orabona C, Bianchi R, Belladonna ML, Volpi C, Santamaria P, Fioretti MC, Puccetti P (2006). The combined effects of tryptophan starvation and tryptophan catabolites down-regulate T cell receptor zeta-chain and induce a regulatory phenotype in naive T cells. J Immunol 176(11): 6752-6761. doi: 10.4049/jimmunol.176.11.6752
- Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, Mellor AL (2005). GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity 22(5): 633-642. doi: 10.1016/j.immuni.2005.03.013
- Zamanakou M, Germenis AE, Karanikas V (2007). Tumor immune escape mediated by indoleamine 2,3-dioxygenase. Immunol Lett 111(2): 69-75. doi: 10.1016/j.imlet.2007.06.001
- Mondanelli G, Ugel S, Grohmann U, Bronte V (2017). The immune regulation in cancer by the amino acid metabolizing enzymes ARG and IDO. Curr Opin Pharmacol 35: 30-39. doi: 10.1016/j.coph.2017.05.002
- Tang D, Yue L, Yao R, Zhou L, Yang Y, Lu L, Gao W (2017). P53 prevent tumor invasion and metastasis by down-regulating IDO in lung cancer. Oncotarget 8(33): 54548-54557. doi: 10.18632/oncotarget.17408
- Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, Boon T, Van den Eynde BJ (2003). Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med 9(10): 1269-1274. doi: 10.1038/nm934
- Orabona C, Pallotta MT, Grohmann U (2012). Different partners, opposite outcomes: a new perspective of the immunobiology of indoleamine 2,3-dioxygenase. Mol Med 18: 834-842. doi: 10.2119/molmed.2012.00029
- Mbongue JC, Nicholas DA, Zhang K, Kim NS, Hamilton BN, Larios M, Zhang G, Umezawa K, Firek AF, Langridge WH (2015). Induction of indoleamine 2, 3-dioxygenase in human dendritic cells by a cholera toxin B subunit-proinsulin vaccine. PLoS One 10(2): e0118562. doi: 10.1371/journal.pone.0118562
- Tas SW, Vervoordeldonk MJ, Hajji N, Schuitemaker JH, van der Sluijs KF, May MJ, Ghosh S, Kapsenberg ML, Tak PP, de Jong EC (2007). Noncanonical NF-kappaB signaling in dendritic cells is required for indoleamine 2,3-dioxygenase (IDO) induction and immune regulation. Blood 110(5): 1540-1549. doi: 10.1182/blood-2006-11-056010
- Hanahan D, Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144(5): 646-674. doi: 10.1016/j.cell.2011.02.013
- Bozza PT, Viola JP (2010). Lipid droplets in inflammation and cancer. Prostaglandins Leukot Essent Fatty Acids 82(4-6): 243-250. doi: 10.1016/j.plefa.2010.02.005
- de Gonzalo-Calvo D, Lopez-Vilaro L, Nasarre L, Perez-Olabarria M, Vazquez T, Escuin D, Badimon L, Barnadas A, Lerma E, Llorente-Cortes V (2015). Intratumor cholesteryl ester accumulation is associated with human breast cancer proliferation and aggressive potential: a molecular and clinicopathological study. BMC cancer 15: 460. doi: 10.1186/s12885-015-1469-5
- Yue S, Li J, Lee SY, Lee HJ, Shao T, Song B, Cheng L, Masterson TA, Liu X, Ratliff TL, Cheng JX (2014). Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness. Cell Metab 19(3): 393-406. doi: 10.1016/j.cmet.2014.01.019
- O’Sullivan D, van der Windt GJ, Huang SC, Curtis JD, Chang CH, Buck MD, Qiu J, Smith AM, Lam WY, DiPlato LM, Hsu FF, Birnbaum MJ, Pearce EJ, Pearce EL (2014). Memory CD8(+) T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity 41(1): 75-88. doi: 10.1016/j.immuni.2014.06.005
- Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, Karoly ED, Freeman GJ, Petkova V, Seth P, Li L, Boussiotis VA (2015). PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun 6: 6692. doi: 10.1038/ncomms7692
- Patsoukis N, Weaver JD, Strauss L, Herbel C, Seth P, Boussiotis VA (2017). Immunometabolic Regulations Mediated by Coinhibitory Receptors and Their Impact on T Cell Immune Responses. Front Immunol 8: 330. doi: 10.3389/fimmu.2017.00330
- Warner TD, Mitchell JA (2004). Cyclooxygenases: new forms, new inhibitors, and lessons from the clinic. FASEB J 18(7): 790-804. doi: 10.1096/fj.03-0645rev
- Berod L, Friedrich C, Nandan A, Freitag J, Hagemann S, Harmrolfs K, Sandouk A, Hesse C, Castro CN, Bahre H, Tschirner SK, Gorinski N, Gohmert M, Mayer CT, Huehn J, Ponimaskin E, Abraham WR, Muller R, Lochner M, Sparwasser T (2014). De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med 20(11): 1327-1333. doi: 10.1038/nm.3704
- Lochner M, Berod L, Sparwasser T (2015). Fatty acid metabolism in the regulation of T cell function. Trends Immunol 36(2): 81-91. doi: 10.1016/j.it.2014.12.005
- Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR, Xiao B, Worley PF, Powell JD (2011). The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol 12(4): 295-303. doi: 10.1038/ni.2005
- Zeng H, Yang K, Cloer C, Neale G, Vogel P, Chi H (2013). mTORC1 couples immune signals and metabolic programming to establish T(reg)-cell function. Nature 499(7459): 485-490. doi: 10.1038/nature12297
- Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, Sullivan SA, Nichols AG, Rathmell JC (2011). Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol 186(6): 3299-3303. doi: 10.4049/jimmunol.1003613
- Al-Khami AA, Zheng L, Del Valle L, Hossain F, Wyczechowska D, Zabaleta J, Sanchez MD, Dean MJ, Rodriguez PC, Ochoa AC (2017). Exogenous lipid uptake induces metabolic and functional reprogramming of tumor-associated myeloid-derived suppressor cells. Oncoimmunology 6(10): e1344804. doi: 10.1080/2162402X.2017.1344804
- Hossain F, Al-Khami AA, Wyczechowska D, Hernandez C, Zheng L, Reiss K, Valle LD, Trillo-Tinoco J, Maj T, Zou W, Rodriguez PC, Ochoa AC (2015). Inhibition of Fatty Acid Oxidation Modulates Immunosuppressive Functions of Myeloid-Derived Suppressor Cells and Enhances Cancer Therapies. Cancer Immunol Res 3(11): 1236-1247. doi: 10.1158/2326-6066.CIR-15-0036
- Zhang Y, Kurupati R, Liu L, Zhou XY, Zhang G, Hudaihed A, Filisio F, Giles-Davis W, Xu X, Karakousis GC, Schuchter LM, Xu W, Amaravadi R, Xiao M, Sadek N, Krepler C, Herlyn M, Freeman GJ, Rabinowitz JD, Ertl HCJ (2017). Enhancing CD8(+) T Cell Fatty Acid Catabolism within a Metabolically Challenging Tumor Microenvironment Increases the Efficacy of Melanoma Immunotherapy. Cancer Cell 32(3): 377-391 e379. doi: 10.1016/j.ccell.2017.08.004
ACKNOWLEDGMENTS
We thank Isabel Gregoire for English editing.
COPYRIGHT
© 2019

Regulation of T cell antitumor immune response by tumor induced metabolic stress by Chalmin et al. is licensed under a Creative Commons Attribution 4.0 International License.


