Reviews:
Cell Stress, Vol. 3, No. 4, pp. 118 - 135; doi: 10.15698/cst2019.04.183
Constitutional Mosaic Epimutations – a hidden cause of cancer?

1 K.G.Jebsen Center for Genome Directed Cancer Therapy, Department of Clinical Science, University of Bergen, Norway.
2 Department of Oncology, Haukeland University Hospital, Bergen, Norway.
Keywords: epimutations, promoter methylation, cancer risk, BRCA1, MLH1.
Abbreviatons:
HGSOC – high-grade serous ovarian cancer,
HRD – homologous repair deficiency,
LOH - loss-of-heterozygosity,
MSI – microsatellite-instability,
OR – odds ratio,
PARP - Poly-ADP-Ribose-Polymerase,
SNP – single nucleotide polymorphism,
WBC – white blood cell.
Received originally: 05/01/2019 Received in revised form: 04/03/2019
Accepted: 05/03/2019
Published: 22/03/2019
Correspondence:
Per E. Lønning, per.lonning@helse-bergen.no
Conflict of interest statement: None of the authors declare any conflicts of interest related to this work.
Please cite this article as: Per E. Lønning, Hans P. Eikesdal, Inger M. Løes and Stian Knappskog (2019). Constitutional Mosaic Epimutations - a hidden cause of cancer? Cell Stress 3(4): 118-135. doi: 10.15698/cst2019.04.183
Abstract
Silencing of tumor suppressor genes by promoter hypermethylation is a key mechanism to facilitate cancer progression in many malignancies. While promoter hypermethylation can occur at later stages of the carcinogenesis process, constitutional methylation of key tumor suppressors may be an initiating event whereby cancer is started. Constitutional BRCA1 methylation due to cis-acting germline genetic variants is associated with a high risk of breast and ovarian cancer. However, this seems to be a rare event, restricted to a very limited number of families. In contrast, mosaic constitutional BRCA1 methylation is detected in 4-7% of newborn females without germline BRCA1 mutations. While the cause of such methylation is poorly understood, mosaic normal tissue BRCA1 methylation is associated with a 2-3 fold increased risk of high-grade serous ovarian cancer (HGSOC). As such, BRCA1 methylation may be the cause of a significant number of ovarian cancers. Given the molecular similarities between HGSOC and basal-like breast cancer, the findings with respect to HGSOC suggest that constitutional BRCA1 methylation could be a risk factor for basal-like breast cancer as well. Similar to BRCA1, some specific germline variants in MLH1 and MSH2 are associated with promoter methylation and a high risk of colorectal cancers in rare hereditary cases of the disease. However, as many as 15% of all colorectal cancers are of the microsatellite instability (MSI) “high” subtype, in which commonly the tumors harbor MLH1 hypermethylation. Constitutional mosaic methylation of MLH1 in normal tissues has been detected but not formally evaluated as a potential risk factor for incidental colorectal cancers. However, the findings with respect to BRCA1 in breast and ovarian cancer raises the question whether mosaic MLH1 methylation is a risk factor for MSI positive colorectal cancer as well. As for MGMT, a promoter variant is associated with elevated methylation across a panel of solid cancers, and MGMT promoter methylation may contribute to an elevated cancer risk in several of these malignancies. We hypothesize that constitutional mosaic promoter methylation of crucial tumor suppressors may trigger certain types of cancer, similar to germline mutations inactivating the same particular genes. Such constitutional methylation events may be a spark to ignite cancer development, and if associated with a significant cancer risk, screening for such epigenetic alterations could be part of cancer prevention programs to reduce cancer mortality in the future.
INTRODUCTION
Malignant tumors are thought to arise through a sequence of genetic disturbances [1]. In some cancers, like colorectal carcinomas, the sequence of key genomic events in general follows a common order [2], while in other cancer types, like breast cancer, the sequence of events in carcinogenesis seems to occur at random [3][4].
–
The identification of genomic aberrations predisposing to cancer have added substantially to our understanding of cancer-inducing events. Importantly, the finding that germline mutations in genes like BRCA1/2, TP53, RB1, CDKN2A and others are associated with an elevated risk of certain cancers indicates that mutations in these genes may act as the initial events in malignant transformation in sporadic cancers as well [5].
–
In addition to gene mutations and rearrangements, somatic epigenetic alterations, i.e. epimutations, affecting gene expression levels can play a pivotal role during carcinogenesis [6]. Further, recent findings have indicated that underlying epimutations of certain genes in the normal tissue are associated with an elevated risk of particular cancer subtypes. This indicates that an epigenetic, and not genetic, event may be the initial step in the carcinogenesis process for these particular cancers (Figure 1). Of notice, epimutations that are widely distributed across normal tissues, predisposing to disease, are defined as constitutional epimutations [7][8][9][10]. By definition, constitutional epimutations involve tissue derived from all the three germ layers [9]. Thus, in order to distinguish constitutional methylation from gene methylations acquired during lifetime, it is important to assess methylation status of the gene in question across tissues derived from more than one of the germ layers (endoderm, ectoderm, as well as mesoderm).
–
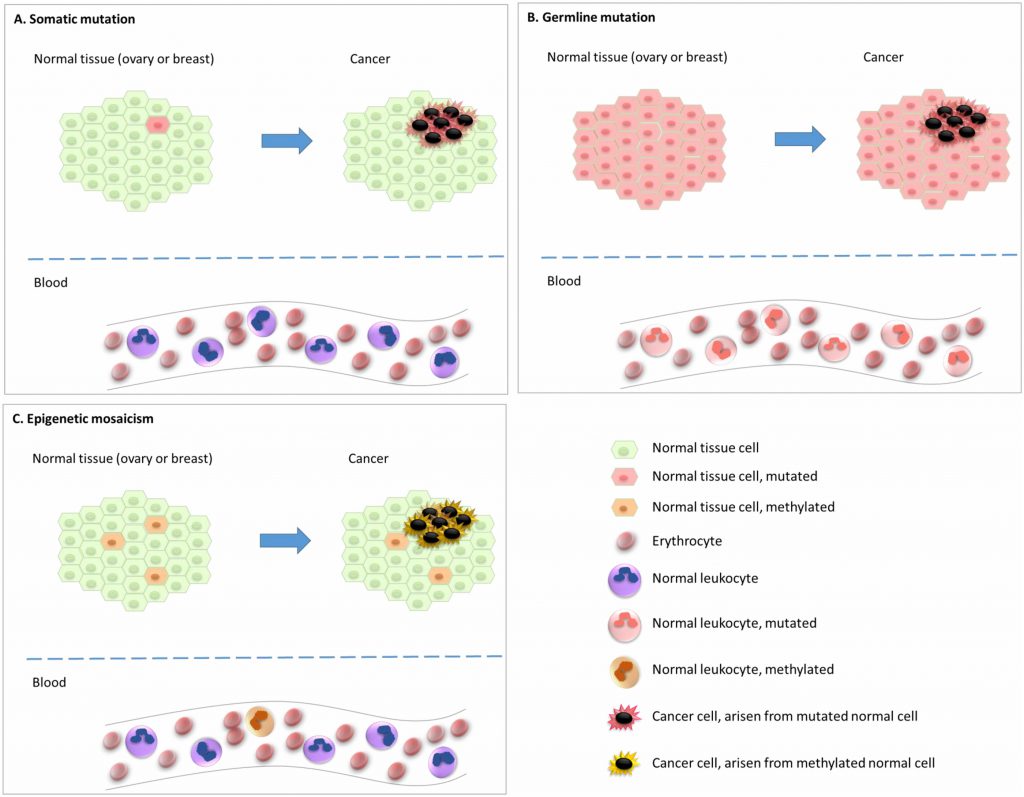 | FIGURE 1: Early events underlying carcinogenesis. (A) Cancer arising from normal cells subject to a somatic driver mutation as the initial event, with subsequent alterations leading to malignant transformation. In such cases the initial event will not be detected in white blood cells (WBC). (B) Cancer arising from normal cells harbouring a germline driver mutation, acting as the initial event, with subsequent alterations leading to malignant transformation. In such cases the “initial event” will be detectable in all WBCs. (C) Cancer arising from a minority of normal cells with a key tumour suppressor methylated from early embryonic life (epigenetic mosaicism). This methylation may act as the initial event, with subsequent alterations leading to malignant transformation. In such cases the initial event / methylation will be detectable also as mosaicism in WBC. |
–
Studies of normal tissues show that epimutations which affect only a fraction of the alleles may still mediate an increased cancer risk. On the other hand, in a malignant tumor arising from a precursor cell carrying an epimutation, we may expect all cells in the resulting tumor to harbour the epigenetic event if it is essential to cancer progression. While epigenetic gene silencing involves several types of modifications, including DNA methylation, histone modifications and RNA interference, in this review we will focus on promoter CpG methylation, the only mechanism that has been explored with respect to cancer predisposition in human studies so far.
–
Evidence linking epimutations to cancer risk has been reported for a limited number of genes. For these particular genes, we will discuss the evidence indicating normal tissue epimutations to infer an elevated cancer risk. Additionally, we will discuss the potential clinical and biological importance of these epimutations with respect to how they may mediate the phenotype of particular cancers. Of notice, if epimutations act as the initial trigger events, we expect such malignancies in general to mirror the phenotype of cancer in the same organ developing due to germline mutations in the same gene. This is in contrast to sporadic cancers where one would expect a more diverse phenotype based on heterogenous genomic events triggering cancer initiation.
–
The Lynch syndrome [11] or hereditary non-polyposis colorectal cancer (HNPCC), is an autosomal dominant genetic condition characterized by an elevated risk of colorectal cancer, with a preponderance for proximal/right-sided colon cancers, as well as an elevated risk of endometrial cancer. The syndrome is caused by defects in DNA mismatch repair due to germline mutations in either MLH1 or MSH2, or, more rarely, in the MSH6, PMS1 or PMS2 genes [12]. Additionally, recent findings have revealed a moderately increased risk of various other solid malignancies as part of the Lynch syndrome, affecting the stomach, small intestine, pancreas, hepatobiliary and upper urinary tract, brain, ovary or breast [11].
–
Due to the mismatch repair gene defects, malignant tumors associated with Lynch syndrome are characterized by a microsatellite-instability (MSI) phenotype [13]. While the Lynch syndrome accounts for only 2-3% of all colorectal cancers [14][15], up to 15% of all colorectal cancers are defined as MSI “high”, where other underlying mechanisms must be at play [16]. Interestingly, the majority of these MSI “high” tumors reveal somatic hypermethylation of the MLH1 promoter region [17][18][19]. Moreover, all malignant tumors with MSI are characterized by a similar phenotype, regardless of which of the mismatch genes that are mutated – this includes MSI “high” cancers due to MLH1 inactivation; whether it is by germline mutations or epigenetic silencing [20]. Compared to other colorectal cancers, MSI high tumors are characterized by a high mutation burden, and they also seem to draw a substantial benefit from immunotherapy [21][22].
–
Characteristics such as MSI and a high mutation load in malignancies from patients with Lynch syndrome or spontaneous cancers harboring MLH1 promoter methylation indicate that epimutations, as well as somatic mutations affecting MLH1, are early events during malignant transformation [23]. Alternatively, for tumors harbouring MLH1 hypermethylation, if the high mutational load occurs at a later stage it must have been selected for through a profound “selective sweep” [24]. Interestingly, Miyakura et al. [19], in addition to analyzing for MLH1 methylation in the tumor tissue, examined MLH1 methylation status in the matching normal colon mucosa, detecting partial MLH1 promoter methylation in one third of the patients. Although no firm conclusions can be drawn from this finding, one may speculate that in a subset of patients the carcinogenesis process may have started by methylation of normal colon mucosal cells.
–
 |
CONSTITUTIONAL MLH1 METHYLATION AND COLORECTAL CANCER RISK
As defined by Hitchins and colleagues [25][26], epimutations may be separated into two major groups; primary epimutations (or promoter methylation) where no DNA alterations are detected, and secondary epimutations, occurring in concert with (and caused by) a local cis-acting DNA alteration. Following the findings by Kane and colleagues [27] demonstrating MLH1 promoter methylation in colorectal tumor tissue, and the report of Gazzoli and colleagues in 2002 [28] demonstrating white blood cell (WBC) DNA methylation of the MLH1 promoter in a young man diagnosed with an MSI positive colon cancer, MLH1 methylation has been detected in circulating leucocytes (WBC) of a subset of patients with sporadic MSI positive colorectal cancers. In some cases, such findings have also been made in probands without a cancer diagnosis. However, less than 50 individuals have been reported in the literature with concurrent colorectal cancer and constitutional (normal tissue) MLH1 methylation so far [29][30][31][32][33][34][35][36][37][38][39][40][41][42]. As for studies reporting the fraction of methylated alleles in the blood of these individuals, this has been in the range of 20-50% [39], with a monoallelic pattern.
–
In 2011, Hitchins and colleagues identified a haplotype harbouring tandem nucleotide substitutions, where a c.-27C>A variant was the likely cause of MLH1 methylation and cancer diagnosis across a family with Lynch syndrome [38]. Additionally, in a few cases, methylation has been detected in concert with larger genomic rearrangements of the MLH1 gene [35][37]. Apart from these individuals, the potential pathogenic contribution of genomic rearrangements to MLH1 methylation remains an open question.
–
Contrasting the “high-level” methylation associated with colorectal cancer mentioned above, low level mosaic WBC methylation of MLH1 in patients with colorectal cancer has also been reported [29][39]. The potential contribution of such low level methylation in MLH1 to colorectal cancer risk remains to be formally assessed. Anecdotally, Sloane and colleagues [43] reported a young male diagnosed with colorectal cancer to harbour constitutional methylation in about 50% of his alleles, while his mother revealed mosaic MLH1 methylation in less than 5% of the alleles. Interestingly, among retinoblastoma patients diagnosed with germline RB1 mutations, in some cases an unaffected parent carried the same mutation at low frequency in her/his WBCs [44][45]. Mosaic gene methylation as a cancer risk factor will be discussed further as part of reviewing BRCA1 methylation data below.
MSH2
While MSH2 methylation was detected in a small subset of colorectal cancers, most importantly it appeared only in subfractions of the malignant cells, with no correlation to gene expression level or MSI status [46]. Constitutional methylation of MSH2 is a rare event, first described by Chan et al. [47] in 2006. In a subsequent study [48], the same family was further characterized together with an additional set of nine Dutch and Chinese families. In summary, patients in these families all revealed loss of MSH2 staining by immunohistochemistry (IHC) and hypermethylation of the MSH2 promoter within the colorectal cancers, as well as methylation of the MSH2 promoter across various normal tissues, although to a variable extent. Importantly, all patients carried a deletion in a gene upstream of MSH2, namely TACSTD1 (encoding Ep-CAM). This deletion resulted in MSH2 promoter methylation and reduced MSH2 transcription in the colon mucosa and subsequent colorectal cancer cells. This finding was confirmed by Niessen and colleagues in another three independent individuals carrying the Lynch syndrome [42].
O-6-METHYLGUANINE-DNA METHYLTRANSFERASE (MGMT)
MGMT is downregulated by promoter methylation in various types of cancers [49][50][51][52][53][54][55][56][57]. Subsequent loss of methylation and re-elevated expression of MGMT has been associated with resistance towards alkylating agents like temozolomide and cyclophosphamide [49][50][55][58][59][60]. While germline mutations in MGMT have not been detected so far, the T-allele of the single nucleotide polymorphism (SNP) rs16906252, located in the first exon of MGMT, close to the transcription start site, has been associated with elevated promoter methylation across a panel of solid malignancies [51][53][54][61][62]. Mirroring findings for MLH1 (see above), Shen and colleagues [52] detected MGMT methylation not only in cancer tissue, but also in normal colon mucosa located 10 cm from the tumor borders. More recently, mosaic MGMT methylation (up to 10% of the alleles) associated with the rs16906252 SNP T-allele has also been detected in WBC [63]. In a large study of germline genotypes (WBC) including a validation cohort, Kuroiwa-Trzmielina and colleagues found the rs16906252 T-allele to be associated with an odds ratio (OR) of 3-4 for harbouring MGMT promoter methylation within a colorectal cancer [56]. In addition, one smaller study found a moderate but significant association between the rs16906252 T-allele of MGMT and glioblastoma risk [61]. Taken together, these studies indicate that the rs16906252 SNP may affect the risk of different cancers by causing increased MGMT promoter methylation.
BRCA1 AND BRCA2 EPIMUTATIONS IN BREAST AND OVARIAN CANCER TISSUE
Women carrying germline pathogenic mutations in BRCA1 and BRCA2 are at high risk of developing breast as well as ovarian cancer [64][65][66][67]. Notably, germline mutations in BRCA1/2 have also been linked to an elevated risk of cancer of the prostate and pancreas [68][69], and germline BRCA2 mutations to a moderately increased risk of several other malignancies [70][71]. With respect to the current review, evidence linking BRCA1/2 methylation to cancer risk has so far only been collected from patients with breast and ovarian cancer.
–
BRCA1 and BRCA2 both participate in homologous DNA repair. BRCA2 is part of the Fanconi complex (FANCD1), whereas BRCA1 has a critical role as a downstream executor of this complex [72][73]. However, the breast cancer phenotypes linked to deficiencies in these two gene varies. As for breast cancers arising in BRCA1 mutation carriers, >80% belongs to the so-called “basal-like” subtype [74], accounting for the majority of triple negative breast cancers [75]. This contrasts spontaneous breast cancers where triple negative tumors account for approximately 15% [76][77]. On the other hand, tumors arising in BRCA2 mutation carriers reveal a phenotype distribution mirroring spontaneous breast cancers [78]. Among basal-like breast cancers, 10-25% are associated with a germline BRCA1 mutation. This rather wide range is due to differences in ethnicity and age distribution at cancer diagnosis in different studies [79][80].
–
While somatic BRCA1/2 mutations in breast cancer previously were thought of as rare, compared to germline mutations, contemporary evidence indicates that one third of BRCA mutations have a somatic origin [4][81][82][83][84][85][86]. Moreover, mutations of BRCA1/2 as well as other crucial DNA repair genes inflict homologous repair deficiency (HRD), which is associated with distinct gene mutation signatures, including copy number variations [84]. Thus, different mutational signatures aiming at predicting HRD have been generated [87][88][89]. Applying such a signature assessment to breast cancers have indicated that HRD may characterize as many as 20% of all cases [87]. The biological and clinical relevance of such signatures are underlined by merging evidence validating their role in predicting sensitivity towards treatment with PARP (Poly-ADP-Ribose-Polymerase) inhibitors as well as certain chemotherapy regimens, resembling what may be seen for patients harbouring germline BRCA1/2 mutations [83][90][91][92][93][94]. The reason for the homologous repair defect in most of these tumors remains unknown, but BRCA1 promoter methylation has been reported in 30-35% of all triple negative breast cancers with germline BRCA1/2 wild-type status, in particular among tumors of the basal-like subtype [95]. Further, BRCA1 promoter methylation has been associated with transcriptional downregulation of BRCA1 [96][97][98][99]. The incidence of BRCA1 methylation is lower (5-25%) among breast cancers that are not basal-like [79][100][101][102][103][104][105][106][107], consistent with the subtype skewness seen for BRCA1 mutation carriers [108][109]. Further, conflicting evidence has indicated similarities with respect to drug sensitivity and outcome between individuals with breast cancers harbouring BRCA1 mutations and those with promoter methylation [83][101][110][111][112][113][114]. In spontaneous breast cancer, the BRCA2 methylation frequencies vary between 0 and 12% [105][107]. Notably, promoter methylation of PALB2, another gene in the Fanconi complex, has been detected in a small number of spontaneous breast cancers as well [115].
–
The reported BRCA1/2 methylation frequencies vary substantially between different clinical studies. Similar to variation in the incidence of BRCA1/2 germline mutations this could be due to ethnic variations or the age distribution in the patient cohort undergoing analysis. However, the reported frequency differences are most likely due to methodological differences.
–
Approximately 50% of patients diagnosed with a high-grade serous ovarian cancer seem to harbour homologous repair deficiencies in the tumor tissue [116]. High-grade serous ovarian cancer is the cancer subtype for which germline BRCA1/2 mutations are detected at the highest frequency with 8-15% carrying a BRCA1 and 4-8% a BRCA2 mutation [98][117][118][119]. In addition, The Cancer Genome Atlas [98] reported somatic BRCA1/2 mutations in a small number of cases. BRCA1 methylation is detected in 9-15% of spontaneous cases of serous ovarian cancer, but does not seem to occur in concert with germline mutations [97][120][121]. Notably, ovarian cancer tissue methylation for the BRCA1 promoter, similar to germline BRCA1 mutation status, was associated with the high-grade serous cancer subtype and young age at diagnosis [122][123]. In contrast to the frequencies reported in BRCA1, methylation of BRCA2 occurs in <1% of ovarian cancers [124][125][126]. In germline mutation carriers, no BRCA2 methylated ovarian cancer has been detected so far [127].
–
While most breast cancers carrying BRCA1 mutations undergo loss-of-heterozygosity (LOH) of their wild-type allele as their second hit, BRCA1 and BRCA2 promoter methylation have also been detected in some tumors without LOH [104][127][128]. However, the methylation profile varies across individual CpG nucleotides [107], and a recent study found LOH for BRCA1 as well as BRCA2 to occur in concert with promoter methylation of the same gene in different subclones of the same tumor [129].
–
In patients with ovarian cancers that are wild-type for BRCA1/2, BRCA1 promoter methylation predicted better outcome to platinum-taxane based therapy [130] as well as PARP inhibition, as compared to patients without such methylation, thus mirroring findings in patients harbouring germline mutations [131][132][133]. Contrasting this are results from the recently published TnT trial, where patients with triple negative metastatic breast cancer and tumor BRCA1 methylation did not respond any better to platinum chemotherapy than those without such epimutations [83]. However, methylation analyses were performed on archival tumor tissue extracted at the time of the first breast cancer diagnosis, which could have skewed the results, compared to an analysis of cancer biopsies taken at screening before entering the trial, but after previous exposure to adjuvant chemotherapy.
NORMAL TISSUE BRCA1/2 METHYLATION AND RISK OF BREAST AND OVARIAN CANCER
Few studies have assessed BRCA2 normal tissue (or WBC) methylation status. To the best of our knowledge, no formal assessment for WBC BRCA2 methylation with respect to breast or ovarian cancer risk has been conducted. Notably, in a recent study using a low detection limit, Peplonska and colleagues detected evidence of WBC BRCA2 and BRCA1 methylation in 18.3% and 21.5%, respectively, among (presumably) cancer-free participants [134]. However, their estimates are unusually high, also for BRCA1 methylation. Until recently, BRCA1/2 hypermethylation was not associated with increased risk of hereditary breast cancer [135][136]. As for the studies presented, most of them contained a limited number of patients, raising the question of potential publication bias (negative studies may not have been reported). Also, methylation frequency within the control populations are at substantial variance across the studies due to different analytical methods and thresholds applied.
–
Notably, an interesting study was presented by Wong and colleagues from Dobrovic’s group [108]. Analyzing a total of 255 women diagnosed with breast cancer below the age of 40 years and without germline BRCA1/2 mutations, they detected BRCA1 promoter methylation in WBC among 31% of patients, revealing strong morphologic characteristics (five or more individual parameters) otherwise associated with a BRCA1 mutation. In contrast, they found peripheral blood methylation in 10% and 5% among those harboring 4 and ≤3 BRCA1 mutation characteristics, respectively. This significantly contrasted a BRCA1 methylation incidence of 4% among unaffected controls.
–
Data on constitutional BRCA1 methylation with respect to ovarian cancer risk has in general been lacking. However, analyzing individuals wild-type for BRCA1/2 germline mutations, Hansmann and colleages [137] identified WBC BRCA1 methylation in 3 out of 39 patients with ovarian cancer (8%) and belonging to families with an elevated risk of ovarian and breast cancer. Among individuals recorded as BRCA1 hypermethylated, methylation affected between 12 and 40% of the WBC alleles – thus indicating mosaic hypermethylation of this gene in the normal tissue. In the same cohort, they also identified RAD51C methylation in one ovarian cancer index patient.
–
In a recent study [138], we examined WBC BRCA1 promoter methylation status among 1688 healthy controls and 925 patients with ovarian cancer (Figure 2). BRCA1 methylation was detected by methylation-specific qPCR in 4.2% of healthy controls. While we recorded a similar methylation frequency among patients diagnosed with non-serous or low-grade serous ovarian cancers, the methylation frequency was as high as 9.6% among patients diagnosed with a high-grade serous ovarian cancer (HGSOC), revealing an OR of 2.91 (CI: 1.85 – 4.56). Our findings were confirmed in a validation cohort containing 607 patients and 1914 controls, revealing an OR for HGSOC of 2.22 (CI 1.40 – 3.52). Among patients testing positive for BRCA1 metylation, the median percentage of methylated alleles was 4.1%, with 21% as the highest level recorded – again pointing to partial/mosaic hypermethylation of the BRCA1 gene. Combining data from the exploratory and validation cohorts, we found an OR for HGSOC of 1.82 for individuals harbouring a methylation level below the median percentage, contrasting an OR of 4.20 for those with BRCA1 methylation levels above the median. Finally, the OR for HGSOC associated with positive BRCA1 methylation was highest in individuals below 50 years of age (OR 4.42). Of notice, although excluded from the formal OR assessments, we detected WBC methylation also among individuals carrying germline BRCA1 and BRCA2 mutations (in 1.5% and 9.0%, respectively). The biological interpretation of this potential difference between BRCA1 and BRCA2 mutation carriers is uncertain due to the low number of individuals analyzed.
–
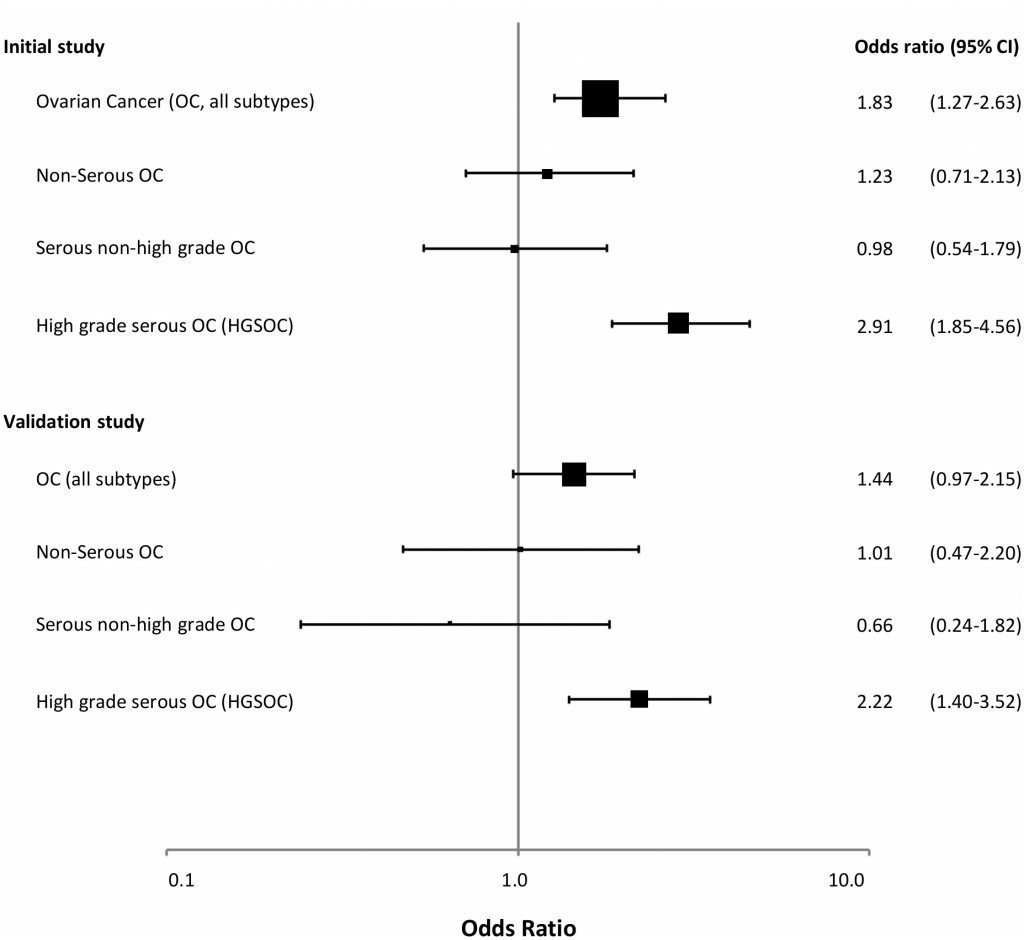 | FIGURE 2: BRCA1 methylation and risk of ovarian cancer. Forest plot illustrating the odds ratio (OR) for ovarian cancer (all subtypes), non-serous, serous non-high grade and high grade serous ovarian cancer (HGSOC) related to BRCA1 promoter methylation, derived from the initial study population and the validation cohort in the recent publication by Lønning et al. [131]. The odds ratios (ORs) were based on analyses of 925 cases and 1688 controls (initial study) and 607 cases and 1914 controls (validation study). Reprint of original figure, with permission from Annals of Internal Medicine. |
–
In the same study, we examined the BRCA1 methylation status in normal as well as ovarian cancer tissue in a subgroup of patients. Notably, we confirmed BRCA1 methylation in various paraffin-embedded normal tissue samples from patients testing positive for WBC BRCA1 methylation. The samples were derived from the endoderm as well as the mesoderm germ layers. While we did not have ectodermal derived samples available, it is unlikely that tissue derived from that germ layer should deviate from the other two; thus, our findings strongly indicate constitutional methylation [8][9]. Among patients testing positive for BRCA1 methylation in WBC, 62% were methylation positive in the tumor tissue, contrasting 12% for patients testing negative for WBC BRCA1 methylation. These data mirror the findings by Dobrovic and colleagues in HGSOC [139]. They analyzed blood and tumor tissue from 154 patients with HGSOC and among 20 patients harbouring WBC BRCA1 methylation, 14 of them (70%) revealed methylation of the tumor DNA as well. The finding of a 60-70% methylation frequency in tumors from individuals carrying a constitutive BRCA1 promoter methylation is in accordance with what is expected. Assuming methylation of a small fraction of normal (including ovarian tissue) BRCA1 alleles to be associated with an OR for HGSOC of 2.0, such a finding should indicate 50% of the cancers may arise from unmethylated cells (like in an individual not carrying any BRCA1 methylated allele). The additional 50% of cancers would then arise from the small fraction of methylated cells. Similarly, in case of an OR of 3.0 for HGSOC, we may envision 67% of the cancers to arise from methylated cells. In our previous study, the OR in the one cohort was 2.9, and in the second cohort 2.2 [138]. Following the assumptions above, these findings correspond well with the finding of BRCA1 tissue methylation in between 60% and 70% of the HGSOC.
–
In addition to the large case-control studies described above, there are also reports of special cases where BRCA1 methylation is strongly linked to cis-acting genetic variants. Importantly, Evans and colleagues [140] reported constitutional BRCA1 methylation in WBC from members of two families characterized by high incidence of breast and ovarian cancer, but testing negative for BRCA1/2 germline mutations. Here, the methylation was associated with a 5′UTR promoter variant and about 50% of the alleles were methylated, indicating complete methylation of affected alleles. Of notice, these findings parallel the recent findings by Hitchins and colleagues described above [38] with respect to MLH1 methylation in a colorectal cancer family. Notably, while BRCA1 promoter variants influencing breast and ovarian cancer risk have been reported earlier [141][142], the finding by Evans et al. is the first to link such variants to BRCA1 methylation status.
BRCA1 PROMOTER HYPERMETHYLATION MAY BE A CONSTITUTIONAL EVENT ARISING IN UTERO
Epigenetic gene silencing is a normal feature during embryonic development. Indeed, recent studies revealed that dramatic epigenetic alterations may occur already at the pre-implantation stage [143]. DNA methylation status varies between individuals and is influenced by genetic as well as environmental factors [48][144][145][146]. The latter is particularly underlined by the fact that methylation patterns change with aging [147][148], and that identical twins reveal much similarity at young age but grow more epigenetically different with time [144]. Assessing BRCA1 promoter methylation in umbilical cord blood of >600 girls [138], we detected BRCA1 methylation among 7% of them. Notably, the methylation profile across the CpG’s mirrored the methylation status in healthy adults as well as ovarian cancer patients, indirectly supporting the hypothesis that BRCA1 methylation is a constitutional event.
–
In order to be a risk factor for cancer, one may assume that methylation must persist over time. Taken together, our findings in newborns and adults are in accordance with the hypothesis that constitutional methylation may arise in utero and persist through life, constituting a cancer risk factor. Thus, such methylation follows a different pattern from methylation related to external influence and senescence [149] mirroring the difference between inherited subclonal mutations and hematological subclones carrying distinct gene mutations arising in response to accumulated genotoxic influence [150][151][152][153].
–
Consistent with our findings, Al-Moghrabi and colleagues, testing 300 newborns, found WBC BRCA1 methylation in 9.9% of their cohort [154]. Moreover, they detected MGMT promoter methylation in 12.3% of newborns, revealing that neonatal methylation of tumor suppressor genes may not be restricted to BRCA1 exclusively.
–
Interestingly, Al-Moghrabi and colleagues reported a potential association between BRCA1 methylation status in mothers and their newborns [154]. While the data did not allow for formal statistical assessment, their explorative analysis indicated a moderate correlation, albeit not in accordance with Mendelian dominant inheritance [155]. Importantly, their findings do not define whether there was a paternal or maternal transfer of methylation. In some cases, transfer could be related to genetic variants (secondary epimutations) but in other cases it could be the transfer of primary epimutations. Considering germline mutations, mosaic mutations have been found related to neurological disorders [156], as well as in families with increased incidence of retinoblastoma [44][45][157]. In the latter case, mosaic mutations have been detected even as subclones in unaffected parents of an affected proband. This probably relates to such mutations arising somatically at the embryonic stage, and subsequently transferred through the gamete to the offspring. Further studies are needed to clarify this topic. Notably, among the patients with ovarian cancer and healthy controls that we examined, BRCA1 methylation occurred independently of the two major haplotypes of the BRCA1 promoter [138]. In line with lack of Mendelian inheritance patterns, this argues against a hypothesis suggesting constitutional methylation to be associated with a cis-acting factor.
–
The cause of BRCA1 promoter methylation occurring among certain newborns is unknown. Yet there is substantial evidence linking prenatal factors to subsequent risk of different types of cancer in adult life, and breast cancer in particular [158][159][160][161][162][163][164][165]. As for methylation status in umbilical cord blood, global methylation patterns are associated with external factors like smoking during pregnancy, folate levels and famine [166][167][168], as well as birth weight [169]. To the best of our knowledge, studies evaluating the association between prenatal external factors and methylation of specific tumor suppressor promoters, such as for BRCA1, are lacking.
POTENTIAL CAVEATS
While some studies applying genome-wide methylation analyses have detected differences in methylation of distinct CpG’s related to incidental cancers [170][171][172], such differences in methylation in general occurred in CpG’s located outside gene promoters; thus, the biological implications of these findings are uncertain. As for studies examining BRCA1 promoter methylation with respect to breast and ovarian cancer, blood samples in general were collected from patients already diagnosed with their cancer. Thus, data assessing the predictive value of BRCA1 promoter methylation to incidental cancers (by collecting blood samples years prior to diagnosis) are lacking. However, the risk of potential tumor DNA contamination, either from plasma free tumor DNA or circulating tumor cells seems negligible since the fraction of circulating tumor cells versus WBC detected in the circulation is estimated to be less than 1 to a million, and the concentration of free tumor DNA in the plasma is far lower than the DNA derived from WBCs [173][174][175].
–
On the other hand, distinct alterations in the WBC global gene methylation profile has been shown in patients with different cancers. This may not be directly linked to the cancer per se, but is probably related to alterations in WBC composition due to a cancer-related inflammatory response in patients with active disease [176][177][178]. This is consistent with the finding that WBC global gene methylation pattern varies between WBC subfractions [145][179][180][181][182].
–
In our study on BRCA1 methylation status and ovarian cancer risk, we performed extensive sensitivity analyses [138]. Here, we examined methylation status as a factor of tumor load, either by FIGO stage, or by examining methylation in patients who had recently had their tumors removed by surgery. Also, we examined methylation status in an additional cohort of ovarian cancer patients who had received chemotherapy. None of these factors influenced WBC BRCA1 methylation status. Notably, we detected a methylation frequency which resembled that of healthy individuals (about 4%) across all subgroups of patients diagnosed with non-HGSOC, contrasting a methylation frequency of 9-10% among all subgroups of patients diagnosed with HGSOC.
–
Variations between WBC subfractions also need to be taken into consideration when comparing methylation among newborns versus adults. However, examining publicly available datasets [179][180] we detected no variation in BRCA1 promoter methylation patterns with respect to WBC subfractions, neither in newborns nor adults [138]. Thus, differences in WBC subfractions between cancer patients and controls is not a likely explanation why BRCA1 methylation is increased among the patients diagnosed with HGSOC.
–
A final limitation relates to the use of conventional methylation-specific PCR (MSP) assessment methods, in as much as such methods do not allow for detailed quantification of the allele fraction being methylated. Neither do they inform whether cells are subject to mono-allelic or bi-allelic methylation. Such problems may be overcome by applying pyrosequencing [183] or contemporary next generation sequencing methodologies. This relates to mosaic methylation affecting a low allele fraction [138] in particular.
WHAT ARE THE IMPLICATIONS OF THESE FINDINGS?
Merging evidence links constitutive methylation to cancer as well as other diseases, such as neurological disorders [184]. Further, we are beginning to learn how prenatal exposure (like smoking and diet) as well as maternal health issues may influence methylation status in the newborn [166][167][185][186]. Most interestingly, experimental evidence has revealed acquired skills, like olfactory experience and sperm epigenetic programming in response to temperature, to be transmitted not only to the offspring, but into the third generation as well [187][188]. Merging evidence indicates trans-generational responses also in humans [189][190].
–
BRCA1, MLH1, MSH2 and MGMT are all pivotal in DNA repair. With the exception of MSH2, all these genes are methylated in a significant fraction of certain cancer types. As for MSH2, we should recall the mechanism causing promoter methylation (deletion in the upstream Ep-CAM gene), making this mechanism unique in comparison to the others. As for other DNA repair genes for which WBC promoter methylation has not been linked to cancer, such as BRCA2, somatic methylation is a rare event in breast as well as ovarian cancer. Thus, it may well be that larger cohorts are needed in order to detect BRCA2 methylation as a risk factor.
–
In colorectal cancers carrying MLH1 tissue methylation, as well as breast and ovarian cancers carrying BRCA1 methylation, a provoking question is whether these are acquired events occurring at some stage during tumor evolution, or if they may act as the primary event in the process of carcinogenesis. And in the latter case, could small groups of normal tissue cells that are methylated in utero act as cancer precursors? Importantly, mosaic germline mutations, likely to have occurred early during embryogenesis, have been detected in multiple genes related to different disease conditions in affected individuals (see [191] for additional details), including tumor suppressor genes like TP53, RB1, genes involved in neurofibromatosis type- 1 and -2, the Fanconi syndrome as well as BRCA1 [44][156][192][193][194][195][196].
–
Notably, DNA methylation status, similar to somatic mutations, continuously evolve during cancer progression [6][197][198]. Postulating BRCA1, MGMT or MLH1 promoter methylation to be a “first event” in carcinogenesis in some individuals by no means exclude the possibility that it may occur as a secondary event at a later stage in other individuals. While pathogenic germline mutations of TP53 in Li Fraumeni syndrome patients are likely to represent the initial event in breast cancers of such patients (as well as other neoplasia in these patients), recent studies revealed that somatic TP53 mutations may arise at later stages of tumor evolution in many non-hereditary breast cancers [3][4]. Accordingly, if a methylated tumor reveals a genomic signature mirroring the signature of a tumor arising in a germline mutation carrier with respect to secondary genomic events (BRCA-ness signature in breast cancers and MSI in colorectal cancers) the epigenetic event is likely to have occurred at a very early stage of tumor evolution. Alternatively, these tumors may have undergone selective sweeps [24] in response to an epigenetic event occurring at a later stage.
–
Taken together, we believe there is substantial evidence indicating cis-acting mutations to be associated with promoter methylation in some cancer-prone families (secondary constitutive epimutations; Figure 3). In addition, the findings regarding partial BRCA1 methylation in particular, but supported by similar findings for MLH1 and MGMT, may indicate mosaic epimutations to be far more frequent than previously appreciated. Furthermore, such primary constitutive epimutations could contribute to a substantial number of cancer cases. The clinical implications of such findings, if confirmed, is substantial. There may be a rationale for offering routine testing of methylation status as part of general health control programs in adult healthy individuals. For instance, women carrying BRCA1 WBC methylation could be offered regular ovarian surveillance by ultrasound exams from an age of 50 years. Secondly, the current findings should stimulate further research into the mechanisms by which such methylation arises, looking for potential pathogenic environmental influences or preventive strategies avoiding such events. While we lack selective drugs that may reverse gene-specific methylation as of today, such possibilities may become available in the future.
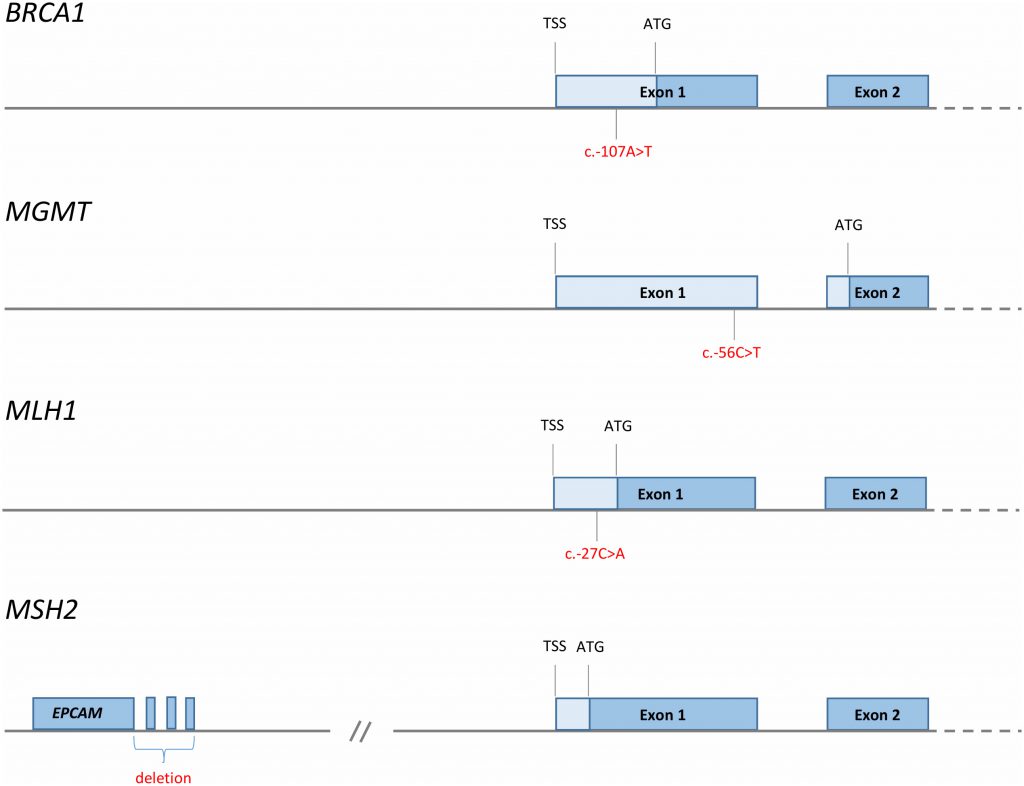 | FIGURE 3: Reported cis-acting factors (red font) causing tumour suppressor promoter methylation and cancer risk [35][43][58][133]. TSS; transcription start site, ATG; translational start site, blue boxes; protein coding regions, pale blue boxes; 5′UTR. x |
–
REFERENCES
- Hanahan D, Weinberg RA (2011). Hallmarks of Cancer: The Next Generation. Cell 144(5): 646-674. doi: 10.1016/j.cell.2011.02.013
- Fearon ER, Vogelstein B (1990). A genetic model for colorectal tumorigenesis. Cell 61(5): 759-767. doi: 10.1016/0092-8674(90)90186-i
- Yates LR, Gerstung M, Knappskog S, Desmedt C, Gundem G, Van Loo P, Aas T, Alexandrov LB, Larsimont D, Davies H, Li YL, Ju YS, Ramakrishna M, Haugland HK, Lilleng PK, Nik-Zainal S, McLaren S, Butler A, Martin S, Glodzik D, Menzies A, Raine K, Hinton J, Jones D, Mudie LJ, Jiang B, Vincent D, Greene-Colozzi A, Adnet PY, Fatima A, et al. (2015). Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat Med 21(7): 751-9. doi: 10.1038/nm.3886
- Yates LR, Knappskog S, Wedge D, Farmery JHR, Gonzalez S, Martincorena I, Alexandrov LB, Van Loo P, Haugland HK, Lilleng PK, Gundem G, Gerstung M, Pappaemmanuil E, Gazinska P, Bhosle SG, Jones D, Raine K, Mudie L, Latimer C, Sawyer E, Desmedt C, Sotiriou C, Stratton MR, Sieuwerts AM, Lynch AG, Martens JW, Richardson AL, Tutt A, Lonning PE, Campbell PJ (2017). Genomic Evolution of Breast Cancer Metastasis and Relapse. Cancer Cell 32(2): 169-184. doi: 10.1016/j.ccell.2017.07.005
- Lønning PE, Knappskog S (2013). Mapping genetic alterations causing chemoresistance in cancer; identifying the roads by tracking the drivers. Oncogene 32(46): 5315-5330. doi: 10.1038/onc.2013.48
- Mazor T, Pankov A, Johnson BE, Hong CB, Hamilton EG, Bell RJA, Smirnov IV, Reis GF, Phillips JJ, Barnes MJ, Idbaih A, Alentorn A, Kloezeman JJ, Lamfers MLM, Bollen AW, Taylor BS, Molinaro AM, Olshen AB, Chang SM, Song JS, Costello JF (2015). DNA Methylation and Somatic Mutations Converge on the Cell Cycle and Define Similar Evolutionary Histories in Brain Tumors. Cancer Cell 28(3): 307-317. doi: 10.1016/j.ccell.2015.07.012
- Snell C, Krypuy M, Wong EM, Loughrey MB, Dobrovic A (2008). BRCA1 promoter methylation in peripheral blood DNA of mutation negative familial breast cancer patients with a BRCA1 tumour phenotype. Breast Cancer Res 10(1): 1-8. doi: 10.1186/bcr1858
- Hitchins MP (2015). Constitutional epimutation as a mechanism for cancer causality and heritability? Nature Rev Cancer 15(10): 181-194. doi: 10.1038/nrc4001
- Sloane MA, Ward RL, Hesson LB (2016). Defining the criteria for identifying constitutional epimutations. Clin Epigenetics 8: 39. doi: 10.1186/s13148-016-0207-4
- Dobrovic A, Kristensen LS (2009). DNA methylation, epimutations and cancer predisposition. Int J Biochem Cell Biol 41(1): 34-39. doi: 10.1016/j.biocel.2008.09.006
- Lynch HT, Snyder CL, Shaw TG, Heinen CD, Hitchins MP (2015). Milestones of Lynch syndrome: 1895-2015. Nat Rev Cancer 15(3): 181-194. doi: 10.1038/nrc3878
- Marmol I, Sanchez-De-Diego C, Dieste AP, Cerrada E, Yoldi MJR (2017). Colorectal Carcinoma: A General Overview and Future Perspectives in Colorectal Cancer. Int J Mol Sci 18(1): E197. doi: 10.3390/ijms18010197
- Roberts ME, Jackson SA, Susswein LR, Zeinomar N, Ma XR, Marshall ML, Stettner AR, Milewski B, Xu ZX, Solomon BD, Terry MB, Hruska KS, Klein RT, Chung WK (2018). MSH6 and PMS2 germ-line pathogenic variants implicated in Lynch syndrome are associated with breast cancer. Genet Med 20(10): 1167-1174. doi: 10.1038/gim.2017.254
- Lynch HT, de la Chapelle A (2003). Genomic medicine – Hereditary colorectal cancer. N Engl J Med 348(10): 919-932. doi: 10.1056/NEJMra012242
- Umar A, Boland CR, Terdiman JP, Syngal S, de la Chapelle A, Ruschoff J, Fishel R, Lindor NM, Burgart LJ, Hamelin R, Hamilton SR, Hiatt RA, Jass J, Lindblom A, Lynch HT, Peltomaki P, Ramsey SD, Rodriguez-Bigas MA, Vasen HFA, Hawk ET, Barrett JC, Freedman AN, Srivastava S (2004). Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst 96(4): 261-268. doi: 10.1093/jnci/djh034
- Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA, Fodde R, Ranzani GN, Srivastava S (1998). A National Cancer Institute Workshop on Microsatellite Instability for Cancer Detection and Familial Predisposition: Development of International Criteria for the Determination of Microsatellite Instability in Colorectal Cancer. Cancer Res 58(22): 5248-5257. PMID: 9823339
- Herman JG, Umar A, Polyak K, Graff JR, Ahuja N, Issa JPJ, Markowitz S, Willson JKV, Hamilton SR, Kinzler KW, Kane MF, Kolodner RD, Vogelstein B, Kunkel TA, Baylin SB (1998). Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci U S A 95(12): 6870-6875. doi: 10.1073/pnas.95.12.6870
- Cunningham JM, Christensen ER, Tester DJ, Kim CY, Roche PC, Burgart LJ, Thibodeau SN (1998). Hypermethylation of the hMLH1 promoter in colon cancer with microsatellite instability. Cancer Res 58(15): 3455-3460. PMID: 9699680
- Miyakura Y, Sugano K, Akasu T, Yoshida T, Maekawa M, Saitoh S, Sasaki H, Nomizu T, Konishi F, Fujita S, Moriya Y, Nagai H (2004). Extensive but Hemiallelic Methylation of the hMLH1 Promoter Region in Early-Onset Sporadic Colon Cancers With Microsatellite Instability. Clin Gastroenterol Hepatol 2(2): 147-156. doi: 10.1053/s1542-3565(03)00314-8
- Haraldsdottir S, Hampel H, Wu C, Weng DY, Shields PG, Frankel WL, Pan XL, de la Chapelle A, Goldberg RM, Bekaii-Saab T (2016). Patients with colorectal cancer associated with Lynch syndrome and MLH1 promoter hypermethylation have similar prognoses. Genet Med 18(9): 863-868. doi: 10.1038/gim.2015.184
- Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, Biedrzycki B, Donehower RC, Zaheer A, Fisher GA, Crocenzi TS, Lee JJ, Duffy SM, Goldberg RM, de la Chapelle A, Koshiji M, Bhaijee F, Huebner T, Hruban RH, Wood LD, Cuka N, Pardoll DM, Papadopoulos N, Kinzler KW, Zhou S, Cornish TC, et al. (2015). PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med 372(26): 2509-2520. doi: 10.1056/NEJMoa1500596
- Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, Lu S, Kemberling H, Wilt C, Luber BS, Wong F, Azad NS, Rucki AA, Laheru D, Donehower R, Zaheer A, Fisher GA, Crocenzi TS, Lee JJ, Greten TF, Duffy AG, Ciombor KK, Eyring AD, Lam BH, Joe A, Kang SP, Holdhoff M, Danilova L, Cope L, Meyer C, et al. (2017). Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357(6349): 409-413. doi: 10.1126/science.aan6733
- Mensenkamp AR, Vogelaar IP, van Zelst-Stams WAG, Goossens M, Ouchene H, Hendriks-Cornelissen SJB, Kwint MP, Hoogerbrugge N, Nagtegaal ID, Ligtenberg MJL (2014). Somatic Mutations in MLH1 and MSH2 Are a Frequent Cause of Mismatch-Repair Deficiency in Lynch Syndrome-Like Tumors. Gastroenterology 146(3): 643-646. doi: 10.1053/j.gastro.2013.12.002
- Sottoriva A, Kang H, Ma ZC, Graham TA, Salomon MP, Zhao JS, Marjoram P, Siegmund K, Press MF, Shibata D, Curtis C (2015). A Big Bang model of human colorectal tumor growth. Nat Genet 47(3): 209-16. doi: 10.1038/ng.3214
- Hitchins MP (2013). The role of epigenetics in Lynch syndrome. Fam Cancer 12(2): 189-205. doi: 10.1007/s10689-013-9613-3
- Hesson LB, Hitchins MP, Ward RL (2010). Epimutations and cancer predisposition: importance and mechanisms. Curr Opin Genet Dev 20(3): 290-298. doi: 10.1016/j.gde.2010.02.005
- Kane MF, Loda M, Gaida GM, Lipman J, Mishra R, Goldman H, Jessup JM, Kolodner R (1997). Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res 57(5): 808-811. PMID: 9041175
- Gazzoli I, Loda M, Garber J, Syngal S, Kolodner RD (2002). A hereditary nonpolyposis colorectal carcinoma case associated with hypermethylation of the MLH1 gene in normal tissue and loss of heterozygosity of the unmethylated allele in the resulting microsatellite instability-high tumor. Cancer Res 62(14): 3925-3928. PMID: 12124320
- Auclair J, Vaissiere T, Desseigne F, Lasset C, Bonadona V, Giraud S, Saurin JC, Joly MO, Leroux D, Faivre L, Audoynaud C, Montmain G, Ruano E, Herceg Z, Puisieux A, Wang Q (2011). Intensity-Dependent Constitutional MLH1 Promoter Methylation Leads to Early Onset of Colorectal Cancer by Affecting Both Alleles. Genes Chromosomes Cancer 50(3): 178-185. doi: 10.1002/gcc.20842
- Hitchins M, Williams R, Cheong K, Halani N, Lin VA, Packham D, Ku S, Buckle A, Hawkins N, Burn J, Gallinger S, Goldblatt J, Kirk J, Tomlinson I, Scott R, Spigelman A, Suter C, Martin D, Suthers G, Ward R (2005). MLH1 germline epimutations as a factor in hereditary nonpolyposis colorectal cancer. Gastroenterology 129(5): 1392-1399. doi: 10.1053/j.gastro.2005.09.003
- Suter CM, Martin DIK, Ward RL (2004). Germline epimutation of MLH1 in individuals with multiple cancers. Nat Genet 36(5): 497-501. doi: 10.1038/ng1342
- Hitchins MP, Wong JJL, Suthers G, Suter CM, Martin DIK, Hawkins NJ, Ward RL (2007). Brief report: Inheritance of a cancer-associated MLH1 germ-line epimutation. N Engl J Med 356(7): 697-705. doi: 10.1056/NEJMoa064522
- Goel A, Nguyen T-P, Leung H-CE, Nagasaka T, Rhees J, Hotchkiss E, Arnold M, Banerji P, Koi M, Kwok C-T, Packham D, Lipton L, Boland CR, Ward RL, Hitchins MP (2011). De novo constitutional MLH1 epimutations confer early-onset colorectal cancer in two new sporadic Lynch syndrome cases, with derivation of the epimutation on the paternal allele in one. Int J Cancer 128(4): 869-878. doi: 10.1002/ijc.25422
- Morak M, Schackert HK, Rahner N, Betz B, Ebert M, Walldorf C, Royer-Pokora B, Schulmann K, von Knebel-Doeberitz M, Dietmaier W, Keller G, Kerker B, Leitner G, Holinski-Feder E (2008). Further evidence for heritability of an epimutation in one of 12 cases with MLH1 promoter methylation in blood cells clinically displaying HNPCC. Eur J Hum Genet 16(7): 804-811. doi: ejhg200825 [pii]10.1038/ejhg.2008.25
- Morak M, Koehler U, Schackert HK, Steinke V, Royer-Pokora B, Schulmann K, Kloor M, Hochter W, Weingart J, Keiling C, Massdorf T, Holinski-Feder E, German HC (2011). Biallelic MLH1 SNP cDNA expression or constitutional promoter methylation can hide genomic rearrangements causing Lynch syndrome. J Med Genet 48(8): 513-519. doi: 10.1136/jmedgenet-2011-100050
- Morak M, Ibisler A, Keller G, Jessen E, Laner A, Gonzales-Fassrainer D, Locher M, Massdorf T, Nissen AM, Benet-Pages A, Holinski-Feder E (2018). Comprehensive analysis of the MLH1 promoter region in 480 patients with colorectal cancer and 1150 controls reveals new variants including one with a heritable constitutional MLH1 epimutation. J Med Genet 55(4): 240-248. doi: 10.1136/jmedgenet-2017-104744
- Gylling A, Ridanpaa M, Vierimaa O, Aittomaki K, Avela K, Kaariainen H, Laivuori H, Poyhonen M, Sallinen SL, Wallgren-Pettersson C, Jarvinen HJ, Mecklin JP, Peltomaki P (2009). Large genomic rearrangements and germline epimutations in Lynch syndrome. Int J Cancer 124(10): 2333-2340. doi: 10.1002/ijc.24230
- Hitchins MP, Rapkins RW, Kwok CT, Srivastava S, Wong JJL, Khachigian LM, Polly P, Goldblatt J, Ward RL (2011). Dominantly Inherited Constitutional Epigenetic Silencing of MLH1 in a Cancer-Affected Family Is Linked to a Single Nucleotide Variant within the 5 ‘ UTR. Cancer Cell 20(2): 200-213. doi: 10.1016/j.ccr.2011.07.003
- Ward RL, Dobbins T, Lindor NLM, Rapkins RW, Hitchins MP (2013). Identification of constitutional MLH1 epimutations and promoter variants in colorectal cancer patients from the Colon Cancer Family Registry. Genet Med 15(1): 25-35. doi: 10.1038/gim.2012.91
- Pinto D, Pinto C, Guerra J, Pinheiro M, Santos R, Vedeld HM, Yohannes Z, Peixoto A, Santos C, Pinto P, Lopes P, Lothe R, Lind GE, Henrique R, Teixeira MR (2018). Contribution of MLH1 constitutional methylation for Lynch syndrome diagnosis in patients with tumor MLH1 downregulation. Cancer Med 7(2): 433-444. doi: 10.1002/cam4.1285
- Crepin M, Dieu MC, Lejeune S, Escande F, Boidin D, Porchet N, Morin G, Manouvrier S, Mathieu M, Buisine MP (2012). Evidence of constitutional MLH1 epimutation associated to transgenerational inheritance of cancer susceptibility. Hum Mutat 33: 180-188. doi: 10.1002/humu.21617
- Niessen RC, Hofstra RMW, Westers H, Ligtenberg MJL, Kooi K, Jager POJ, de Groote ML, Dijkhuizen T, Olderode-Berends MJW, Hollema H, Kleibeuker JH, Sijmons RH (2009). Germline Hypermethylation of MLH1 and EPCAM Deletions Are a Frequent Cause of Lynch Syndrome. Genes Chromosomes Cancer 48(8): 737-744. doi: 10.1002/gcc.20678
- Sloane MA, Nunez AC, Packham D, Kwok CT, Suthers G, Hesson LB, Ward RL (2015). Mosaic Epigenetic Inheritance as a Cause of Early-Onset Colorectal Cancer. JAMA Oncology 1(7): 953-957. doi: 10.1001/jamaoncol.2015.1484
- Sippel KC, Fraioli RE, Smith GD, Schalkoff ME, Sutherland J, Gallie BL, Dryja TP (1998). Frequency of somatic and germ-line mosaicism in retinoblastoma: Implications for genetic counseling. Am J Hum Genet 62(3): 610-619. doi: 10.1086/301766
- Rushlow D, Piovesan B, Zhang K, Prigoda-Lee NL, Marchong MN, Clark RD, Gallie BL (2009). Detection of Mosaic RB1 Mutations in Families with Retinoblastoma. Hum Mutat 30(5): 842-851. doi: 10.1002/humu.20940
- Jensen LH, Kuramochi H, Cruger DG, Lindebjerg J, Kolvraa S, Danenberg P, Danenberg K, Jakobsen A (2011). Gene expression of the mismatch repair gene MSH2 in primary colorectal cancer. Tumour Biol 32(5): 977-983. doi: 10.1007/s13277-011-0199-9
- Chan TL, Yuen ST, Kong CK, Chan YW, Chan ASY, Ng WF, Tsui WY, Lo MWS, Tam WY, Li VSW, Leung SY (2006). Heritable germline epimutation of MSH2 in a family with hereditary nonpolyposis colorectal cancer. Nat Genet 38(10): 1178-1183. doi: 10.1038/ng1866
- Ligtenberg MJL, Kuiper RP, Chan TL, Goossens M, Hebeda KM, Voorendt M, Lee TYH, Bodmer D, Hoenselaar E, Hendriks-Cornelissen SJB, Tsui WY, Kong CK, Brunner HG, van Kessel AG, Yuen ST, van Krieken J, Leung SY, Hoogerbrugge N (2009). Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3 ‘ exons of TACSTD1. Nat Genet 41(1): 112-117. doi: 10.1038/ng.283
- Hegi ME, Diserens A, Gorlia T, Hamou M, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, Bromberg JEC, Hau P, Mirimanoff RO, Cairncross JG, Janzer RC, Stupp R (2005). MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 352(10): 997-1003. doi: 10.1056/NEJMoa043331
- Wiewrodt D, Nagel G, Dreimueller N, Hundsberger T, Perneczky A, Kaina B (2008). MGMT in primary and recurrent human glioblastomas after radiation and chemotherapy and comparison with p53 status and clinical outcome. Int J Cancer 122(6): 1391-1399. doi: 10.1002/ijc.23219
- Esteller M, Hamilton SR, Burger PC, Baylin SB, Herman JG (1999). Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res 59(4): 793-797. PMID: 10029064
- Shen LL, Kondo Y, Rosner GL, Xiao LC, Hernandez NS, Vilaythong J, Houlihan PS, Krouse RS, Prasad AR, Einspahr JG, Buckmeier J, Alberts DS, Hamilton SR, Issa JPJ (2005). MGMT promoter methylation and field defect in sporadic colorectal cancer. J Natl Cancer Inst 97(18): 1330-1338. doi: 10.1093/jnci/dji275
- Kristensen LS, Treppendahl MB, Asmar F, Girkov MS, Nielsen HM, Kjeldsen TE, Ralfkiaer E, Hansen LL, Gronbaek K (2013). Investigation of MGMT and DAPK1 methylation patterns in diffuse large B-cell lymphoma using allelic MSP-pyrosequencing. Sci Rep 3: 2789. doi: 10.1038/srep02789
- Kristensen LS, Nielsen HM, Hager H, Hansen LL (2011). Methylation of MGMT in malignant pleural mesothelioma occurs in a subset of patients and is associated with the T allele of the rs16906252 MGMT promoter SNP. Lung Cancer 71(2): 130-136. doi: 10.1016/j.lungcan.2010.05.008
- Isono S, Fujishima M, Azumi T, Hashimoto Y, Komoike Y, Yukawa M, Watatani M (2014). O-6-methylguanine-DNA methyltransferase as a prognostic and predictive marker for basal-like breast cancer treated with cyclophosphamide-based chemotherapy. Oncol Lett 7(6): 1778-1784. doi: 10.3892/ol.2014.1985
- Kuroiwa-Trzmielina J, Wang F, Rapkins RW, Ward RL, Buchanan DD, Win AK, Clendenning M, Rosty C, Southey MC, Winship IM, Hopper JL, Jenkins MA, Olivier J, Hawkins NJ, Hitchins MP (2016). SNP rs16906252C > T Is an Expression and Methylation Quantitative Trait Locus Associated with an Increased Risk of Developing MGMT-Methylated Colorectal Cancer. Clin Cancer Res 22(24): 6266-6277. doi: 10.1158/1078-0432.Ccr-15-2765
- Fumagalli C, Pruneri G, Possanzini P, Manzotti M, Barile M, Feroce I, Colleoni M, Bonanni B, Maisonneuve P, Radice P, Viale G, Barberis M (2012). Methylation of O-6-methylguanine-DNA methyltransferase (MGMT) promoter gene in triple-negative breast cancer patients. Breast Cancer Res Treat 134(1): 131-137. doi: 10.1007/s10549-011-1945-9
- Esteller M, Garcia-Foncillas J, Andion E, Goodman SN, Hidalgo OF, Vanaclocha V, Baylin SB, Herman JG (2000). Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med 343(19): 1350-1354. doi: 10.1056/nejm200011093431901
- Kitange GJ, Carlson BL, Schroeder MA, Grogan PT, Lamont JD, Decker PA, Wu WT, James CD, Sarkaria JN (2009). Induction of MGMT expression is associated with temozolomide resistance in glioblastoma xenografts. Neuro Oncol 11(3): 281-291. doi: 10.1215/15228517-2008-090
- Mattern J, Eichhorn U, Kaina B, Volm M (1998). O-6-methylguanine-DNA methyltransferase activity and sensitivity to cyclophosphamide and cisplatin in human lung tumor xenografts. Int J Cancer 77(6): 919-922. doi: 10.1002/(sici)1097-0215(19980911)77:6<919::Aid-ijc20>3.0.Co;2-v
- Rapkins RW, Wang F, Nguyen HN, Cloughesy TF, Lai A, Ha W, Nowak AK, Hitchins MP, McDonald KL (2015). The MGMT promoter SNP rs16906252 is a risk factor for MGMT methylation in glioblastoma and is predictive of response to temozolomide. Neuro Oncol 17(12): 1589-1598. doi: 10.1093/neuonc/nov064
- Leng SG, Bernauer AM, Hong CB, Do KC, Yingling CM, Flores KG, Tessema M, Tellez CS, Willink RP, Burki EA, Picchi MA, Stidley CA, Prados MD, Costello JF, Gilliland FD, Crowell RE, Belinsky SA (2011). The A/G Allele of Rs16906252 Predicts for MGMT Methylation and Is Selectively Silenced in Premalignant Lesions from Smokers and in Lung Adenocarcinomas. Clin Cancer Res 17(7): 2014-2023. doi: 10.1158/1078-0432.Ccr-10-3026
- Candiloro ILM, Dobrovic A (2009). Detection of MGMT Promoter Methylation in Normal Individuals Is Strongly Associated with the T Allele of the rs16906252 MGMT Promoter Single Nucleotide Polymorphism. Cancer Prev Res 2(10): 862-867. doi: 10.1158/1940-6207.Capr-09-0056
- Mavaddat N, Peock S, Frost D, Ellis S, Platte R, Fineberg E, Evans DG, Izatt L, Eeles RA, Adlard J, Davidson R, Eccles D, Cole T, Cook J, Brewer C, Tischkowitz M, Douglas F, Hodgson S, Walker L, Porteous ME, Morrison PJ, Side LE, Kennedy MJ, Houghton C, Donaldson A, Rogers MT, Dorkins H, Miedzybrodzka Z, Gregory H, Eason J, et al. (2013). Cancer Risks for BRCA1 and BRCA2 Mutation Carriers: Results From Prospective Analysis of EMBRACE. J Natl Cancer Inst 105(11): 812-822. doi: 10.1093/jnci/djt095
- Brohet RM, Velthuizen ME, Hogervorst FBL, Meijers-Heijboer HEJ, Seynaeve C, Collee MJ, Verhoef S, Ausems M, Hoogerbrugge N, van Asperen CJ, Garcia EG, Menko F, Oosterwijk JC, Devilee P, van’t Veer LJ, van Leeuwen FE, Easton DF, Rookus MA, Antoniou AC, Resource H (2014). Breast and ovarian cancer risks in a large series of clinically ascertained families with a high proportion of BRCA1 and BRCA2 Dutch founder mutations. J Med Genet 51(2): 98-107. doi: 10.1136/jmedgenet-2013-101974
- Alsop K, Fereday S, Meldrum C, deFazio A, Emmanuel C, George J, Dobrovic A, Birrer MJ, Webb PM, Stewart C, Friedlander M, Fox S, Bowtell D, Mitchell G (2012). BRCA Mutation Frequency and Patterns of Treatment Response in BRCA Mutation-Positive Women With Ovarian Cancer: A Report From the Australian Ovarian Cancer Study Group. J Clin Oncol 30(21): 2654-2663. doi: 10.1200/jco.2011.39.8545
- Rebbeck TR, Friebel TM, Friedman E, Hamann U, Huo DZ, Kwong A, Olah E, Olopade OI, Solano AR, Teo SH, Thomassen M, Weitzel JN, Chan TL, Fergus J, Goldgar DE, Kruse TA, Edenir I, Park SK, Torres D, van Rensburg EJ, McGuffog L, Parsons MT, Leslie G, Aalfs CM, Abugattas J, Adlard J, Agata S, Aittomaki K, Andrews L, Andrulis IL, et al. (2018). Mutational spectrum in a worldwide study of 29,700 families with BRCA1 or BRCA2 mutations. Hum Mutat 39(5): 593-620. doi: 10.1002/humu.23406
- Hu CL, Hart SN, Polley EC, Gnanaolivu R, Shimelis H, Lee KY, Lilyquist J, Na J, Moore R, Antwi SO, Bamlet WR, Chaffee KG, DiCarlo J, Wu Z, Samara R, Kasi PM, McWilliams RR, Petersen GM, Couch FJ (2018). Association Between Inherited Germline Mutations in Cancer Predisposition Genes and Risk of Pancreatic Cancer. JAMA 319(23): 2401-2409. doi: 10.1001/jama.2018.6228
- Zhen JT, Syed J, Nguyen KA, Leapman MS, Agarwal N, Brierley K, Llor X, Hofstatter E, Shuch B (2018). Genetic Testing for Hereditary Prostate Cancer: Current Status and Limitations. Cancer 124(15): 3105-3117. doi: 10.1002/cncr.31316
- Breast Canc Linkage C (1999). Cancer risks in BRCA2 mutation carriers. J Natl Cancer Inst 91(15): 1310-1316. doi: 10.1093/jnci/91.15.1310
- van Asperen CJ, Brohet RM, Meijers-Heijboer EJ, Hoogerbrugge N, Verhoef S, Vasen HFA, Ausems M, Menko FH, Garcia EBG, Klijn JGM, Hogervorst FBL, van Houwelingen JC, van’t Veer LJ, Rookus MA, van Leeuwen FE, Netherlands Collaborative Grp H (2005). Cancer risks in BRCA2 families: estimates for sites other than breast and ovary. J Med Genet 42(9): 711-719. doi: 10.1136/jmg.2004.028829
- Valeri A, Martinez S, Casado JA, Bueren JA (2011). Fanconi anaemia: from a monogenic disease to sporadic cancer. Clin Transl Oncol 13(4): 215-221. doi: 10.1007/s12094-011-0645-6
- Garcia MJ, Benitez J (2008). The Fanconi anaemia/BRCA pathway and cancer susceptibility. Searching for new therapeutic targets. Clin Transl Oncol 10(2): 78-84. doi: 10.1007/s12094-008-0160-6
- Hedenfalk I, Duggan D, Chen YD, Radmacher M, Bittner M, Simon R, Meltzer P, Gusterson B, Esteller M, Kallioniemi OP, Wilfond B, Borg A, Trent J (2001). Gene-expression profiles in hereditary breast cancer. N Engl J Med 344(8): 539-548. doi: 10.1056/nejm200108233450822
- Lonning PE (2010). Molecular basis for therapy resistance. Mol Oncol 4(3): 284-300. doi: 10.1016/j.molonc.2010.04.005
- Sørlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, vandeRijn M, Jeffrey SS, Thorsen T, Quist H, Matese JC, Brown PO, Botstein D, Lønning PE, BorresenDale AL (2001). Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Nat Acad Sci 98(19): 10869-10874. doi: 10.1073/pnas.191367098
- Sørlie T, Tibshirani R, Parker J, Hastie T, Marron JS, Nobel A, Deng S, Johnsen H, Pesich R, Geisler S, Demeter J, Perou CM, Lonning PE, Brown PO, Borresen-Dale AL, Botstein D (2003). Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci U S A 100(14): 8418-8423. doi: 10.1073/pnas.0932692100
- Honrado E, Osorio A, Palacios J, Benitez J (2006). Pathology and gene expression of hereditary breast tumors associated with BRCA1, BRCA2 and CHEK2 gene mutations. Oncogene 25(43): 5837-5845. doi: 10.1038/sj.onc.1209875
- Brianese RC, Nakamura KDD, de Almeida F, Ramalho RF, Barros BDD, Ferreira ENE, Formiga MND, de Andrade VP, de Lima VCC, Carraro DM (2018). BRCA1 deficiency is a recurrent event in early-onset triple-negative breast cancer: a comprehensive analysis of germline mutations and somatic promoter methylation. Breast Cancer Res Treat 167(3): 803-814. doi: 10.1007/s10549-017-4552-6
- Young SR, Pilarski RT, Donenberg T, Shapiro C, Hammond LS, Miller J, Brooks KA, Cohen S, Tenenholz B, Desai D, Zandvakili I, Royer R, Li S, Narod SA (2009). The prevalence of BRCA1 mutations among young women with triple-negative breast cancer. Bmc Cancer 9: 86. doi: 10.1186/1471-2407-9-86
- Rechsteiner M, Dedes K, Fink D, Pestalozzi B, Sobottka B, Moch H, Wild P, Varga Z (2018). Somatic BRCA1 mutations in clinically sporadic breast cancer with medullary histological features. J Cancer Res Clin Oncol 144(5): 865-874. doi: 10.1007/s00432-018-2609-5
- Gonzalez-Angulo AM, Timms KM, Liu SY, Chen HQ, Litton JK, Potter J, Lanchbury JS, Stemke-Hale K, Hennessy BT, Arun BK, Hortobagyi GN, Do KA, Mills GB, Meric-Bernstam F (2011). Incidence and Outcome of BRCA Mutations in Unselected Patients with Triple Receptor-Negative Breast Cancer. Clin Cancer Res 17(5): 1082-1089. doi: 10.1158/1078-0432.Ccr-10-2560
- Tutt A, Tovey H, Cheang MCU, Kernaghan S, Kilburn L, Gazinska P, Owen J, Abraham J, Barrett S, Barrett-Lee P, Brown R, Chan S, Dowsett M, Flanagan JM, Fox L, Grigoriadis A, Gutin A, Harper-Wynne C, Hatton MQ, Hoadley KA, Parikh J, Parker P, Perou CM, Roylance R, Shah V, Shaw A, Smith IE, Timms KM, Wardley AM, Wilson G, et al. (2018). Carboplatin in BRCA1/2-mutated and triple-negative breast cancer BRCAness subgroups: the TNT Trial. Nat Med 24(5): 628-637. doi: 10.1038/s41591-018-0009-7
- Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, Martincorena I, Alexandrov LB, Martin S, Wedge DC, Van Loo P, Ju YS, Smid M, Brinkman AB, Morganella S, Aure MR, Lingjaerde OC, Langerod A, Ringner M, Ahn SM, Boyault S, Brock JE, Broeks A, Butler A, Desmedt C, Dirix L, Dronov S, Fatima A, Foekens JA, Gerstung M, et al. (2016). Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 534(7605): 47-54. doi: 10.1038/nature17676
- Meric-Bernstam F, Brusco L, Daniels M, Wathoo C, Bailey AM, Strong L, Shaw K, Lu K, Qi Y, Zhao H, Lara-Guerra H, Litton J, Arun B, Eterovic AK, Aytac U, Routbort M, Subbiah V, Janku F, Davies MA, Kopetz S, Mendelsohn J, Mills GB, Chen K (2016). Incidental germline variants in 1000 advanced cancers on a prospective somatic genomic profiling protocol. Ann Oncol 27(5): 795-800. doi: 10.1093/annonc/mdw018
- Winter C, Nilsson MP, Olsson E, George AM, Chen Y, Kvist A, Torngren T, Vallon-Christersson J, Hegardt C, Hakkinen J, Jonsson G, Grabau D, Malmberg M, Kristoffersson U, Rehn M, Gruvberger-Saal SK, Larsson C, Borg A, Loman N, Saal LH (2016). Targeted sequencing of BRCA1 and BRCA2 across a large unselected breast cancer cohort suggests that one-third of mutations are somatic. Ann Oncol 27(8): 1532-1538. doi: 10.1093/annonc/mdw209
- Davies H, Glodzik D, Morganella S, Yates LR, Staaf J, Zou XQ, Ramakrishna M, Martin S, Boyault S, Sieuwerts AM, Simpson PT, King TA, Raine K, Eyfjord JE, Kong G, Borg A, Birney E, Stunnenberg HG, van de Vijver MJV, Borresen-Dale AL, Martens JWM, Span PN, Lakhani SR, Vincent-Salomon A, Sotiriou C, Tutt A, Thompson AM, Van Laere S, Richardson AL, Viari A, et al. (2017). HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat Med 23(4): 517-525. doi: 10.1038/nm.4292
- Lips EH, Laddach N, Savola SP, Vollebergh MA, Oonk AMM, Imholz ALT, Wessels LFA, Wesseling J, Nederlof PM, Rodenhuis S (2011). Quantitative copy number analysis by Multiplex Ligation-dependent Probe Amplification (MLPA) of BRCA1-associated breast cancer regions identifies BRCAness. Breast Cancer Res 13(5): R107. doi: 10.1186/bcr3049
- Polak P, Kim J, Braunstein LZ, Karlic R, Haradhavala NJ, Tiao G, Rosebrock D, Livitz D, Kubler K, Mouw KW, Kamburov A, Maruvka YE, Leshchiner I, Lander ES, Golub TR, Zick A, Orthwein A, Lawrence MS, Batra RN, Caldas C, Haber DA, Laird PW, Shen H, Ellisen LW, D’Andrea AD, Chanock SJ, Foulkes WD, Getz G (2017). A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat Genet 49(10): 1476-1485. doi: 10.1038/ng.3934
- Severson TM, Wolf DM, Yau C, Peeters J, Wehkam D, Schouten PC, Chin SF, Majewski IJ, Michaut M, Bosma A, Pereira B, Bismeijer T, Wessels L, Caldas C, Bernards R, Simon IM, Glas AM, Linn S, van ‘T Veer L (2017). The BRCA1ness signature is associated significantly with response to PARP inhibitor treatment versus control in the I-SPY 2 randomized neoadjuvant setting. Breast Cancer Res 19(1): 99. doi: 10.1186/s13058-017-0861-2
- Schouten PC, Gluz O, Harbeck N, Mohrmann S, Diallo-Danebrock R, Pelz E, Kruizinga J, Velds A, Nieuwland M, Kerkhoven RM, Liedtke C, Frick M, Kates R, Linn SC, Nitz U, Marme F (2016). BRCA1-like profile predicts benefit of tandem high dose epirubicin-cyclophospamide-thiotepa in high risk breast cancer patients randomized in the WSG-AM01 trial. Int J Cancer 139(4): 882-889. doi: 10.1002/ijc.30078
- Schouten PC, Marme F, Aulmann S, Sinn HP, van Essen HF, Ylstra B, Hauptmann M, Schneeweiss A, Linn SC (2015). Breast Cancers with a BRCA1-like DNA Copy Number Profile Recur Less Often Than Expected after High-Dose Alkylating Chemotherapy. Clin Cancer Res 21(4): 763-770. doi: 10.1158/1078-0432.ccr-14-1894
- Isakoff SJ, Mayer EL, He L, Traina TA, Carey LA, Krag KJ, Rugo HS, Liu MC, Stearns V, Come SE, Timms KM, Hartman AR, Borger DR, Finkelstein DM, Garber JE, Ryan PD, Winer EP, Goss PE, Ellisen LW (2015). TBCRC009: A Multicenter Phase II Clinical Trial of Platinum Monotherapy With Biomarker Assessment in Metastatic Triple-Negative Breast Cancer. J Clin Oncol 33(17): 1902-U1967. doi: 10.1200/jco.2014.57.6660
- Vollebergh MA, Lips EH, Nederlof PM, Wessels LFA, Wesseling J, Vijver MJV, de Vries EGE, Van Tinteren H, Jonkers J, Hauptmann M, Rodenhuis S, Linn SC (2014). Genomic patterns resembling BRCA1- and BRCA2-mutated breast cancers predict benefit of intensified carboplatin-based chemotherapy. Breast Cancer Res 16(3): R47. doi: 10.1186/bcr3655
- Jonsson G, Staaf J, Vallon-Christersson J, Ringner M, Gruvberger-Saal SK, Saal LH, Holm K, Hegardt C, Arason A, Fagerholm R, Persson C, Grabau D, Johnsson E, Lovgren K, Magnusson L, Heikkila P, Agnarsson BA, Johannsson OT, Malmstrom P, Ferno M, Olsson H, Loman N, Nevanlinna H, Barkardottir RB, Borg A (2012). The Retinoblastoma Gene Undergoes Rearrangements in BRCA1-Deficient Basal-like Breast Cancer. Cancer Res 72(16): 4028-4036. doi: 10.1158/0008-5472.Can-12-0097
- Esteller M, Silva JM, Dominguez G, Bonilla F, Matias-Guiu X, Lerma E, Bussaglia E, Prat J, Harkes IC, Repasky EA, Gabrielson E, Schutte M, Baylin SB, Herman JG (2000). Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J Natl Cancer Inst 92(7): 564-569. doi: 10.1093/jnci/92.7.564
- Cunningham JM, Cicek MS, Larson NB, Davila J, Wang C, Larson MC, Song H, Dicks EM, Harrington P, Wick M, Winterhoff BJ, Hamidi H, Konecny GE, Chien J, Bibikova M, Fan JB, Kalli KR, Lindor NM, Fridley BL, Pharoah PPD, Goode EL (2014). Clinical Characteristics of Ovarian Cancer Classified by BRCA1, BRCA2, and RAD51C Status. Sci Rep 4: 4026. doi: 10.1038/srep04026
- Bell D, Berchuck A, Birrer M, Chien J, Cramer DW, Dao F, Dhir R, DiSaia P, Gabra H, Glenn P, Godwin AK, Gross J, Hartmann L, Huang M, Huntsman DG, Iacocca M, Imielinski M, Kalloger S, Karlan BY, Levine DA, Mills GB, Morrison C, Mutch D, Olvera N, Orsulic S, Park K, Petrelli N, Rabeno B, Rader JS, Sikic BI, et al. (2011). Integrated genomic analyses of ovarian carcinoma. Nature 474(7353): 609-615. doi: 10.1038/nature10166
- Rice JC, Futscher BW (2000). Transcriptional repression of BRCA1 by aberrant cytosine methylation, histone hypoacetylation and chromatin condensation of the BRCA1 promoter. Nucleic Acids Res 28(17): 3233-3239. doi: 10.1093/nar/28.17.3233
- Joosse SA, Brandwijk KIM, Mulder L, Wesseling J, Hannemann J, Nederlof PM (2011). Genomic Signature of BRCA1 Deficiency in Sporadic Basal-Like Breast Tumors. Genes Chromosomes Cancer 50(2): 71-81. doi: 10.1002/gcc.20833
- Xu Y, Diao L, Chen Y, Liu Y, Wang C, Ouyang T, Li J, Wang T, Fan Z, Fan T, Lin B, Deng D, Narod SA, Xie Y (2013). Promoter methylation of BRCA1 in triple-negative breast cancer predicts sensitivity to adjuvant chemotherapy. Ann Oncol 24(6): 1498-1505. doi: 10.1093/annonc/mdt011
- Lips EH, Mulder L, Oonk A, van der Kolk LE, Hogervorst FBL, Imholz ALT, Wesseling J, Rodenhuis S, Nederlof PM (2013). Triple-negative breast cancer: BRCAness and concordance of clinical features with BRCA1-mutation carriers. Br J Cancer 108(10): 2172-2177. doi: 10.1038/bjc.2013.144
- Jacot W, Thezenas S, Senal R, Viglianti C, Laberenne AC, Lopez-Crapez E, Bibeau F, Bleuse JP, Romieu G, Lamy PJ (2013). BRCA1 promoter hypermethylation, 53BP1 protein expression and PARP-1 activity as biomarkers of DNA repair deficit in breast cancer. Bmc Cancer 13: 523. doi: 10.1186/1471-2407-13-523
- Esteller M, Fraga MF, Guo MZ, Garcia-Foncillas J, Hedenfalk I, Godwin AK, Trojan J, Vaurs-Barriere C, Bignon YJ, Ramus S, Benitez J, Caldes T, Akiyama Y, Yuasa Y, Launonen V, Canal MJ, Rodriguez R, Capella G, Peinado MA, Borg A, Aaltonen LA, Ponder BA, Baylin SB, Herman JG (2001). DNA methylation patterns in hereditary human cancers mimic sporadic tumorigenesis. Hum Mol Genet 10(26): 3001-3007. doi: 10.1093/hmg/10.26.3001
- Kontorovich T, Cohen Y, Nir U, Friedman E (2009). Promoter methylation patterns of ATM, ATR, BRCA1, BRCA2 and P53 as putative cancer risk modifiers in Jewish BRCA1/BRCA2 mutation carriers. Breast Cancer Res Treat 116(1): 195-200. doi: 10.1007/s10549-008-0121-3
- Severson TM, Peeters J, Majewski I, Michaut M, Bosma A, Schouten PC, Chin SF, Pereira B, Goldgraben MA, Bismeijer T, Kluin RJC, Muris JJF, Jirstrom K, Kerkhoven RM, Wessels L, Caldas C, Bernards R, Simon IM, Linn S (2015). BRCA1-like signature in triple negative breast cancer: Molecular and clinical characterization reveals subgroups with therapeutic potential. Mol Oncol 9(8): 1528-1538. doi: 10.1016/j.molonc.2015.04.011
- Vos S, Moelans CB, van Diest PJ (2017). BRCA promoter methylation in sporadic versus BRCA germline mutation-related breast cancers. Breast Cancer Res 19(1): 64. doi: 10.1186/s13058-017-0856-z
- Wong EM, Southey MC, Fox SB, Brown MA, Dowty JG, Jenkins MA, Giles GG, Hopper JL, Dobrovic A (2011). Constitutional Methylation of the BRCA1 Promoter Is Specifically Associated with BRCA1 Mutation-Associated Pathology in Early-Onset Breast Cancer. Cancer Prev Res 4(1): 23-33. doi: 10.1158/1940-6207.capr-10-0212
- Yamashita N, Tokunaga E, Kitao H, Hitchins M, Inoue Y, Tanaka K, Hisamatsu Y, Taketani K, Akiyoshi S, Okada S, Oda Y, Saeki H, Oki E, Maehara Y (2015). Epigenetic Inactivation of BRCA1 Through Promoter Hypermethylation and Its Clinical Importance in Triple-Negative Breast Cancer. Clin Breast Cancer 15(6): 498-504. doi: 10.1016/j.clbc.2015.06.009
- Veeck J, Ropero S, Setien F, Gonzalez-Suarez E, Osorio A, Benitez J, Herman JG, Esteller M (2010). BRCA1 CpG Island Hypermethylation Predicts Sensitivity to Poly(Adenosine Diphosphate)-Ribose Polymerase Inhibitors. J Clin Oncol 28(29): E563-E564. doi: 10.1200/jco.2010.30.1010
- Stefansson OA, Villanueva A, Vidal A, Marti L, Esteller M (2012). BRCA1 epigenetic inactivation predicts sensitivity to platinum-based chemotherapy in breast and ovarian cancer. Epigenetics 7(11): 1225-1229. doi: 10.4161/epi.22561
- Cai FF, Ge I, Wang MH, Biskup E, Lin XY, Zhong XY (2014). Pyrosequencing analysis of BRCA1 methylation level in breast cancer cells. Tumour Biol 35(4): 3839-3844. doi: 10.1007/s13277-013-1508-2
- Xu XR, Gammon MD, Zhang YJ, Bestor TH, Zeisel SH, Wetmur JG, Wallenstein S, Bradshaw PT, Garbowski G, Teitelbaum SL, Neugut AI, Santella RM, Chen J (2009). BRCA1 promoter methylation is associated with increased mortality among women with breast cancer. Breast Cancer Res Treat 115(2): 397-404. doi: 10.1007/s10549-008-0075-5
- Chen YL, Zhou J, Xu Y, Li ZS, Wen XZ, Yao LH, Xie YT, Deng DJ (2009). BRCA1 promoter methylation associated with poor survival in Chinese patients with sporadic breast cancer. Cancer Sci 100(9): 1663-1667. doi: 10.1111/j.1349-7006.2009.01225.x
- Potapova A, Hoffman AM, Godwin AK, Al-Saleem T, Cairns P (2008). Promoter hypermethylation of the PALB2 susceptibility gene in inherited and sporadic breast and ovarian cancer. Cancer Res 68(4): 998-1002. doi: 10.1158/0008-5472.can-07-2418
- Konstantinopoulos PA, Ceccaldi R, Shapiro GI, D’Andrea AD (2015). Homologous Recombination Deficiency: Exploiting the Fundamental Vulnerability of Ovarian Cancer. Cancer Discov 5(11): 1137-1154. doi: 10.1158/2159-8290.cd-15-0714
- Risch HA, McLaughlin JR, Cole DEC, Rosen B, Bradley L, Kwan E, Jack E, Vesprini DJ, Kuperstein G, Abrahamson JLA, Fan I, Wong B, Narod SA (2001). Prevalence and penetrance of germline BRCA1 and BRCA2 mutations in a population series of 649 women with ovarian cancer. Am J Hum Genet 68(3): 700-710. doi: 10.1086/318787
- Pal T, Permuth-Wey J, Betts JA, Krischer JP, Fiorica J, Arango H, LaPolla J, Hoffman M, Martino MA, Wakeley K, Wilbanks G, Nicosia S, Cantor A, Sutphen R (2005). BRCA1 and BRCA2 mutations account for a large proportion of ovarian carcinoma cases. Cancer 104(12): 2807-2816. doi: 10.1002/cncr.21536
- Hoberg-Vetti H, Bjorvatn C, Fiane BE, Aas T, Woie K, Espelid H, Rusken T, Eikesdal HP, Listol W, Haavind MT, Knappskog PM, Haukanes BI, Steen VM, Hoogerbrugge N (2016). BRCA1/2 testing in newly diagnosed breast and ovarian cancer patients without prior genetic counselling: the DNA-BONus study. Eur J Hum Genet 24(6): 881-888. doi: 10.1038/ejhg.2015.196
- Baldwin RL, Nemeth E, Tran H, Shvartsman H, Cass I, Narod S, Karlan BY (2000). BRCA1 promoter region hypermethylation in ovarian carcinoma: A population-based study. Cancer Res 60(19): 5329-5333. PMID: 11034065
- Geisler JP, Hatterman-Zogg MA, Rathe JA, Buller RE (2002). Frequency of BRCA1 dysfunction in ovarian cancer. J Natl Cancer Inst 94(1): 61-67. doi: 10.1093/jnci/94.1.61
- Sun TT, Ruscito I, Dimitrova D, Chekerov R, Kulbe H, Baron U, Blanchard R, Panici PB, Darb-Esfahani S, Sehouli J, Olek S, Braicu EI (2017). Genetic Versus Epigenetic BRCA1 Silencing Pathways: Clinical Effects in Primary Ovarian Cancer Patients A Study of the Tumor Bank Ovarian Cancer Consortium. Int J Gynecol Cancer 27(8): 1658-1665. doi: 10.1097/igc.0000000000001071
- Ruscito I, Dimitrova D, Vasconcelos I, Gellhaus K, Schwachula T, Bellati F, Zeillinger R, Benedetti-Panici P, Vergote I, Mahner S, Cacsire-Tong D, Concin N, Darb-Esfahani S, Lambrechts S, Sehouli J, Olek S, Braicu EI (2014). BRCA1 gene promoter methylation status in high-grade serous ovarian cancer patients – A study of the tumour Bank ovarian cancer (TOC) and ovarian cancer diagnosis consortium (OVCAD). Eur J Cancer 50(12): 2090-2098. doi: 10.1016/j.ejca.2014.05.001
- Gras E, Cortes J, Diez O, Alonso C, Matias-Guiu X, Baiget M, Prat J (2001). Loss of heterozygosity on chromosome 13q12-q14, BRCA-2 mutations and lack of BRCA-2 promoter hypermethylation in sporadic epithelial ovarian tumors. Cancer 92(4): 787-795. doi: 10.1002/1097-0142(20010815)92:4<787::Aid-cncr1384>3.0.Co;2-4
- Hilton JL, Geisler JP, Rathe JA, Hattermann-Zogg MA, DeYoung B, Buller RE (2002). Inactivation of BRCA1 and BRCA2 in ovarian cancer. Journal Natl Cancer Inst 94(18): 1396-1406. doi: 10.1093/jnci/94.18.1396
- Goodheart MJ, Rose SL, Hattermann-Zogg M, Smith BJ, De Young BR, Buller RE (2009). BRCA2 alteration is important in clear cell carcinoma of the ovary. Clin Genet 76(2): 161-167. doi: 10.1111/j.1399-0004.2009.01207.x
- Vos S, van Diest PJ, Moelans CB (2018). A systematic review on the frequency of BRCA promoter methylation in breast and ovarian carcinomas of BRCA germline mutation carriers: Mutually exclusive, or not? Crit Rev Oncol Hematol 127: 29-41. doi: 10.1016/j.critrevonc.2018.05.008
- Dworkin AM, Spearman AD, Tseng SY, Sweet K, Toland AE (2009). Methylation not a frequent “second hit” in tumors with germline BRCA mutations. Fam Cancer 8(4): 339-346. doi: 10.1007/s10689-009-9240-1
- Van Heetvelde M, Van Bockstal M, Poppe B, Lambein K, Rosseel T, Atanesyan L, Deforce D, Van Den Berghe I, De Leeneer K, Van Dorpe J, Vral A, Claes KBM (2018). Accurate detection and quantification of epigenetic and genetic second hits in BRCA1 and BRCA2-associated hereditary breast and ovarian cancer reveals multiple co-acting second hits. Cancer Lett 425: 125-133. doi: 10.1016/j.canlet.2018.03.026
- Ignatov T, Eggemann H, Costa SD, Roessner A, Kalinski T, Ignatov A (2014). BRCA1 promoter methylation is a marker of better response to platinum-taxane-based therapy in sporadic epithelial ovarian cancer. J Cancer Res Clin Oncol 140(9): 1457-1463. doi: 10.1007/s00432-014-1704-5
- Kondrashova O, Topp M, Nesic K, Lieschke E, Ho GY, Harrell MI, Zapparoli GV, Hadley A, Holian R, Boehm E, Heong V, Sanij E, Pearson RB, Krais JJ, Johnson N, McNally O, Ananda S, Alsop K, Hutt KJ, Kaufmann SH, Lin KK, Harding TC, Traficante N, deFazio A, McNeish LA, Bowtell DD, Swisher EM, Dobrovic A, Wakefield MJ, Scott CL, et al. (2018). Methylation of all BRCA1 copies predicts response to the PARP inhibitor rucaparib in ovarian carcinoma. Nat Commun 9: 3970. doi: 10.1038/s41467-018-05564-z
- Mirza MR, Monk BJ, Herrstedt J, Oza AM, Mahner S, Redondo A, Fabbro M, Ledermann JA, Lorusso D, Vergote I, Ben-Baruch NE, Marth C, Madry R, Christensen RD, Berek JS, Dorum A, Tinker AV, du Bois A, Gonzalez-Martin A, Follana P, Benigno B, Rosenberg P, Gilbert L, Rimel BJ, Buscema J, Balser JP, Agarwal S, Matulonis UA, Investigators E-ON (2016). Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N Engl J Med 375(22): 2154-2164. doi: 10.1056/NEJMoa1611310
- Gorodnova TV, Sokolenko AP, Ivantsov AO, Iyevleva AG, Suspitsin EN, Aleksakhina SN, Yanus GA, Togo AV, Maximov SY, Imyanitov EN (2015). High response rates to neoadjuvant platinum-based therapy in ovarian cancer patients carrying germ-line BRCA mutation. Cancer Lett 369(2): 363-367. doi: 10.1016/j.canlet.2015.08.028
- Peplonska B, Bukowska A, Wieczorek E, Przybek M, Zienolddiny S, Reszka E (2017). Rotating night work, lifestyle factors, obesity and promoter methylation in BRCA1 and BRCA2 genes among nurses and midwives. Plos One 12(6): e0178792. doi: 10.1371/journal.pone.0178792
- Rodriguez-Balada M, Roig B, Mele M, Salvat M, Martorell L, Borras J, Guma J (2018). Germline promoter hypermethylation in BRCA1 and BRCA2 genes is not present in hereditary breast cancer patients. Clin Transl Oncol 20(9): 1226-1231. doi: 10.1007/s12094-018-1837-0
- Chen Y, Toland AE, McLennan J, Fridlyand J, Crawford B, Costello JF, Ziegler JL (2006). Lack of germ-line promoter methylation in BRCA1-negative families with familial breast cancer. Genet Test 10(4): 281-284. doi: 10.1089/gte.2006.10.281
- Hansmann T, Pliushch G, Leubner M, Kroll P, Endt D, Gehrig A, Preisler-Adams S, Wieacker P, Haaf T (2012). Constitutive promoter methylation of BRCA1 and RAD51C in patients with familial ovarian cancer and early-onset sporadic breast cancer. Hum Mol Genet 21(21): 4669-4679. doi: 10.1093/hmg/dds308
- Lønning PE, Berge EO, Bjornslett M, Minsaas L, Chrisanthar R, Hoberg-Vetti H, Dulary C, Busato F, Bjorneklett S, Eriksen C, Kopperud R, Axcrona U, Davidson B, Bjorge L, Evans G, Howell A, Salvesen HB, Janszky I, Hveem K, Romundstad PR, Vatten LJ, Tost J, Dorum A, Knappskog S (2018). White Blood Cell BRCA1 Promoter Methylation Status and Ovarian Cancer Risk. Ann Intern Med 168(5): 326-334. doi: 10.7326/m17-0101
- Dobrovic A, Mikeska T, Alsop K, Candiloro I, George J, Mitchell G, Bowtell D (2014). Constitutional BRCA1 methylation is a major predisposition factor for high-grade serous ovarian cancer. Cancer Res 74(19): 290. doi: 10.1158/1538-7445.Am2014-290
- Evans DGR, van Veen EM, Byers HJ, Wallace AJ, Ellingford JM, Beaman G, Santoyo-Lopez J, Aitman TJ, Eccles DM, Lalloo FI, Smith MJ, Newman WG (2018). A Dominantly Inherited 5′ UTR Variant Causing Methylation-Associated Silencing of BRCA1 as a Cause of Breast and Ovarian Cancer. Am J Hum Genet 103(2): 213-220. doi: 10.1016/j.ajhg.2018.07.002
- Chan KYK, Liu W, Long JR, Yip SP, Chan SY, Shu XO, Chua DTT, Cheung ANY, Ching JCY, Cai H, Au GKH, Chan M, Foo W, Ngan HYS, Gao YT, Ngan ESW, Garcia-Barcelo MM, Zheng W, Khoo US (2009). Functional polymorphisms in the BRCA1 promoter influence transcription and are associated with decreased risk for breast cancer in Chinese women. J Med Genet 46(1): 32-39. doi: 10.1136/jmg.2007.057174
- Bielinska B, Gaj P, Kluska A, Nowakowska D, Balabas A, Dabrowska M, Niwinska A, Gruchota J, Zub R, Skasko E, Steffen J, Ostrowski J, Siedlecki JA (2013). Association of the BRCA1 promoter polymorphism rs11655505 with the risk of familial breast and/or ovarian cancer. Fam Cancer 12(4): 691-698. doi: 10.1007/s10689-013-9647-6
- Li L, Guo F, Gao Y, Ren YX, Yuan P, Yan LY, Li R, Lian Y, Li JY, Hu BQ, Gao JP, Wen L, Tang FC, Qiao J (2018). Single-cell multi-omics sequencing of human early embryos. Nat Cell Biol 20(7): 847-858. doi: 10.1038/s41556-018-0123-2
- Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestart ML, Heine-Suner D, Cigudosa JC, Urioste M, Benitez J, Boix-Chornet M, Sanchez-Aguilera A, Ling C, Carlsson E, Poulsen P, Vaag A, Stephan Z, Spector TD, Wu YZ, Plass C, Esteller M (2005). Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A 102(30): 10604-10609. doi: 10.1073/pnas.0500398102
- Ollikainen M, Smith KR, Joo EJH, Ng HK, Andronikos R, Novakovic B, Aziz NKA, Carlin JB, Morley R, Saffery R, Craig JM (2010). DNA methylation analysis of multiple tissues from newborn twins reveals both genetic and intrauterine components to variation in the human neonatal epigenome. Hum Mol Genet 19(21): 4176-4188. doi: 10.1093/hmg/ddq336
- Galetzka D, Hansmann T, El Hajj N, Weis E, Irmscher B, Ludwig M, Schneider-Ratzke B, Kohlschmidt N, Beyer V, Bartsch O, Zechner U, Spix C, Haaf T (2012). Monozygotic twins discordant for constitutive BRCA1 promoter methylation, childhood cancer and secondary cancer. Epigenetics 7(1): 47-54. doi: 10.4161/epi.7.1.18814
- Jones MJ, Goodman SJ, Kobor MS (2015). DNA methylation and healthy human aging. Aging Cell 14(6): 924-932. doi: 10.1111/acel.12349
- Pal S, Tyler JK (2016). Epigenetics and aging. Sci Adv 2(7): e1600584. doi: 10.1126/sciadv.1600584
- Xie WB, Kagiampakis I, Pan LX, Zhang YW, Murphy L, Tao Y, Kong XQ, Kang B, Xia LM, Carvalho FLF, Sen S, Yen RWC, Zahnow CA, Ahuja N, Baylin SB, Easwaran H (2018). DNA Methylation Patterns Separate Senescence from Transformation Potential and Indicate Cancer Risk. Cancer Cell 33(2): 309-321. doi: 10.1016/j.ccell.2018.01.008
- Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, Chambert K, Mick E, Neale BM, Fromer M, Purcell SM, Svantesson O, Landen M, Hoglund M, Lehmann S, Gabriel SB, Moran JL, Lander ES, Sullivan PF, Sklar P, Gronberg H, Hultman CM, McCarroll SA (2014). Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. N Engl J Med 371(26): 2477-2487. doi: 10.1056/NEJMoa1409405
- Coombs CC, Zehir A, Devlin SM, Kishtagari A, Syed A, Jonsson P, Hyman DM, Solit DB, Robson ME, Baselga J, Arcila ME, Ladanyi M, Tallman MS, Levine RL, Berger MF (2017). Therapy-Related Clonal Hematopoiesis in Patients with Non-hematologic Cancers Is Common and Associated with Adverse Clinical Outcomes. Cell Stem Cell 21(3): 374-382. doi: 10.1016/j.stem.2017.07.010
- Weber-Lassalle K, Harter P, Hauke J, Ernst C, Kommoss S, Marme F, Weber-Lassalle N, Prieske K, Dietrich D, Borde J, Pohl-Rescigno E, Reuss A, Ataseven B, Engel C, Stingl JC, Schmutzler RK, Hahnen E (2018). Diagnosis of Li-Fraumeni Syndrome: Differentiating TP53 germline mutations from clonal hematopoiesis Results of the observational AGO-TR1 trial. Hum Mutat 39(12): 2040-2046. doi: 10.1002/humu.23653
- Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, Higgins JM, Moltchanov V, Kuo FC, Kluk MJ, Henderson B, Kinnunen L, Koistinen HA, Ladenvall C, Getz G, Correa A, Banahan BF, Gabriel S, Kathiresan S, Stringham HM, McCarthy MI, Boehnke M, Tuomilehto J, Haiman C, Groop L, Atzmon G, et al. (2014). Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N Engl J Med 371(26): 2488-2498. doi: 10.1056/NEJMoa1408617
- Al-Moghrabi N, Al-Showimi M, Al-Yousef N, Al-Shahrani B, Karakas B, Alghofaili L, Almubarak H, Madkhali S, Al Humaidan H (2018). Methylation of BRCA1 and MGMT genes in white blood cells are transmitted from mothers to daughters. Clin Epigenetics 10(1): 99. doi: 10.1186/s13148-018-0529-5
- Lønning PE, Knappskog S (2018). BRCA1 methylation in newborns: genetic disposition, maternal transfer, environmental influence, or by chance only? Clin Epigenetics 10(1): 128. doi: 10.1186/s13148-018-0566-0
- Evans GR, Ramsden RT, Shenton A, Gokhale C, Bowers NL, Huson SM, Pichert G, Wallace A (2007). Mosaicism in neurofibromatosis type 2: an update of risk based on uni/bilaterality of vestibular schwannoma at presentation and sensitive mutation analysis including multiple ligation-dependent probe amplification. J Med Genet 44(7): 424-428. doi: 10.1136/jmg.2006.047753
- Amitrano S, Marozza A, Somma S, Imperatore V, Hadjistilianou T, De Francesco S, Toti P, Galimberti D, Meloni I, Cetta F, Piu P, Di Marco C, Dosa L, Lo Rizzo C, Carignani G, Mencarelli MA, Mari F, Renieri A, Ariani F (2015). Next generation sequencing in sporadic retinoblastoma patients reveals somatic mosaicism. Eur J Hum Genet 23(11): 1523-1530. doi: 10.1038/ejhg.2015.6
- Mucci LA, Hjelmborg JB, Harris JR, Czene K, Havelick DJ, Scheike T, Graff RE, Holst K, Moller S, Unger RH, McIntosh C, Nuttall E, Brandt I, Penney KL, Hartman M, Kraft P, Parmigiani G, Christensen K, Koskenvuo M, Holm NV, Heikkila K, Pukkala E, Skytthe A, Adami HO, Kaprio J, Nordic Twin Study Canc N (2016). Familial Risk and Heritability of Cancer Among Twins in Nordic Countries. JAMA 315(1): 68-76. doi: 10.1001/jama.2015.17703
- Swerdlow AJ, DeStavola BL, Swanwick MA, Maconochie NES (1997). Risks of breast and testicular cancers in young adult twins in England and Wales: Evidence on prenatal and genetic aetiology. Lancet 350(9093): 1723-1728. doi: 10.1016/s0140-6736(97)05526-8
- Vatten LJ, Nilsen TI, Tretli S, Trichopoulos D, Romundstad PR (2005). Size at birth and risk of breast cancer: prospective population-based study. Int J Cancer 114(3): 461-464. doi: 10.1002/ijc.20726
- Qiu L, Onoyama S, Low HP, Chang CI, Strohsnitter WC, Norwitz ER, Lopresti M, Edmiston K, Lambe M, Trichopoulos D, Lagiou P, Hsieh CC (2015). Effect of preeclampsia on umbilical cord blood stem cells in relation to breast cancer susceptibility in the offspring. Carcinogenesis 36(1): 94-98. doi: 10.1093/carcin/bgu231
- Sandvei MS, Lagiou P, Romundstad PR, Trichopoulos D, Vatten LJ (2015). Size at birth and risk of breast cancer: update from a prospective population-based study. Eur J Epidemiol 30(6): 485-492. doi: 10.1007/s10654-015-0045-2
- Ekbom A, Hsieh CC, Lipworth L, Adami HO, Trichopoulos D (1997). Intrauterine environment and breast cancer risk in women: A population-based study. J Natl Cancer Inst 89(1): 71-76. doi: 10.1093/jnci/89.1.71
- Ekbom A, Erlandsson G, Hsieh CC, Trichopoulos D, Adami HO, Cnattingius S (2000). Risk of breast cancer in prematurely born women. J Natl Cancer Inst 92(10): 840-841. doi: 10.1093/jnci/92.10.840
- Troisi R, Grotmol T, Jacobsen J, Tretli S, Toft-Sorensen H, Gissler M, Kaaja R, Potischman N, Ekbom A, Hoover RN, Stephansson O (2013). Perinatal characteristics and breast cancer risk in daughters: a Scandinavian population-based study. J Dev Orig Health Dis 4(1): 35-41. doi: 10.1017/s2040174412000645
- Joubert BR, Haberg SE, Nilsen RM, Wang X, Vollset SE, Murphy SK, Huang Z, Hoyo C, Midttun O, Cupul-Uicab LA, Ueland PM, Wu MC, Nystad W, Bell DA, Peddada SD, London SJ (2012). 450K Epigenome-Wide Scan Identifies Differential DNA Methylation in Newborns Related to Maternal Smoking during Pregnancy. Environ Health Perspect 120(10): 1425-1431. doi: 10.1289/ehp.1205412
- Pauwels S, Ghosh M, Duca RC, Bekaert B, Freson K, Huybrechts I, Langie SAS, Koppen G, Devlieger R, Godderis L (2017). Dietary and supplemental maternal methyl-group donor intake and cord blood DNA methylation. Epigenetics 12(1): 1-10. doi: 10.1080/15592294.2016.1257450
- Tobi EW, Lumey L, Talens RP, Kremer D, Putter H, Stein AD, Slagboom PE, Heijmans BT (2009). DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Hum Mol Genet 18(21): 4046-4053. doi: 10.1093/hmg/ddp353
- Engel SM, Joubert BR, Wu MC, Olshan AF, Haberg SE, Ueland PM, Nystad W, Nilsen RM, Vollset SE, Peddada SD, London SJ (2014). Neonatal Genome-Wide Methylation Patterns in Relation to Birth Weight in the Norwegian Mother and Child Cohort. Am J Epidemiol 179(7): 834-842. doi: 10.1093/aje/kwt433
- Sandanger TM, Nost TH, Guida F, Rylander C, Campanella G, Muller DC, van Dongen J, Boomsma DI, Johansson M, Vineis P, Vermeulen R, Lund E, Chadeau-Hyam M (2018). DNA methylation and associated gene expression in blood prior to lung cancer diagnosis in the Norwegian Women and Cancer cohort. Sci Rep 8: 16714. doi: 10.1038/s41598-018-34334-6
- Barry KH, Moore LE, Sampson J, Yan LY, Meyer A, Oler AJ, Chung CC, Wang ZM, Yeager M, Amundadottir L, Berndt SI (2014). DNA Methylation Levels at Chromosome 8q24 in Peripheral Blood Are Associated with 8q24 Cancer Susceptibility Loci. Cancer Prev Res 7(12): 1282-1292. doi: 10.1158/1940-6207.Capr-14-0132
- FitzGerald LM, Naeem H, Makalic E, Schmidt DF, Dowty JG, Joo JE, Jung CH, Bassett JK, Dugue PA, Chung J, Lonie A, Milne RL, Wong EM, Hopper JL, English DR, Severi G, Baglietto L, Pedersen J, Giles GG, Southey MC (2017). Genome-Wide Measures of Peripheral Blood Dna Methylation and Prostate Cancer Risk in a Prospective Nested Case-Control Study. Prostate 77(5): 471-478. doi: 10.1002/pros.23289
- Liggett TE, Melnikov AA, Marks JR, Levenson VV (2011). Methylation patterns in cell-free plasma DNA reflect removal of the primary tumor and drug treatment of breast cancer patients. Int J Cancer 128(2): 492-499. doi: 10.1002/ijc.25363
- Rack B, Schindlbeck C, Andergassen U, Lorenz R, Zwingers T, Schneeweiss A, Lichtenegger W, Beckmann MW, Sommer HL, Pantel K, Friese K, Janni W (2010). Prognostic relevance of circulating tumor cells in the peripheral blood of primary breast cancer patients. Ca Res 70(Suppl 24): S6-5. doi: 10.1158/0008-5472.sabcs10-s6-5
- Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, Thornton K, Agrawal N, Sokoll L, Szabo SA, Kinzler KW, Vogelstein B, Diaz LA (2008). Circulating mutant DNA to assess tumor dynamics. Nat Med 14(9): 985-990. doi: 10.1038/nm.1789
- Teschendorff AE, Menon U, Gentry-Maharaj A, Ramus SJ, Gayther SA, Apostolidou S, Jones A, Lechner M, Beck S, Jacobs IJ, Widschwendter M (2009). An Epigenetic Signature in Peripheral Blood Predicts Active Ovarian Cancer. Plos One 4(12): e8274. doi: 10.1371/journal.pone.0008274
- Koestler DC, Usset J, Christensen BC, Marsit CJ, Karagas MR, Kelsey KT, Wiencke JK (2017). DNA Methylation-Derived Neutrophil-to-Lymphocyte Ratio: An Epigenetic Tool to Explore Cancer Inflammation and Outcomes. Cancer Epidemiol Biomarkers Prev 26(3): 328-338. doi: 10.1158/1055-9965.epi-16-0461
- Marsit CJ, Koestler DC, Christensen BC, Karagas MR, Houseman EA, Kelsey KT (2011). DNA Methylation Array Analysis Identifies Profiles of Blood-Derived DNA Methylation Associated With Bladder Cancer. J Clin Oncol 29(9): 1133-1139. doi: 10.1200/jco.2010.31.3577
- Gervin K, Page CM, Aass HCD, Jansen MA, Fjeldstad HE, Andreassen BK, Duijts L, van Meurs JB, van Zelm MC, Jaddoe VW, Nordeng H, Knudsen GP, Magnus P, Nystad W, Staff AC, Felix JF, Lyle R (2016). Cell type specific DNA methylation in cord blood: A 450K-reference data set and cell count-based validation of estimated cell type composition. Epigenetics 11(9): 690-698. doi: 10.1080/15592294.2016.1214782
- Reinius LE, Acevedo N, Joerink M, Pershagen G, Dahlen SE, Greco D, Soderhall C, Scheynius A, Kere J (2012). Differential DNA Methylation in Purified Human Blood Cells: Implications for Cell Lineage and Studies on Disease Susceptibility. Plos One 7(7; e41361): 1-13. doi: 10.1371/journal.pone.0041361
- Fridley BL, Armasu SM, Cicek MS, Larson MC, Wang C, Winham SJ, Kalli KR, Koestler DC, Rider DN, Shridhar V, Olson JE, Cunningham JM, Goode EL (2014). Methylation of leukocyte DNA and ovarian cancer: relationships with disease status and outcome. Bmc Medical Genomics 7(21): 1-12. doi: 10.1186/1755-8794-7-21
- Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, Wiencke JK, Kelsey KT (2012). DNA methylation arrays as surrogate measures of cell mixture distribution. Bmc Bioinformatics 13 (86): 1-16. doi: 10.1186/1471-2105-13-86
- Tost J, Gut IG (2007). DNA methylation analysis by pyrosequencing. Nat Protoc 2(9): 2265-2275. doi: 10.1038/nprot.2007.314
- Barbosa M, Joshi RS, Garg P, Martin-Trujillo A, Patel N, Jadhav B, Watson CT, Gibson W, Chetnik K, Tessereau C, Mei H, De Rubeis S, Reichert J, Lopes F, Vissers L, Kleefstra T, Grice DE, Edelmann L, Soares G, Maciel P, Brunner HG, Buxbaum JD, Gelb BD, Sharp AJ (2018). Identification of rare de novo epigenetic variations in congenital disorders. Nat Commun 9: 2064. doi: 10.1038/s41467-018-04540-x
- Steegers-Theunissen RP, Obermann-Borst SA, Kremer D, Lindemans J, Siebel C, Steegers EA, Slagboom PE, Heijmans BT (2009). Periconceptional Maternal Folic Acid Use of 400 mu g per Day Is Related to Increased Methylation of the IGF2 Gene in the Very Young Child. Plos One 4(11): e7845. doi: 10.1371/journal.pone.0007845
- Chen P, Piaggi P, Traurig M, Bogardus C, Knowler WC, Baier LJ, Hanson RL (2017). Differential methylation of genes in individuals exposed to maternal diabetes in utero. Diabetologia 60(4): 645-655. doi: 10.1007/s00125-016-4203-1
- Dias BG, Ressier KJ (2014). Parental olfactory experience influences behavior and neural structure in subsequent generations. Nat Neurosci 17(1): 89-96. doi: 10.1038/nn.3594
- Sun WF, Dong H, Becker AS, Dapito DH, Modica S, Grandl G, Opitz L, Efthymiou V, Straub LG, Sarker G, Balaz M, Balazova L, Perdikari A, Kiehlmann E, Bacanovic S, Zellweger C, Peleg-Raibstein D, Pelczar P, Reik W, Burger IA, von Meyenn F, Wolfrum C (2018). Cold-induced epigenetic programming of the sperm enhances brown adipose tissue activity in the offspring. Nat Med 24(9): 1372-1383. doi: 10.1038/s41591-018-0102-y
- Yehuda R, Daskalakis NP, Bierer LM, Bader HN, Klengel T, Holsboer F, Binder EB (2016). Holocaust Exposure Induced Intergenerational Effects on FKBP5 Methylation. Biol Psychiatry 80(5): 372-380. doi: 10.1016/j.biopsych.2015.08.005
- Vagero D, Pinger PR, Aronsson V, van den Berg GJ (2018). Paternal grandfather’s access to food predicts all-cause and cancer mortality in grandsons. Nat Commun 9: 5124. doi: 10.1038/s41467-018-07617-9
- Youssoufian H, Pyeritz RE (2002). Mechanisms and consequences of somatic mosaicism in humans. Nat Rev Genet 3(10): 748-758. doi: 10.1038/nrg906
- Zhang JH, Walsh MF, Wu G, Edmonson MN, Gruber TA, Easton J, Hedges D, Ma XT, Zhou X, Yergeau DA, Wilkinson MR, Vadodaria B, Chen X, McGee RB, Hines-Dowell S, Nuccio R, Quinn E, Shurtleff SA, Rusch M, Patel A, Becksfort JB, Wang SG, Weaver MS, Ding L, Mardis ER, Wilson RK, Gajjar A, Ellison DW, Pappo AS, Pui CH, et al. (2015). Germline Mutations in Predisposition Genes in Pediatric Cancer. N Engl J Med 373(24): 2336-2346. doi: 10.1056/NEJMoa1508054
- Friedman E, Efrat N, Soussan-Gutman L, Dvir A, Kaplan Y, Ekstein T, Nykamp K, Powers M, Rabideau M, Sorenson J, Topper S (2015). Low-level constitutional mosaicism of a de novoBRCA1 gene mutation. Br J Cancer 112(4): 765-768. doi: 10.1038/bjc.2015.14
- Delon I TA, Molenda A, Drummond J, Oakhill K, Girling A, Liu H, Whittaker J, Treacy R, Tischkowitz M. (2012 ). A germline mosaic BRCA1 exon deletion in a woman with bilateral basal-like breast cancer. Clin Genet 84(3):297-9. doi: 10.1111/cge.12057
- Ainsworth PJ, Chakraborty PK, Weksberg R (1997). Example of somatic mosaicism in a series of de novo neurofibromatosis type 1 cases due to a maternally derived deletion. Hum Mutat 9(5): 452-457. doi: 10.1002/(sici)1098-1004(1997)9:5<452::Aid-humu12>3.3.Co;2-v
- LoTenFoe JR, Kwee ML, Rooimans MA, Oostra AB, Veerman AJP, vanWeel M, Pauli RM, Shahidi NT, Dokal I, Roberts I, Altay C, Gluckman E, Gibson RA, Mathew CG, Arwert F, Joenje H (1997). Somatic mosaicism in Fanconi anemia: Molecular basis and clinical significance. Eur J Hum Genet 5(3): 137-148. PMID: 9272737
- Mazor T, Pankov A, Song JS, Costello JF (2016). Intratumoral Heterogeneity of the Epigenome. Cancer Cell 29(4): 440-451. doi: 10.1016/j.ccell.2016.03.009
- Wu JM, Fackler MJ, Halushka MK, Molavi DW, Taylor ME, Teo WW, Griffin C, Fetting J, Davidson NE, De Marzo AM, Hicks JL, Chitale D, Ladanyi M, Sukumar S, Argani P (2008). Heterogeneity of breast cancer metastases: Comparison of therapeutic target expression and promoter methylation between primary tumors and their multifocal metastases. Clin Cancer Res 14(7): 1938-1946. doi: 10.1158/1078-0432.Ccr-07-4082
COPYRIGHT
© 2019

Constitutional Mosaic Epimutations – a hidden cause of cancer? by Lønning et al. is licensed under a Creative Commons Attribution 4.0 International License.


