Reviews:
Cell Stress, Vol. 5, No. 1, pp. 1 - 17; doi: 10.15698/cst2021.01.239
Improving glucose and lipids metabolism: drug development based on bile acid related targets

1 School of Pharmacy, Fudan University, Shanghai 201203, China.
2 Shanghai Key Laboratory of Complex Prescriptions and MOE Key Laboratory for Standardization of Chinese Medicines, Institute of Chinese Materia Medica, Shanghai University of Traditional Chinese Medicine, Shanghai, 201203, China.
3 Department of Diabetes Complications and Metabolism, Institute of Diabetes and Metabolism Research Center, Beckman Research Institute, City of Hope National Medical Center, 1500 E. Duarte Road, Duarte, CA 91010, USA.
4 Shanghai Institute of Endocrine and Metabolic Diseases, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200025, China.
Keywords: bile acid, FXR, TGR5, GPBAR1, bile salt hydrolase, CYP8B1.
Abbreviatons:
6-ECDCA – 6α-ethyl-chenodeoxycholic acid;
BSH – bile salt hydrolase;
BTA – betulinic acid;
CA – cholic acid;
CDCA – chenodesoxycholic acid;
DHA – dihydroartemisinin;
DPP-4 – dipeptidyl peptidase-4;
FXR – farnesoid X receptor;
GLP-1 – glucagon-like peptide-1;
HFD – high-fat diet;
LCA – lithocholic acid;
MCA – muricholic acid;
miR-26a – microRNA-26a;
NAFLD – nonalcoholic fatty liver disease;
NASH – nonalcoholic steatohepatitis;
OA – oleanolic acid;
PBC – primary biliary cholangitis;
PEG – polyethylene glycol;
PYY – peptide YY;
RYGB – Roux-en-Y gastric bypass;
SAR – structure-activity relationship;
TβMCA – tauro-β-muricholic acid;
TCA – tauro-cholic acid;
TGR5 – Takeda G-protein-coupled receptor 5;
TLCA – taurolithocholic acid;
UDCA – ursodeoxycholic acid;
VSG – vertical sleeve gastrectomy.
Received originally: 19/10/2020 Received in revised form: 04/12/2020
Accepted: 07/12/2020
Published: 05/01/2021
Correspondence:
Jingyan Tian, Shanghai Institute of Endocrine and Metabolic Diseases, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200025, China; tianjypaper@163.com
Yang Wang, School of Pharmacy, Fudan University, Shanghai 201203, China; wangyang@shmu.edu.cn
Wendong Huang, Department of Diabetes Complications and Metabolism, Institute of Diabetes and Metabolism Research Center, Beckman Research Institute, City of Hope National Medical Center, 1500 E. Duarte Road, Duarte, CA 91010, USA; whuang@coh.org
Conflict of interest statement: The authors declare no conflicts of interest.
Please cite this article as: Hanchen Shen, Lili Ding, Mehdi Baig, Jingyan Tian, Yang Wang and Wendong Huang (2021). Improving glucose and lipids metabolism: drug development based on bile acid related targets. Cell Stress 5(1): 1-18. doi: 10.15698/cst2021.01.239
Abstract
Bariatric surgery is one of the most effective treatment options for severe obesity and its comorbidities. However, it is a major surgery that poses several side effects and risks which impede its clinical use. Therefore, it is urgent to develop alternative safer pharmacological approaches to mimic bariatric surgery. Recent studies suggest that bile acids are key players in mediating the metabolic benefits of bariatric surgery. Bile acids can function as signaling molecules by targeting bile acid nuclear receptors and membrane receptors, like FXR and TGR5 respectively. In addition, the composition of bile acids is regulated by either the hepatic sterol enzymes such as CYP8B1 or the gut microbiome. These bile acid related targets all play important roles in regulating metabolism. Drug development based on these targets could provide new hope for patients without the risks of surgery and at a lower cost. In this review, we summarize the most updated progress on bile acid related targets and development of small molecules as drug candidates based on these targets.
INTRODUCTION
The incidence of obesity continues to rise rapidly in industrialized countries, including the US. Obesity is defined by an excessive amount of body fat that impairs health. While mortality of patients is not directly caused by obesity itself, patients who develop comorbidities are likely to have a much higher mortality rate. These comorbidities include cardiovascular disease, diabetes mellitus, kidney related diseases, certain cancers, anxiety, depression, and degenerative joint disorders [1]. Bariatric surgeries, including Roux-en-Y gastric bypass (RYGB), vertical sleeve gastrectomy (VSG), adjustable gastric banding (AGB) and biliopancreatic diversion with duodenal switch (BPDDS), are the most effective therapies for the treatment of severe obesity (Fig. 1) [2]. It can be done for sickly obese patients who have no response to pharmacological or behavioral treatment [3]. Nowadays, the most common bariatric surgeries are RYGB and VSG, both of which change people's digestive system to reduce weight and improve glucose tolerance [4]. Despite its efficiency, the clinical use of bariatric surgery is still impeded by cardiovascular risk and other serious complications [2]. For this reason, there is an urgent need to find less-invasive therapeutic approaches to mimic metabolic surgery.
–
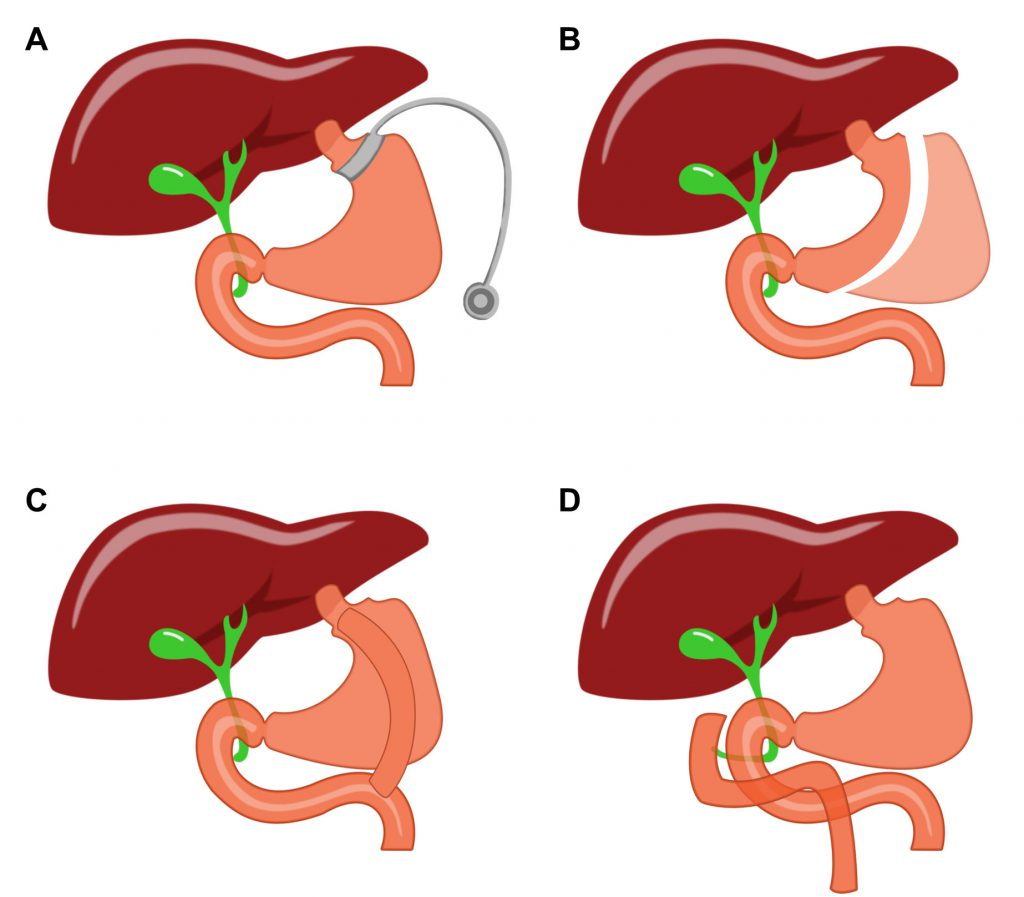 | FIGURE 1: Contemporary procedures for metabolic surgery. (A) adjustable gastric banding (AGB); (B) vertical sleeve gastrectomy (VSG); (C) Roux-en-Y gastric bypass (RYGB); and (D) biliopancreatic diversion with duodenal switch (BPDDS). |
–
Traditionally, bariatric surgeries were perceived to show their efficacy by manipulating the anatomy of the gastrointestinal tract, and consequently limiting nutrient absorption. Different surgeries like RYGB and VSG have been able to target weight loss in their own ways. It can be noted that these differing surgical techniques are able to achieve similar metabolic results. This would suggest that altering the anatomy of the stomach and intestine is not solely responsible for the weight loss. Rather, there are supplementary physiological phenomena involved, other than simply restricting the amount of food absorbed by the body [5][6]. Recent studies showed that the primary mechanism could be because of the alternation in secretion and activity of hormones and neurotransmitters [4][7][8][9]. This molecular component, rather than the mechanical restriction, can explain the beneficial impact of bariatric surgeries. Developing drugs or related therapies based on the molecular mechanism is thus a promising way to treat obese patients without surgical trauma.
–
Among those mechanisms, bile acids play a central role. Several studies have focused on the elevated serum bile acid levels after bariatric surgeries, which led to better glucose and lipids metabolism [10][11]. Bile acids are detergent molecules, and are required for absorption of fats, steroids, and lipid-soluble vitamins in the intestine [12]. They are also signaling molecules that activate nuclear and membrane bile acid receptors, through which bile acids can modulate lipid and glucose metabolism [13]. Bile acids can also behave as antibacterial agents in the gut. They are involved in a complex interaction with the gut microbiota, and are essential for normal gastrointestinal function [14]. In spite of the fact that they are vital for human health, bile acids are highly toxic when they accumulate in the liver and other peripheral tissues [15]. Due to the prominent physiological effects of bile acids, developing drugs based on bile acid related targets are actively pursued by pharmaceutical companies.
NUCLEAR AND MEMBRANE BILE ACID RECEPTORS
In addition to digestive function, bile acids are also versatile signaling molecules that activate multiple signaling pathways [13]. The main functions of bile acids depend on the activation of two receptors: nuclear farnesoid X receptors (FXR) and membrane G-protein-coupled Takeda G-protein-coupled receptor 5 (TGR5) [16]. These two receptors are distributed differently in tissues and have a wide range of physiological functions. Thus, they became promising pharmacological targets in treating diseases such as primary biliary cholangitis (PBC), primary sclerosing cholangitis (PSC), cholestatic diseases, and other metabolic diseases [17].
–
Nuclear farnesoid X receptor, FXR
FXR is mainly expressed in various tissues including the liver, intestine, kidney, and adrenal gland, and it plays crucial roles in the regulation of bile acid synthesis, secretion, and transport [18]. FXR is important for lipid and glucose metabolism; the activation of FXR has shown beneficial effects on various metabolic diseases such as nonalcoholic fatty liver disease (NAFLD), type 2 diabetes, dyslipidemia, and obesity [19]. Ryan et al. demonstrated that FXR is required to for the metabolic improvements of VSG [8]. These results indicate that FXR agonism plays an essential role in “bariatric-mimetic” technology.
–
As a nuclear receptor of bile acids, FXR can be activated by several bile acids at physiological concentrations. CDCA (chenodesoxycholic acid) is the most potent ligand among all the bile acids in human, with EC50 values ranging between 2-5 μM[20]. Because of its high FXR activating potency, the chemical manipulation of the CDCA scaffold offers many compounds with improved potency, efficacy, and metabolic stability. Pellicciari et al. synthesized a series of 6α-alkyl-substituted analogues of CDCA and evaluated their binding potency to FXR [21]. 6α-ethyl-chenodeoxycholic acid (6-ECDCA) was shown to be a very potent and selective FXR agonist (Fig. 2). With FXR agonism EC50 = 0.99 μM, 6-ECDCA showed anticholeretic activity in an in vivo rat model of cholestasis. 6-ECDCA, also known as obeticholic acid or INT-747, is now the most promising FXR agonist for clinical use. It has been approved for the treatment of ursodeoxycholic acid (UDCA) resistant patients in PBC [22]. Additionally, 6-ECDCA is now advancing through Phase III clinical trials in nonalcoholic steatohepatitis (NASH) patients (NCT02548351). Further research was conducted to improve the potency, and physicochemical properties of CDCA derivates. Pellicciari et al. state that modified hydroxyl groups on steroidal nucleus can affect the properties and behavior of different bile acids [23]. Based on docking studies of hydroxyl derivatives of 6-ECDCA, they designed and synthesized a series of semisynthetic bile acid derivatives. Of those, TC-100 not only boasted the highest FXR agonist activity with an EC50 of 0.14 μM, but also had improved physicochemical profiles compared to that of 6-ECDCA (Fig. 2). However, many FXR agonists, including 6-ECDCA, also have TGR5 agonist activity which can cause adverse effects like pruritus. To solve this problem, Sepe et al. conducted chemical modifications on the C3-hydroxyl group of the steroidal core? [24]. This strategy generated a potent FXR agonist BAR704 (EC50 = 0.95 μM) with inhibitory activity against TGR5 (Fig. 2). Xiao et al. further found that modification on the side chains can change the properties of bile acid derivates. They substituted carboxylic tail with amide groups generating compound 1 with good efficacy, FXR selectivity, and pharmacokinetic properties compared to 6-ECDCA (Fig. 2) [25].
–
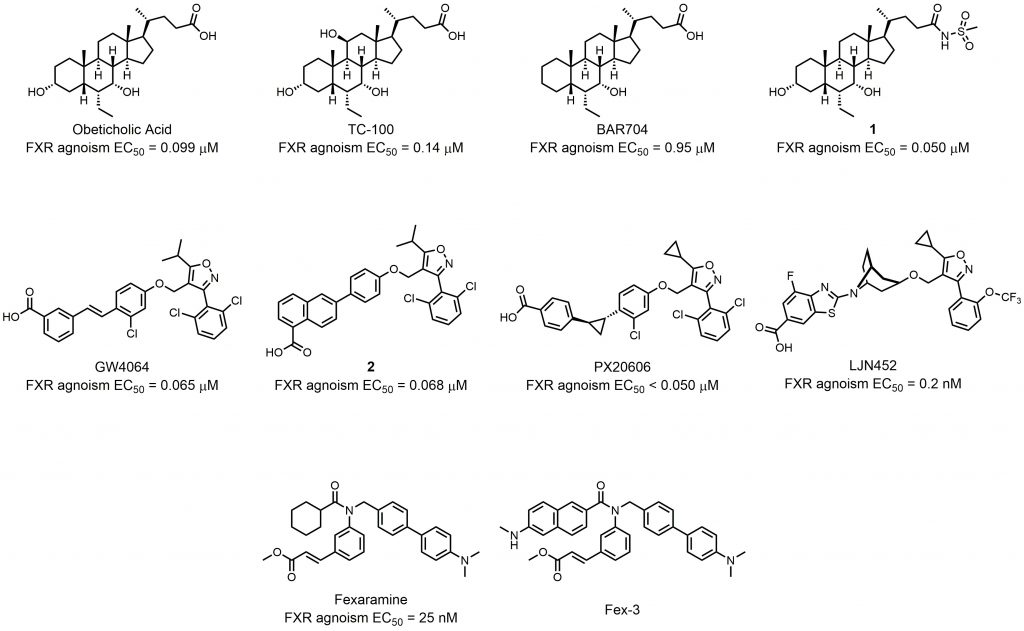 | FIGURE 2: Semisynthetic bile acid derivatives as FXR agonists and synthetic FXR nonsteroidal. |
–
The FXR agonists discussed above all contain a steroidal core?. However, the steroidal bile acid-like chemical structure suffer some limitations such as poor aqueous solubility, poor bioavailability, unattractive pharmacokinetic profile, and low FXR selectivity [26]. For these reasons, research of synthetic nonsteroidal FXR agonists has been gaining attention in recent years. Maloney et al. utilized a cell free ligand-sensing assay to identify an isoxazole compound as the lead compound from a combinatorial library of 9,900 stilbene carboxylic acids [27]. Further structure-activity-relationship (SAR) studies helped researchers to find GW4064, the first high-affinity nonsteroidal FXR agonist (Fig. 2). Despite the high potency, the clinical use of GW4064 was limited because of toxicity, poor pharmacokinetic properties, and stilbene-mediated photo-instability. Akwabi-Ameyaw et al. designed and synthesized compound 2 (EC50 = 0.068 μM), a stilbene replacement of GW4064 (EC50 = 0.065 μM; Fig. 2) [28]. Compound 2 exhibited comparable efficacy and selectivity to GW4064, and could reduce the severity of cholestasis in the a-naphthyl-isothiocyanate (ANIT) acute cholestatic rat model. However, the oral bioavailability of compound 2 in rodents was poor. Kinzel et al. studied the SAR of terminal carboxylic acid-bearing aryl or heteroaryl moiety and linker group of GW4064 derivates [29]. Through the SAR study, they found that PX20606 was a potent FXR agonist (EC50< 0.1 μM) with improved aqueous solubility and metabolic stability (Fig. 2). PX20606 demonstrated beneficial effects in two animal models of prehepatic and intrahepatic portal hypertension [30]. To date, two phase I studies of PX20606 have been completed (NCT01998659, NCT01998672), showing potential for further clinical use. Tullu et al. conducted a thorough SAR study of isoxazole derivates, paying attention to potency and pharmacokinetic properties at the same time [31]. By introducing a bicyclic nortropine-substituted benzothiazole carboxylic acid moiety, the researchers synthesized LJN452 with an EC50 of 0.2 nM (Fig. 2). LJN452 was found to be generally safe and well-tolerated at pharmacologically active doses in healthy volunteers. Now LJN452 has completed two phase II studies for the treatment of patients with PBC (NCT02516605) and NASH (NCT02855164). Besides isoxazole derivates, many compounds with other structures can also act as starting compound in the development of synthetic nonsteroidal FXR agonists. Downes et al. screened for hit compounds from a combinatorial library of ≈ 10,000 benzopyran-based compounds; further modification and screening generated the potent FXR agonist fexaramine (EC50 = 25 nM) (Fig. 2) [32]. Fexaramine was poorly absorbed into circulation when delivered orally, and its intestinal-restricted property showed the possibility of a promising and safer treatment [33]. For this reason, Wang et al. synthesized Fex-3, which targets FXR in the ileum and has a better selectivity [34].
–
Besides semisynthetic bile acid derivatives and synthetic nonsteroidal FXR agonists, many natural products also show FXR agonist activity. Interestingly, these natural products have greater structural diversity. Studies focusing on natural FXR agonists helped researchers gain more insights into the structures of FXR ligands and can lead to innovative drug design and synthesis. Lu et al. showed that hedragonic acid, isolated from the stem and root of Celastrus orbiculatus Thunb, is a selective FXR agonist with anti-inflammatory and liver protection activity (Fig. 3) [35]. Although hedragonic acid belongs to oleanane-type triterpene, whose structure is similar to the steroidal core of bile acids, FXR has a unique binding mode with it. Hedragonic acid occupies a novel binding pocket in FXR, which is different from the classic binding position. In vivo experiments demonstrated outstanding therapeutic effects of hedragonic acid in liver diseases, suggesting its potential for clinical use. Curcumin exhibits a wide range of biological activities, including anticancer, antimicrobial, anti-inflammatory, and antioxidation activities (Fig. 3) [36]. Danning tablet, a Chinese patent medicine preparation, has been clinically used to treat human liver and gallbladder diseases. Yang et al. found that curcumin, one major compound derived from danning tablet, exerted the therapeutic effect [37]. Binding to FXR, curcumin could reduce hepatic bile acids accumulation, normalized the imbalance of bile acid homeostasis, and reduced inflammatory responses. Through molecular docking experiments, the researchers found that curcumin could theoretically bind to the FXR protein at the same site as GW4064, suggesting more structure flexibility at this binding site. Silymarin, consisting of seven flavonolignans, is a natural herbal extraction from the fruit and seeds of the Silybum marianum (Fig. 3). Silymarin displays a wide range of bioactivities, including antioxidant, anti-inflammatory, anti-proliferative, and immunomodulatory effects [38]. Gu et al. reported that silymarin could generate benefits in insulin resistance, hyperlipidemia, and inflammation progress in Diet-induced obesity (DIO) mice model, and silybin A and B are major constituent of silymarin, showing higher activity [39]. Molecular docking showed that the 6-ECDCA binding site of FXR could be validated as the binding pocket for silybin, suggesting a structural basis for silybin activity. Altenusin is a nonsteroidal fungal metabolite isolated from Penicillium sp (Fig. 3) [40]. Zheng et al. used a GAL4-FXR-LBD chimeric receptor to screen for altenusin as a novel FXR agonist (EC50 = 3.4 ± 0.2 μM) [41]. Evaluation in the same luciferase reported assay showed that this compound has a potency and efficacy similar to those of CDCA (EC50 = 3.8 ± 0.3 μM). In vivo experiments demonstrated that administration of altenusin to high-fat diet (HFD)-induced obese mice produced metabolic benefits. Molecular docking analysis proved that altenusin can bind to FXR at the FXR ligand-binding domain (LBD), which is the binding site between CDCA and FXR. Artemisinin is a δ-sesquiterpene lactone endoperoxide isolated and identified from Artemisia annua L by more than 500 Chinese scientists [42]. Dihydroartemisinin (DHA) is a semisynthetic derivative of artemisinin, and it was identified as a FXR ligand (Fig. 3) [43]. DHA showed desirable therapeutic action on alcohol-induced liver injury, inflammation, and steatosis in rat via an FXR-dependent mechanism.
–
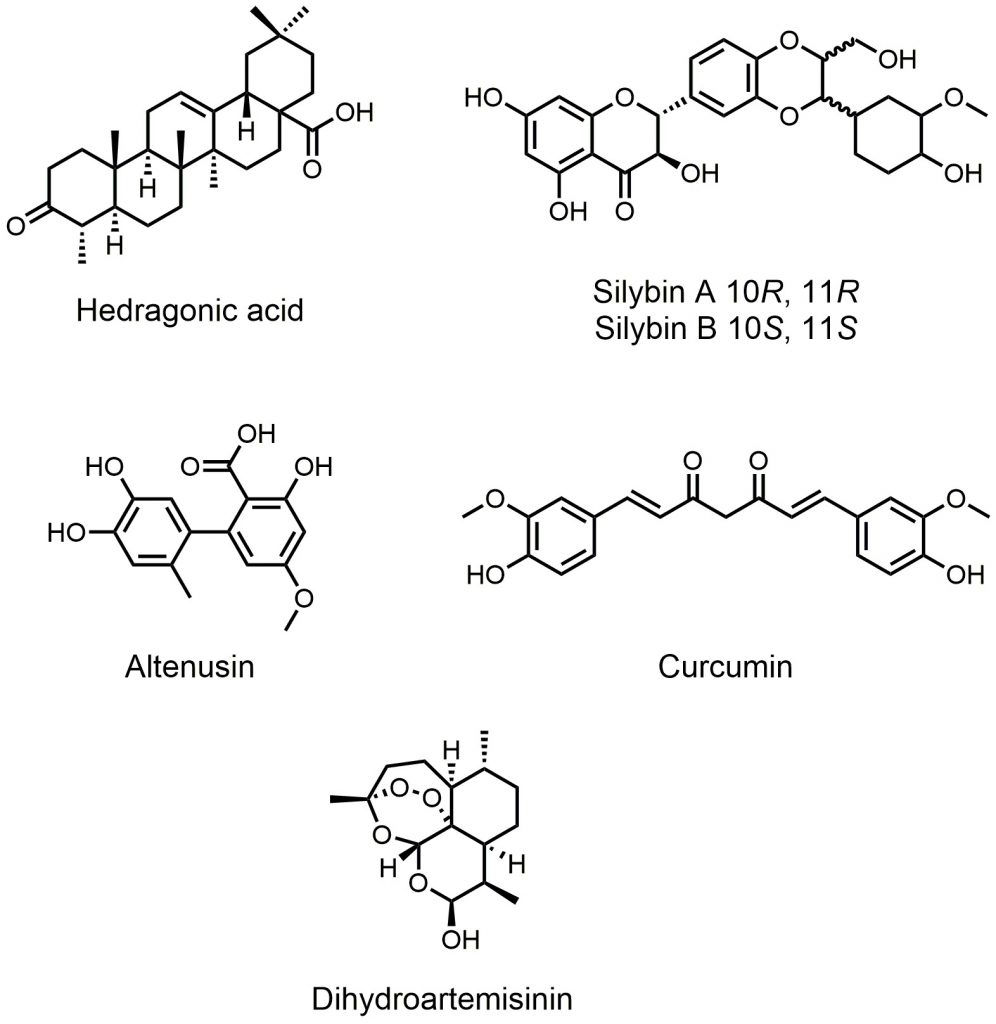 | FIGURE 3. Natural FXR agonists. |
–
Membrane G-protein-coupled Takeda G-protein-coupled receptor TGR5
TGR5, also called G-protein bile acid-activated receptor (GPBAR1), is widely expressed in many tissues, including liver, gallbladder, brown adipose tissue and the intestine [44]. In the intestine, TGR5 promotes the secretion of glucagon-like peptide 1 (GLP-1) and anorectic hormone peptide YY (PYY) by L-cells [45]. GLP-1 improves glucose metabolism by inducing glucose-dependent stimulation of insulin secretion, suppressing glucagon secretion, and stimulating the proliferation and differentiation of insulin-secreting beta cells. PYY, on the other hand, has been shown to reduce appetite, and is helpful for overweight and obese patients. TGR5 activation also increases energy expenditure in tissues like brown adipose tissue and human skeletal muscles [46]. TGR5 activation converts inactive thyroxine (T4) into the active iodothyronine (T3) form by triggering the thyroid hormone-activating enzyme type 2 iodothyronine deiodinase (D2). TGR5 also plays important roles in cell signaling [47]. When activated by bile acids with different affinities, TGR5 can upregulate levels of cyclic adenosine monophosphate (cAMP) by interacting with G protein αS (GαS). Several downstream signaling pathways could thus be stimulated, thereby performing a wide range of physiological functions. Chen et al. reported that when TGR5 was activated by oleanolic acid (OA), the expression of microRNA-26a (miR-26a) in macrophages was strongly increased [48]. MiR-26a can bring beneficial effects in regulating insulin sensitivity and metabolism of glucose and lipids, and is a potential target for the treatment of type 2 diabetes [49]. These results showed a miRNA-mediated mechanism of metabolic improvement through TGR5 activation. Ding et al. reported that the expression of TGR5 increased significantly after VSG in a mouse model [4]. With increased levels of bile acids caused by VSG, TGR5 signaling in the ileum and brown adipose tissues were significantly enhanced, showing improved glucose control and increased energy expenditure. It is thus possible for TGR5 agonism by small molecules to provide metabolic benefits to mimic bariatric surgery.
–
The endogenous natural agonists of TGR5 are bile acids, with lithocholic acid (LCA) and taurolithocholic acid (TLCA) being the most potent endogenous agonists [50]. The EC50 of LCA and TLCA are 0.53 μM and 0.33 μM, respectively. Pellicciari et al. found that 23(S)-methyl substitution on a CDCA side chain afforded TGR5 selectivity to bile acids [51]. According to the ideal pharmacokinetic properties and favorable metabolic profiles of cholic acid? (CA), they generated 6R-ethyl-23(S)methylcholic acid (INT-777) as a novel potent and selective TGR5 agonist with enhanced in vivo activity (EC50 = 0.82 μM) (Fig. 4). INT-777 efficiently increased GLP-1 secretion in vivo, and it could thus increase energy expenditure, reduce hepatic steatosis, and resulted in a significant reduction in weight gain and adiposity [52]. Kumar et al. recently demonstrated that administration of INT-777 to db/db mice improved pancreatic β-cell proliferation, insulin synthesis, and insulin release, showing its potential in treating diabetes mellitus and obesity [53]. Festa et al. further conducted detailed SAR studies, discovering the relationship between the ligand selectivity and the modification of the side chain and the cholane scaffold [54]. This study led to the identification of BAR501, an alcohol derivative of UDCA with potent and selective TGR5 activity (EC50 = 1.03 μM; Fig. 4). BAR501 could reverse insulin resistance, ameliorate liver histology, promote browning of epididymal white adipose tissue (epWAT), and increase energy expenditure in a rodent model of NASH [55]. Besides these semisynthetic bile acids, other natural products also demonstrate TGR5 agonist activity. OA is an active triterpene from Olea europaea and the first and best studied example of a natural TGR5 ligand (Fig. 4). It has an TGR5 agonist activity comparable with that of LCA [56]. Kumar et al. found that TGR5 activation in pancreatic β cells by OA could improve glucose tolerance and insulin release, highlighting the importance of targeting TGR5 in the control of glucose homeostasis [57]. Betulinic acid (BTA) is another identified active triterpene able to activate TGR5 (Fig. 4). It was found by screening a collection of natural triterpenoids [58]. In mice model fed a HFD, BTA increased insulin and leptin levels and decrease blood glucose and lipid levels [59]. However, whether the beneficial effects were related to TGR5 remains unknown. Structural modifications of the C-3 alcohol, the C-17 carboxylic acid, and the C-20 alkene on the BTA scaffold resulted in the finding of RG-239 (Fig. 4) [58]. RG-239 boasted a largely increased potency to activate TGR5 (EC50 = 0.12 μM) compared to that of BTA (EC50 = 1.04 μM) and OA (EC50 = 2.25 μM). Despite its high potency, however, RG-239 had a poor result tested for its antidiabetic properties in vivo. This result underlines the importance of suitable pharmacokinetic properties.
–
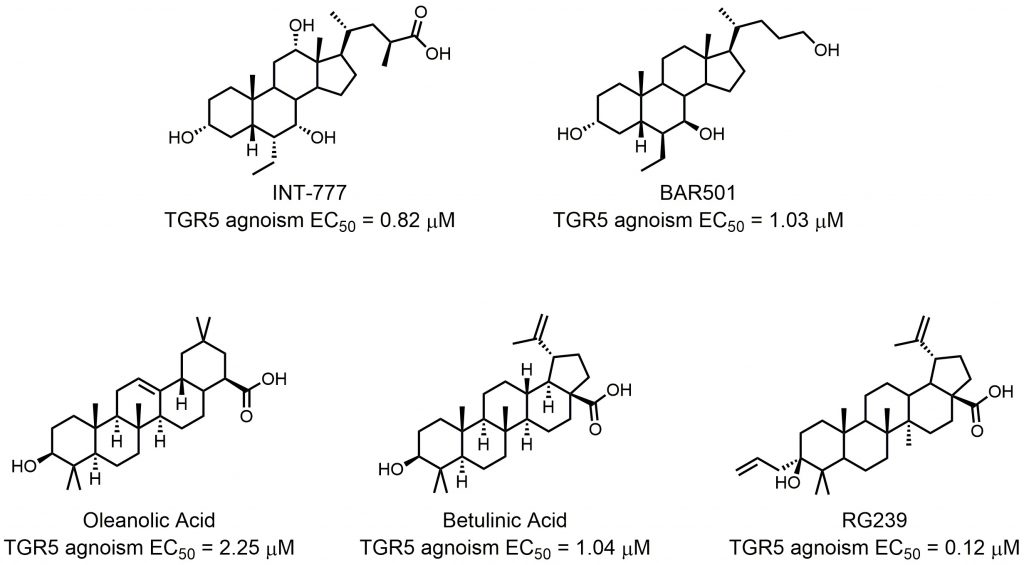 | FIGURE 4: Semisynthetic TGR5 agonists. |
–
In order to avoid potential side effects from the steroidal structure and to explore the structural diversity of TGR5 agonists, several examples of nonsteroidal compounds have been reported as TGR5 agonists by various pharmaceutical companies and research groups. Szewczyk et al. disclosed a series of bis-phenyl sulfonamide TGR5 agonists in 2007 [60]. Among those agonists, SB-756050 is a selective TGR5 agonist (EC50 = 1.3 μM), and its use in humans has been investigated and published (NCT00733577; Fig. 5) [61]. Although chronic enteral administration of SB-756050 in diabetic rats could increase plasma GLP-1 levels and improve glucose metabolism [60], the same effects could hardly be shown in humans because the pharmacodynamic effects of SB-756050 in humans were complex and inconsistent [61]. This result indicates that differences exist between rat and human TGR5 targeting. TRC210258 was found by Zambad et al. by screening ~400 compounds for their ability to activate TGR5 in vitro (Fig. 5) [62]. In diet-induced obesity (DIO) mice and hamsters, TGR5 activation by TRC210258 could reduce cardiovascular risks by improving metabolic parameters, such as GLP-1 secretion, energy expenditure, cholesterol and glucose metabolism.
–
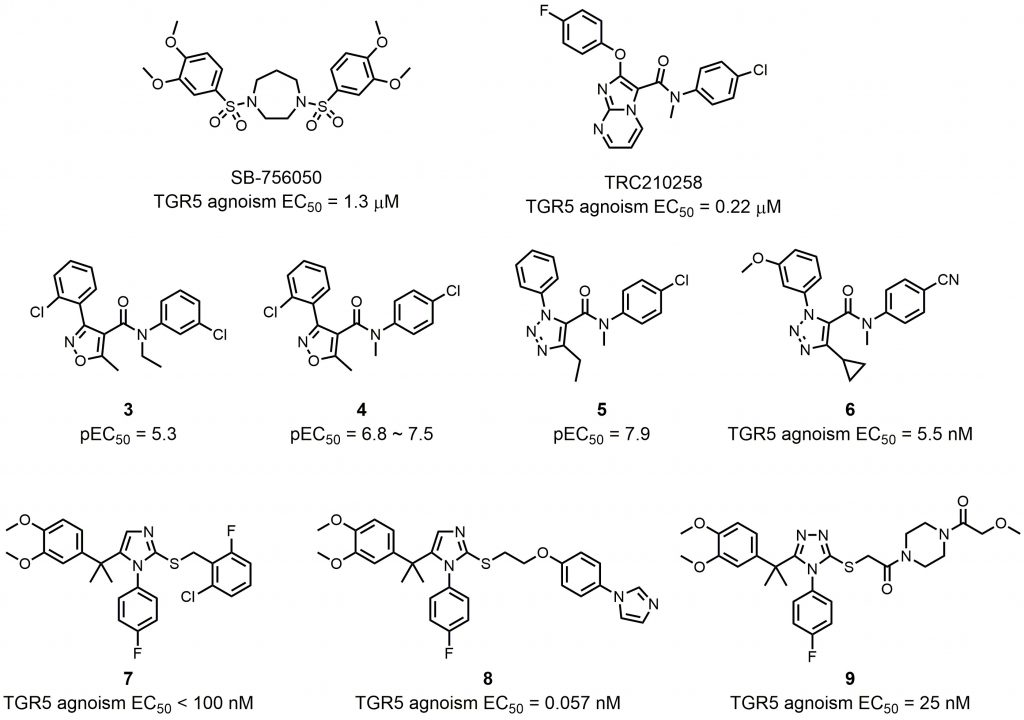 | FIGURE 5: Synthetic nonsteroidal TGR5 agonists. |
–
Some other compounds were identified and modified as chemical templates. Evans et al. employed a high-throughput screen using a BacMam transduced human osteosarcoma cell line (U2-OS) to identify isoxazole compound 3 as a TGR5 agonist with a pEC50 of 5.3 and 100% maximum response (Fig. 5) [63]. SAR optimization of the amide phenyl ring produced compound 4, showing improved potency in the U2-OS cell assay (pEC50 = 6.8) and in melanophore cells (pEC50 = 7.5) (Fig. 5). In a conscious dog model, coadministration of compound 4 and glucose afforded a significant improvement in portal vein GLP-1 secretion and glucose reduction, showing its potential to be a useful therapeutic for metabolic disorders. However, high intrinsic clearance in rat liver microsomes limited further application of compound 4. Budzik et al. found that replacement of the isoxazole with a 1,2,3-triazole could reduce intrinsic clearance, showing promising improvements in the in vitro metabolic stability profile [64]. The generated compound 5 maintain good in vitro potency (pEC50 = 7.9) (Fig. 5). Using compound 5 as starting point because of its favorable physicochemical properties (molecular weight = 341, elogD14 = 2.9, and topological polar surface area = 51) and conformational rigidity (number of rotatable bonds = 6), Futatsugi et al. further optimized triazole based compound 5 in order to identify orally available compounds by maximizing TGR5 potency and improving metabolic stability [65]. Although compound 6 was not the most potent TGR5 agonist in this literature, it showed the best balance of TGR5 activity and in vitro clearance (Fig. 5).
–
In 2010, Bollu et al. from Exelixis reported a series of triazole and imidazole TGR5 agonists [66]. Compound 7 showed receptor activation with EC50 values less than 100 nM in both the TGR5 cAMP assay and the TGR5/CRE-luciferase assay (Fig. 5). Agarwal et al. utilized Exelixis compound 7 as starting point for generating novel TGR5 agonists [67]. Molecular docking studies based on a TGR5 homology model showed that a linker with hydrogen bond acceptor and conformational flexibility could be beneficial for the interaction between small molecules and TGR5.
–
Further substitution on the terminal phenyl ring generated potent TGR5 agonist compound 8 (EC50 = 0.057 nM), which had outstanding TGR5 selectivity (Fig. 5). Besides potency, compound 8 also had better improved pharmacokinetic profile, rendering it an orally efficacious drug candidate. In order to expand the chemical pool with novel compounds for potential TGR5 agonist candidate, Agarwal et al. continued to investigate substituents on the right-hand side of the molecule, which were linked to the imidazole or triazole core with an amide linker [68]. Compound 9 with a triazole core had the highest hydrophilicity, which ensured compound 9 a low systematic exposure and minor side effects, while having an excellent TGR5 agonistic activity at the same time (Fig. 5). Molecular docking studies demonstrated that the tail piperazine ring and the fluoro-phenyl group of compound 9 could form hydrogen bonds and π-π stacking interactions with TGR5, explaining its high affinity to TGR5 (EC50 = 25 nM).
–
Activation of TGR5 has a wide range of bioactivities within different cells and tissues [47]. Systemic TGR5 agonists may trigger unwanted effects such as gallbladder swelling, itching, or cardiovascular issues [65][69][70]. For this reason, it was hypothesized that localized activation of TGR5 within the intestine could help minimize side effects during therapy. Different chemical modulation strategies have been utilized to produce intestinally-targeted TGR5 agonists. Lasalle et al. used the compound 8 derivate compound 10 as a chemical template to design and synthesize topical intestinal agonists (Fig. 6) [71]. Through combining a “pharmacophore” (compound 10) bearing the pharmacological activity and a “kinetophore” (sulfonate group) controlling the pharmacokinetic properties, compound 11 had a much-reduced cellular permeability, while its activation of TGR5 remained good, which was essential for its excellent potency (Fig. 6). The combination of other pharmacophores and kinetophores was also reported by different groups. Compound 12 is an orally efficacious TGR5 agonist based on 4-phenoxynicotinamide (EC50 = 0.72 nM) (Fig. 6) [72]. While exposure of compound 12 to the intestine caused the secretion of GLP-1, its exposure to other tissues such as gallbladder and heart still resulted in unwanted side effects. In order to reduce the passive molecular transporting through membranes, Duan et al. designed and synthesized (polyethylene glycol) PEG-containing derivatives of compound 12[73]. The PEG8 derivative compound 13 exhibited a permeability much lower than that of the small molecule TGR5 agonist 12 in Caco-2 permeability assays, and compound 13 showed satisfactory potency both in vitro (EC50 = 25 nM) and in vivo (Fig. 6). Compound 12 and its derivates underwent other chemical modulation. Bile acid sequestrants, as cholesterol-lowering polymer drugs, can bind to bile acids in the intestine. They are barely absorbed in the gut because of their high molecular weight and positive charge [74]. The quaternary ammonium structure plays an important role in the non-absorbed profile of bile acid sequestrants. Inspired by this, Cao et al. incorporated a quaternary ammonium structure with compound 12 derivates, in which the pyridine ring of 12 was replaced by a thiophene ring, to generate compound 14 (Fig. 6). Compound 14 was a potent TGR5 agonist (EC50 = 4.1 nM), and it was shown to be intestinally targeted through pharmacokinetic studies. Although the gallbladder filling effect of 14 was decreased in mice, however, this side effect was still not eliminated. Chen et al. tuned the performance of TGR5 agonists in the gastrointestinal lumen through systematic SAR optimization [75]. The optimized compound 15 had a similar pharmacophore as compound 12, expect for its thiazolidine structure, and the kinetophore was D-glucamide (Fig. 6). Compound 15 exhibited high potency as shown by in vitro TGR5 agonism (EC50 = 143 nM), and it did not inhibit gallbladder emptying. It could stimulate robust and sustained GLP-1 secretion with minimal TGR5-mediated suppression of gallbladder emptying, which rendered 15 an ideal therapeutic method for patients with metabolic diseases such as type 2 diabetes, NASH, or inflammatory bowel disease. Also, compound 15 could be an ideal tool for elucidating the physiological responses of TGR5 agonism without systemic target engagement.
–
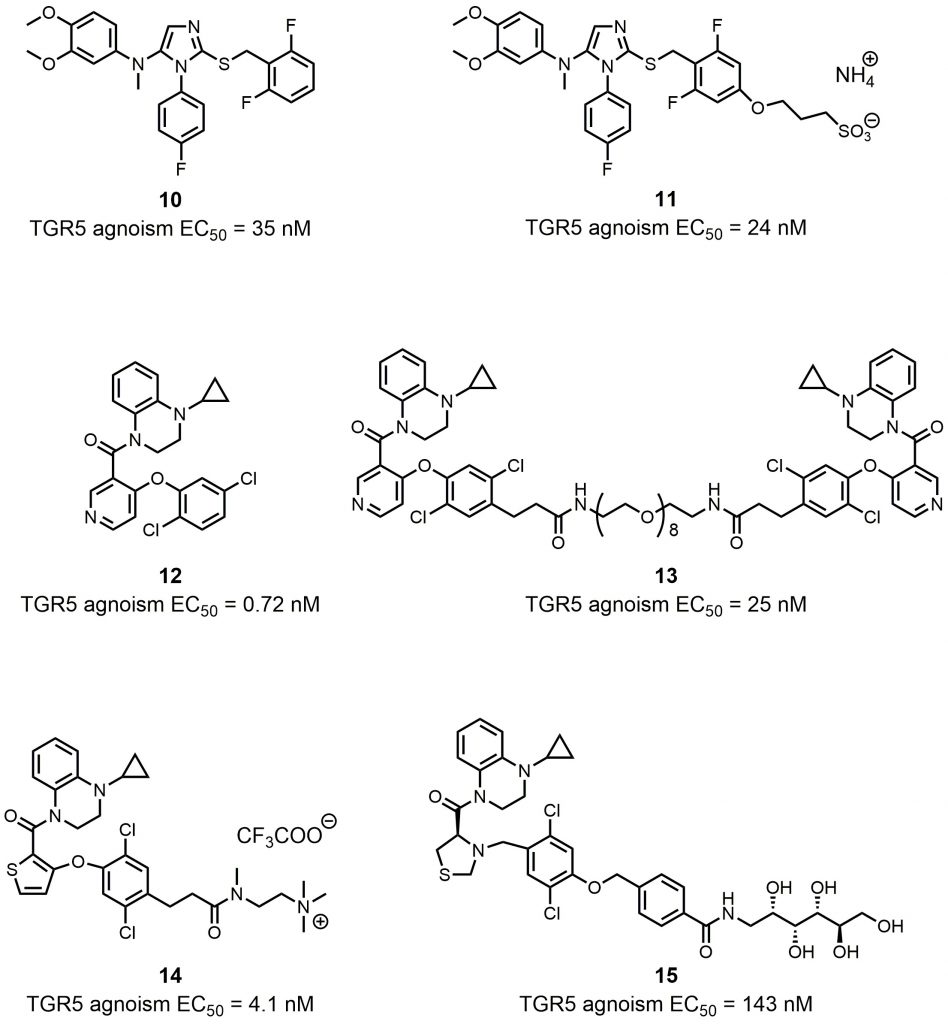 | FIGURE 6: Synthetic intestinally targeted TGR5 agonists. |
–
FXR and TGR5 dual targeting agonists and other dual targeting compounds
Although many efforts have been made to develop selective FXR or TGR agonists with reduced side effects, research shows that dual agonism of FXR and TGR5 is extremely useful in the treatment of diabetes mellitus, obesity [76], NASH [55][77], atherosclerosis [78], and even other diseases like bone loss [79]. These results demonstrate the potential of FXR/TGR5 dual-targeting agonists as a promising therapy.
–
Researchers found that chemical modulation of the side chain of steroidal cores? can change the selectivity of semisynthetic bile acid derivatives. Through introducing a sulfate group at the C-23 position of the 6α-ethylcholane scaffold, Rizzo et al. discovered INT-767, which was not only a very potent FXR agonist but also a potent activator of TGR5 (FXR agonism EC50 = 0.03 μM; TGR5 agonism EC50 = 0.47 μM; Fig. 7) [80]. INT-767 showed low cytotoxic effects and was highly stable to phase I and II enzymatic modifications, suggesting its potential clinical application in the treatment of liver and metabolic diseases as a safe and effective dual-targeting modulator. Currently, INT-767 is the most advanced compound in preclinical trials. BAR502 is another advanced compound that acts as a dual-targeting modulator of FXR/TGR5 (Fig. 7). In their systematic SAR studies, Festa et al. discovered that a hydroxyl group on the C-23 position of the 6α-ethylcholane scaffold generated dual-targeting agonist BAR502 (FXR agonism EC50 = 2 μM; TGR5 agonism EC50 = 0.4 μM) [54]. Although BAR502 was a preferential FXR ligand, its potency to TGR5 agonism was satisfactory as a dual-targeting modulator. Cipriani et al. demonstrated that BAR502 could attenuate liver damage in murine models of cholestasis without inducing itching, suggesting agonisms of both receptors [81]. As mentioned before, active triterpene, like Hedragonic acid and BTA, are another kind of molecules being able to activate FXR and TGR5, so it is possible that FXR/TGR5 dual-targeting agonists can be triterpene. Li et al. screened 35 BTA derivatives using TGR5-dependent cAMP accumulation and FXR response element reporter gene expression [79]. They found that SH-479, which was first synthesized by Xu et al.[82], was a potent TGR5 and FXR dual agonist (Fig. 7). SH-479 abrogated bone loss in a C57BL/6J mouse model through bone remodeling pathways, showing its potential as a therapeutic strategy for osteoporosis.
–
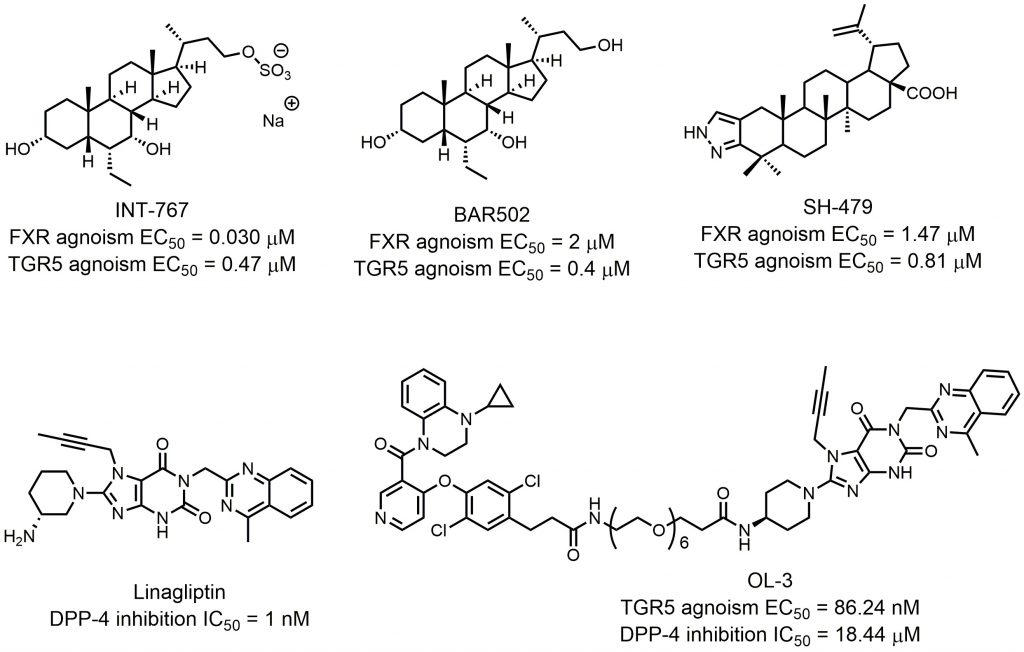 | FIGURE 7: FXR and TGR5 dual targeting agonists and other dual targeting compounds. |
–
Dual modulators for other targets were also developed to treat metabolic diseases. Inspired by the molecular design of compound 16, Ma et al. designed and synthesized OL-3, a novel low-absorbed TGR5 agonist (EC50 = 86.24 nM) with dipeptidyl peptidase-4 (DPP-4) inhibitory activity (IC50 = 18.44 μM; Fig. 7) [83]. Circulating GLP-1 has a short half-life due to the presence of DPP-4, so DPP-4 inhibitors can prevent the inactivation process of GLP-1, thus improving the level of active GLP-1 [84]. Linagliptin is one of the most selective and potent inhibitors of DPP-4 approved by the FDA in 2011 (IC50 = 1 nM; Fig. 7). OL-3 was generated by linking the structure of compound 15 and linagliptin with the PEG6 linker. It showed low cell permeability in Caco-2 cells and low absorption in vivo. Oral administration of OL-3 significantly lowered blood glucose levels but did not cause gallbladder filling, suggesting its high safety when being used.
BILE ACID COMPOSITION AND CYP8B1
Bile acids are the general name of several steroid acids. In humans, the bile acid pool consists of CA, CDCA, Deoxycholic acid (DCA), LCA, and glycine (G)- or taurine (T)-conjugated bile acids [15]. Conversion of cholesterol to bile acids in the liver is the major pathway for catabolism of cholesterol (Fig. 8) [85]. Among those bile acids, CA and CDCA are synthesized in the liver, and are called primary bile acids. CA is more hydrophilic than CDCA, which means that CA can cause higher lipid absorption in the intestine [86]. Furthermore, CA and CDCA can act differently as hormones [87]. As a result, the ratio of CA and CDCA plays an important role in fat absorption and homeostasis. Sterol 12a-hydroxylase (CYP8B1) is the key enzyme for CA synthesis. Research indicates that inhibition of CYP8B1 in mice improves metabolism. Slatis et al. demonstrated that knocking out Cyp8b1 in an ApoE knock out mouse model reduced atherosclerotic plaques [88]. Kaur et al. showed that glucose homeostasis in Cyp8b1-/- mice could be improved with increasing GLP-1 [89]. Chevre et al. reported that in a cholesterol-induced NAFLD mouse model, inhibition of Cyp8b1 expression led to a regression of hepatic steatosis [90]. These studies suggest that human CYP8B1 is a viable therapeutic target. Using a HFD-induced obese C57Bl/6 mouse model, Myronovych et al. found that Cyp8b1 expression was downregulated after VSG [11]. Although VSG mice and sham operation mice achieved weight loss, the VSG mice had a significantly lower hepatic triglyceride content, which suggests that the improved metabolic outcomes were caused by more than just the early weight lost post-surgery. Developing CYP8B1 inhibitors is a promising way for treating glucose and lipid metabolic diseases at a lower cost and with reduced risk.
–
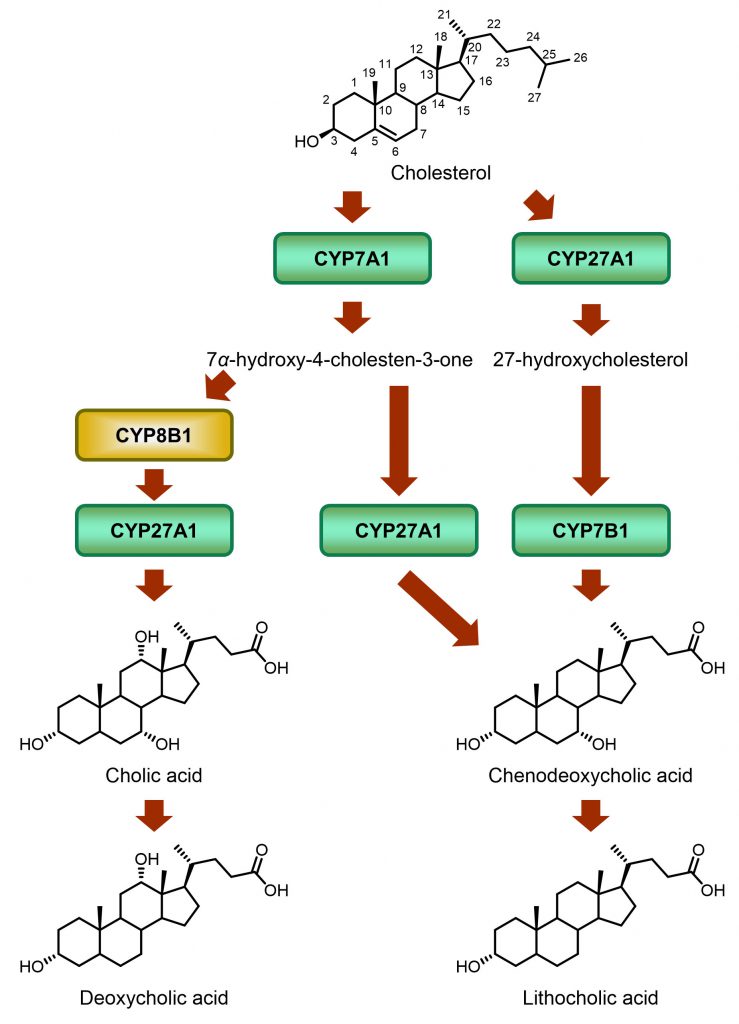 | FIGURE 8: Biosynthesis of bile acids. Bile acids are synthesized in liver through two pathways: the classic pathway and the alternative pathway. CYP7A1 initializes the classic pathway, while CYP27A1 initializes the alternative pathway. CYP8B1 regulates the CA synthesis in the classic pathway, and is the only enzyme catalyzing the CA synthesis. In the intestine, primary bile acid CA and CDCA are metabolized by the gut microbiota to produce the secondary bile acids, DCA and LCA, respectively. |
–
Unlike in humans, the majority of CDCA in mice is typically converted to muricholic acid (MCA) by CYP2c70 which exists in rodents but not humans [13][91][92][93][94]. Therefore, in addition to the slump of CA level, the Cyp8b1 knock out also results in a remarkable increase of MCA levels in mice [95]. Accordingly, the bile acid composition in either wild type or Cyp8b1 knock out mice differs substantially from that in humans. However, both CDCA and MCA belong to non-12α-OH bile acids. Moreover, although MCA is a hydrophilic 6-hydroxylated bile acid, whereas CDCA is more hydrophobic, both have been reported to significantly lower cholesterol absorption as compared to CA [96]. Thus, despite those differences in bile acid composition, lipid absorption may be similarly affected in humans and mice upon CYP8B1 downregulation due to comparable changes in the 12α-OH/non-12α-OH BA ratio.
–
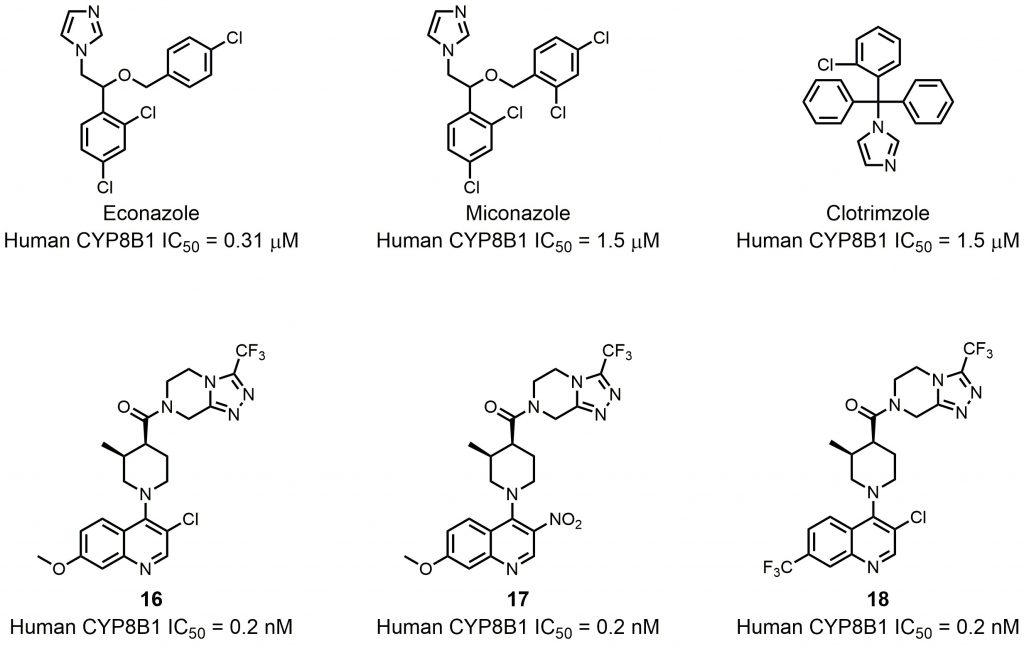 | FIGURE 9: CYP8B1 inhibitors. |
–
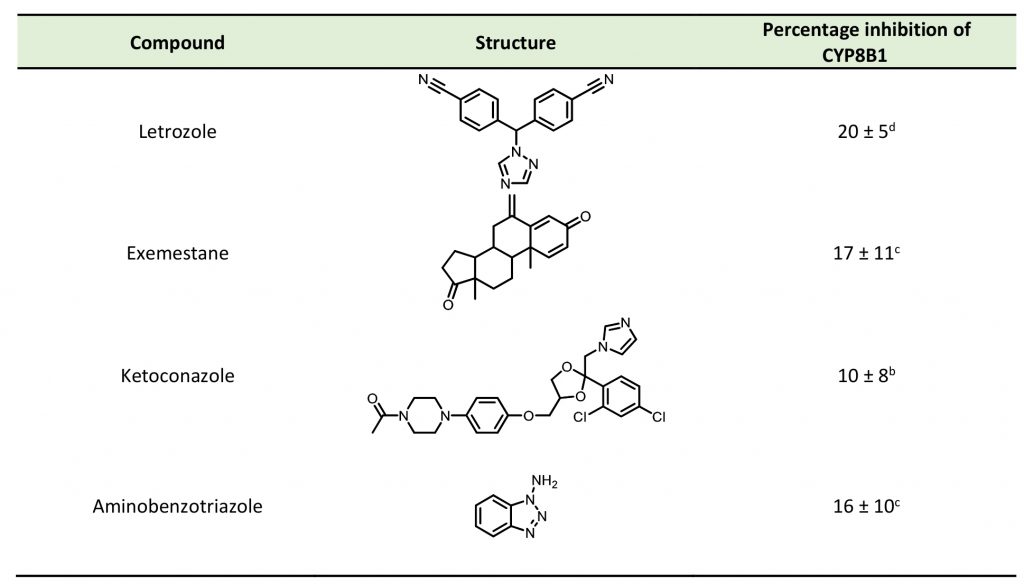
To date, several small molecule inhibitors of CYP8B1 have been reported, which laid a foundation for future drug design. Franchini et al. reported several agents showing CYP8B1 inhibitory activity [97]. Among them, econazole, clotrimazole and miconazole are potent inhibitors exhibiting IC50< 2 μM (Fig. 9). Caldwell et al. reported a series of compounds showing strong CYP8B1 inhibitory activity, with all compounds having a similar core structure (Fig. 9) [98]. The most potent compound 16, 17 and 18 exhibited IC50 = 0.2 nM. Fan et al. established a rapid and convenient CYP8B1 inhibitor test system, and tested six inhibitors of other CYPs using this system [99]. They successfully found ketoconazole, letrozole, exemestane and aminobenzotriazole as potential CYP8B1 inhibitors, which inhibited more than 10% of CYP8B1 at concentrations lower than 10 μM (Table 1). All these inhibitors have not been tested for their selectivity for CYP8B1. Developing inhibitors with high potency and selectivity is a highly challenging work. These compounds might serve as a starting point for further medicinal chemistry work.
–
TABLE 1. Effect of inhibitors on CYP8B1 activity. |
 |
GUT MICROBIOME AND BILE SALT HYDROLASE
Intestinal microbiota disorders can lead to a variety of diseases, such as obesity and colon cancer, implicating the importance of microbes in the human gut in maintaining healthy metabolism [100]. The influence on metabolism brought by intestinal microbiota can be explained by the bidirectional relationship between the bile acids and the gut microbial populations [101]. Different therapeutic strategies have been developed based on the understanding of this relationship.
–
Bile acids synthesized in the liver can regulate intestinal microbiota through a direct or indirect effect. Conjugated bile acids directly alter bacterial membrane architecture and damage DNA, which inhibit growth of bacteria [102][103]. Bile acids can also indirectly affect microbiota via nuclear receptors. Inagaki et al. showed that FXR activation by bile acids can induce the expression of genes that inhibit microbial overgrowth and mucosal damage [104]. These results are consistent with the idea that bile acids are integral in controlling intestinal microbiota, and therefore has significant implications for maintaining equilibrium between human host and intestinal microbiota. According to a systematic study that profiled gut microbiota-related metabolites, Liu et al. found higher serum glutamate concentrations and reduced abundance of Bacteroides thetaiotaomicron in overweight and obese Chinese individuals [105]. Following weight-loss intervention by VSG, analysis of these metabolomes in 23 obese individuals showed a return to baseline levels of both B. thetaiotaomicron and the serum glutamate concentrations. Moreover, gavage of live B. thetaiotaomicron in mice decreased levels of glutamate concentration and alleviated the diet-induced body-weight gain. The alterations in the population of B. thetaiotaomicron in obese individuals after bariatric surgery were possibly caused by the modifications of the bile acid pool. This result highlights the potential for obesity interventions by targeting the gut microbiota.
–
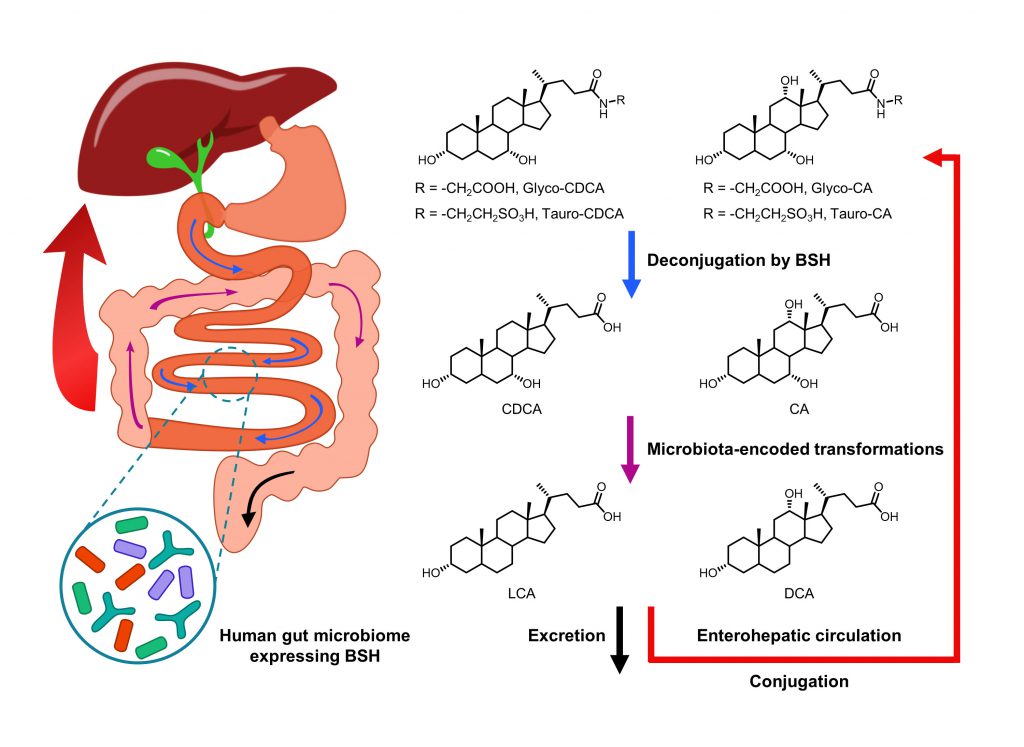 | FIGURE 10: Biotransformation of bile acids by BSH-containing gut microbiota. |
–
On the other hand, intestinal microbiota can change the size and composition of the bile acid pool via biotransformation (Fig. 10). Bile salt hydrolase (BSH) is an enzyme catalyzing the hydrolysis of glycine- and/or taurine-conjugated bile acids into unconjugated bile acids and amino acid residues (Fig. 11) [106]. Conjugated bile acids consist of a hydrophobic steroid core and a hydrophilic amino acid side chain (glycine or taurine). The hydrophilicity of amino acid chains render conjugated bile acids to be better biological detergents than unconjugated bile acid because of their ability to emulsify dietary lipids for fat digestion in the small intestine [107]. For this reason, deconjugation of bile acids by BSHs may induce weight loss in the setting of malabsorption [108]. Inversely, intestinal BSH inhibition may be helpful for energy harvest. In fact, many efforts have been made to develop BSH inhibitors as novel non-antibiotic growth promoters to enhance animal production and health [109]. It is highly possible that small molecule BSH agonists can decrease weight gain in obese patients through the mechanism of malabsorption. However, no compound has been reported that can mediate such an effect. Fortunately, prebiotics or probiotics have been reported to improve glucose and lipid metabolism [110].
–
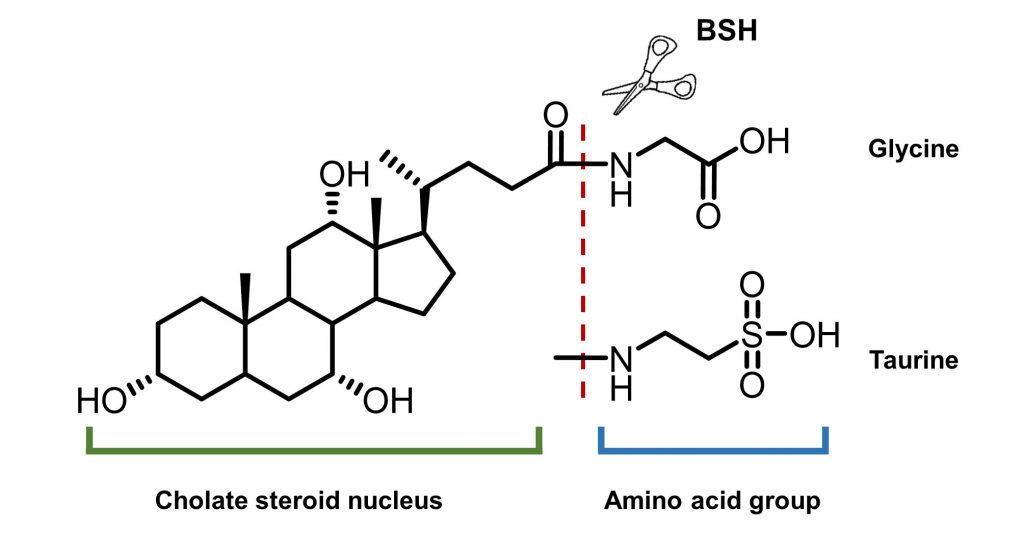 | FIGURE 11: General structures of bile salts and the functions of BSHs. |
–
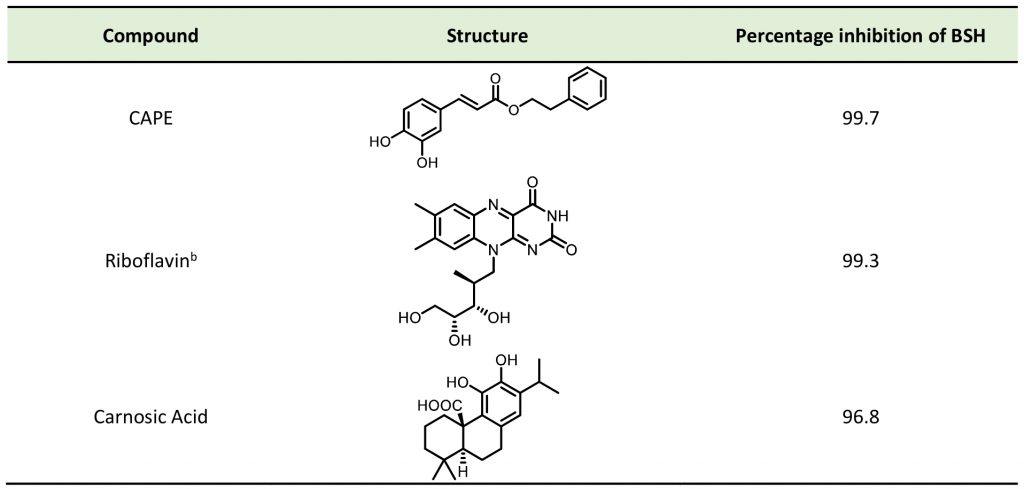
The detergent nature of bile acids creates reason to believe that inhibition of BSH can reduce host weight gain. However, different bile acids can act differently in human bodies as hormones. Under this condition, the relationship between BSH activity and metabolism of the host might change. Yao et al. reported that in gnotobiotic mice mono-colonized with wild type and BSH-deleted B. thetaiotaomicron, the BSH-deleted mice demonstrated significant reduction in weight gain [111]. In mice, the two most abundant murine-conjugated bile acids found were tauro-β-muricholic acid (TβMCA) and tauro-cholic acid (TCA). BSH from B. thetaiotaomicron was able to selectively deconjugate TβMCA, but not TCA. This result demonstrated that inhibition of selective BSHs might benefit the host metabolism. For this reason, development of BSH inhibitors is underway. Wang et al. found that copper (CuCl2) and zinc (ZnSO4) could be potent BSH inhibitors, and they were able to promote food digestion and body weight gain in different animal models [112]. Smith et al. identified three BSH inhibitors (CAPE, Riboflavin and Carnosic Acid) using a high-throughput screening method with a recombinant BSH, which contains broad substrate specificity from a chicken Lactobacillus salivarius, the dominant lactic acid bacterium present in the chicken intestine (Table 2) [113]. However, these inhibitors showed either no inhibitory or moderate inhibitory activity to other BSHs like B. thetaiotaomicron and Bifidobacterium longum BSHs, which largely inhibit their clinical use in humans [114]. Although BSH protein sequences vary largely between different gut strains, all BSHs possess a conserved active site that includes a catalytic cysteine (Cys2) [115]. Based on this property, Adhikari et al. developed a covalent inhibitor, compound 19 (Fig. 12), targeting both B. thetaiotaomicron and B. longum BSHs in a dose-dependent fashion (IC50 = 108-427 nM) [114]. Compound 19 could effectively inhibit deconjugation of bile salts in vitro and in vivo, but did not significantly affect the viability of gut bacteria. This research is meaningful for the investigation of how BSH activity directly affects metabolism in fully colonized hosts, which is more likely to mimic the real situation. Other pan-inhibitors and selective inhibitors of BSHs, which might beneficially affect host physiology, could be developed based on this work.
–
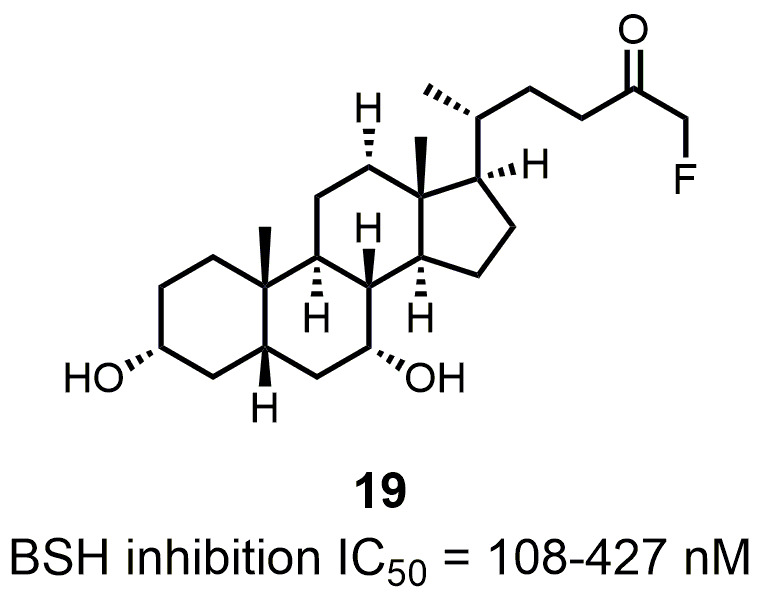 | FIGURE 12: Structure and activity of the covalent pan-inhibitor of BSHs. |
PERSPECTIVES
Among the bile acid related targets discussed above, FXR and TGR5 lead the way in the drug development pipeline. Since FXR and TGR5 may play different roles in different metabolic diseases, it is possible to design small molecules with various potency and selectivity depending on patient profiles. The metabolic role of CYP8B1 has been recently explored; thus, CYP8B1 inhibition may offer a promising therapeutic avenue for treating metabolic diseases. Currently, a few small molecules have been reported to have CYP8B1 inhibitory activity. However, they suffer from low potency and selectivity. More medicinal chemistry studies are required for the development of new CYP8B1 inhibitors. The role of BSHs in metabolism is more dynamic and complex; it is not clear whether BSH agonism or inhibition can be beneficial to the host metabolism. Further studies are warranted to better understand the role of BSHs in regulating metabolism to create an opportunity for drug discovery. On the other hand, through the use of prebiotics, probiotics, and gene editing technologies, more studies can be conducted to improve the overall understanding of BSHs as drug targets.
–
TABLE 2. Effect of inhibitors on BSH activity |
 |
REFERENCES
- Hagan KE, Alasmar A, Exum A, Chinn B, Forbush KT (2020). A systematic review and meta-analysis of attentional bias toward food in individuals with overweight and obesity. Appetite 151: 104710. 10.1016/j.appet.2020.104710
- Pareek M, Schauer PR, Kaplan LM, Leiter LA, Rubino F, Bhatt DL (2018). Metabolic Surgery: Weight Loss, Diabetes, and Beyond. J Am Coll Cardiol 71(6): 670-687. 10.1016/j.jacc.2017.12.014
- Bjorklund G, Semenova Y, Pivina L, Costea DO (2020). Follow-up after bariatric surgery: A review. Nutrition 78: 110831. 10.1016/j.nut.2020.110831
- Ding L, Sousa KM, Jin L, Dong B, Kim BW, Ramirez R, Xiao Z, Gu Y, Yang Q, Wang J, Yu D, Pigazzi A, Schones D, Yang L, Moore D, Wang Z, Huang W (2016). Vertical sleeve gastrectomy activates GPBAR-1/TGR5 to sustain weight loss, improve fatty liver, and remit insulin resistance in mice. Hepatology 64(3): 760-773. 10.1002/hep.28689
- Stefater MA, Wilson-Perez HE, Chambers AP, Sandoval DA, Seeley RJ (2012). All bariatric surgeries are not created equal: insights from mechanistic comparisons. Endocr Rev 33(4): 595-622. 10.1210/er.2011-1044
- Carlin AM, Zeni TM, English WJ, Hawasli AA, Genaw JA, Krause KR, Schram JL, Kole KL, Finks JF, Birkmeyer JD, Share D, Birkmeyer NJ, Michigan Bariatric Surgery C (2013). The comparative effectiveness of sleeve gastrectomy, gastric bypass, and adjustable gastric banding procedures for the treatment of morbid obesity. Ann Surg 257(5): 791-797. 10.1097/SLA.0b013e3182879ded
- Miras AD, le Roux CW (2013). Mechanisms underlying weight loss after bariatric surgery. Nat Rev Gastroenterol Hepatol 10(10): 575-584. 10.1038/nrgastro.2013.119
- Ryan KK, Tremaroli V, Clemmensen C, Kovatcheva-Datchary P, Myronovych A, Karns R, Wilson-Perez HE, Sandoval DA, Kohli R, Backhed F, Seeley RJ (2014). FXR is a molecular target for the effects of vertical sleeve gastrectomy. Nature 509(7499): 183-188. 10.1038/nature13135
- Li K, Zou J, Li S, Guo J, Shi W, Wang B, Han X, Zhang H, Zhang P, Miao Z, Li Y (2020). Farnesoid X receptor contributes to body weight-independent improvements in glycemic control after Roux-en-Y gastric bypass surgery in diet-induced obese mice. Mol Metab 37: 100980. 10.1016/j.molmet.2020.100980
- Steinert RE, Peterli R, Keller S, Meyer-Gerspach AC, Drewe J, Peters T, Beglinger C (2013). Bile acids and gut peptide secretion after bariatric surgery: a 1-year prospective randomized pilot trial. Obesity 21(12): E660-668. 10.1002/oby.20522
- Myronovych A, Kirby M, Ryan KK, Zhang W, Jha P, Setchell KD, Dexheimer PJ, Aronow B, Seeley RJ, Kohli R (2014). Vertical sleeve gastrectomy reduces hepatic steatosis while increasing serum bile acids in a weight-loss-independent manner. Obesity 22(2): 390-400. 10.1002/oby.20548
- Chiang JY (2009). Bile acids: regulation of synthesis. J Lipid Res 50(10): 1955-1966. 10.1194/jlr.R900010-JLR200
- Chiang JY (2013). Bile acid metabolism and signaling. Compr Physiol 3(3): 1191-1212. 10.1002/cphy.c120023
- Ridlon JM, Kang DJ, Hylemon PB (2006). Bile salt biotransformations by human intestinal bacteria. J Lipid Res 47(2): 241-259. 10.1194/jlr.R500013-JLR200
- Chiang JYL, Ferrell JM (2018). Bile Acid Metabolism in Liver Pathobiology. Gene Expr 18(2): 71-87. 10.3727/105221618X15156018385515
- Abrigo J, Gonzalez F, Aguirre F, Tacchi F, Gonzalez A, Meza MP, Simon F, Cabrera D, Arrese M, Karpen S, Cabello-Verrugio C (2020). Cholic acid and deoxycholic acid induce skeletal muscle atrophy through a mechanism dependent on TGR5 receptor. J Cell Physiol 1-13. 10.1002/jcp.29839
- Goldstein J, Levy C (2018). Novel and emerging therapies for cholestatic liver diseases. Liver Int 38(9): 1520-1535. 10.1111/liv.13880
- Teodoro JS, Rolo AP, Palmeira CM (2011). Hepatic FXR: key regulator of whole-body energy metabolism. Trends Endocrinol Metab 22(11): 458-466. 10.1016/j.tem.2011.07.002
- Han CY (2018). Update on FXR Biology: Promising Therapeutic Target? Int J Mol Sci 19(7): 2069. 10.3390/ijms19072069
- Lew JL, Zhao A, Yu J, Huang L, De Pedro N, Pelaez F, Wright SD, Cui J (2004). The farnesoid X receptor controls gene expression in a ligand- and promoter-selective fashion. J Biol Chem 279(10): 8856-8861. 10.1074/jbc.M306422200
- Pellicciari R, Fiorucci S, Camaioni E, Clerici C, Costantino G, Maloney PR, Morelli A, Parks DJ, Willson TM (2002). 6alpha-ethyl-chenodeoxycholic acid (6-ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity. J Med Chem 45(17): 3569-3572. 10.1021/jm025529g
- Sayiner M, Golabi P, Stepanova M, Younossi I, Nader F, Racila A, Younossi ZM (2019). Primary Biliary Cholangitis in Medicare Population: The Impact on Mortality and Resource Use. Hepatology 69(1): 237-244. 10.1002/hep.30174
- Pellicciari R, Passeri D, De Franco F, Mostarda S, Filipponi P, Colliva C, Gadaleta RM, Franco P, Carotti A, Macchiarulo A, Roda A, Moschetta A, Gioiello A (2016). Discovery of 3alpha,7alpha,11beta-Trihydroxy-6alpha-ethyl-5beta-cholan-24-oic Acid (TC-100), a Novel Bile Acid as Potent and Highly Selective FXR Agonist for Enterohepatic Disorders. J Med Chem 59(19): 9201-9214. 10.1021/acs.jmedchem.6b01126
- Sepe V, Festa C, Renga B, Carino A, Cipriani S, Finamore C, Masullo D, Del Gaudio F, Monti MC, Fiorucci S, Zampella A (2016). Insights on FXR selective modulation. Speculation on bile acid chemical space in the discovery of potent and selective agonists. Sci Rep 6: 19008. 10.1038/srep19008
- Xiao H, Li P, Li X, He H, Wang J, Guo F, Zhang J, Wei L, Zhang H, Shi Y, Hou L, Shen L, Chen Z, Du C, Fu S, Zhang P, Hao F, Wang P, Xu D, Liang W, Tian X, Zhang A, Cheng X, Yang L, Wang X, Zhang X, Li J, Chen S (2017). Synthesis and Biological Evaluation of a Series of Bile Acid Derivatives as FXR Agonists for Treatment of NASH. ACS Med Chem Lett 8(12): 1246-1251. 10.1021/acsmedchemlett.7b00318
- Verbeke L, Nevens F, Laleman W (2017). Steroidal or non-steroidal FXR agonists – Is that the question? J Hepatol 66(4): 680-681. 10.1016/j.jhep.2017.01.013
- Maloney PR, Parks DJ, Haffner CD, Fivush AM, Chandra G, Plunket KD, Creech KL, Moore LB, Wilson JG, Lewis MC, Jones SA, Willson TM (2000). Identification of a chemical tool for the orphan nuclear receptor FXR. J Med Chem 43(16): 2971-2974. 10.1021/jm0002127
- Akwabi-Ameyaw A, Bass JY, Caldwell RD, Caravella JA, Chen L, Creech KL, Deaton DN, Jones SA, Kaldor I, Liu Y, Madauss KP, Marr HB, McFadyen RB, Miller AB, Iii FN, Parks DJ, Spearing PK, Todd D, Williams SP, Wisely GB (2008). Conformationally constrained farnesoid X receptor (FXR) agonists: Naphthoic acid-based analogs of GW 4064. Bioorg Med Chem Lett 18(15): 4339-4343. https://doi.org/10.1016/j.bmcl.2008.06.07
- Kinzel O, Steeneck C, Schluter T, Schulz A, Gege C, Hahn U, Hambruch E, Hornberger M, Spalwisz A, Frick K, Perovic-Ottstadt S, Deuschle U, Burnet M, Kremoser C (2016). Novel substituted isoxazole FXR agonists with cyclopropyl, hydroxycyclobutyl and hydroxyazetidinyl linkers: Understanding and improving key determinants of pharmacological properties. Bioorg Med Chem Lett 26(15): 3746-3753. 10.1016/j.bmcl.2016.05.070
- Schwabl P, Hambruch E, Seeland BA, Hayden H, Wagner M, Garnys L, Strobel B, Schubert TL, Riedl F, Mitteregger D, Burnet M, Starlinger P, Oberhuber G, Deuschle U, Rohr-Udilova N, Podesser BK, Peck-Radosavljevic M, Reiberger T, Kremoser C, Trauner M (2017). The FXR agonist PX20606 ameliorates portal hypertension by targeting vascular remodelling and sinusoidal dysfunction. J Hepatol 66(4): 724-733. 10.1016/j.jhep.2016.12.005
- Tully DC, Rucker PV, Chianelli D, Williams J, Vidal A, Alper PB, Mutnick D, Bursulaya B, Schmeits J, Wu X, Bao D, Zoll J, Kim Y, Groessl T, McNamara P, Seidel HM, Molteni V, Liu B, Phimister A, Joseph SB, Laffitte B (2017). Discovery of Tropifexor (LJN452), a Highly Potent Non-bile Acid FXR Agonist for the Treatment of Cholestatic Liver Diseases and Nonalcoholic Steatohepatitis (NASH). J Med Chem 60(24): 9960-9973. 10.1021/acs.jmedchem.7b00907
- Downes M, Verdecia MA, Roecker AJ, Hughes R, Hogenesch JB, Kast-Woelbern HR, Bowman ME, Ferrer J-L, Anisfeld AM, Edwards PA, Rosenfeld JM, Alvarez JGA, Noel JP, Nicolaou KC, Evans RM (2003). A Chemical, Genetic, and Structural Analysis of the Nuclear Bile Acid Receptor FXR. Mol Cell 11(4): 1079-1092. 10.1016/S1097-2765(03)00104-7
- Fang S, Suh JM, Reilly SM, Yu E, Osborn O, Lackey D, Yoshihara E, Perino A, Jacinto S, Lukasheva Y, Atkins AR, Khvat A, Schnabl B, Yu RT, Brenner DA, Coulter S, Liddle C, Schoonjans K, Olefsky JM, Saltiel AR, Downes M, Evans RM (2015). Intestinal FXR agonism promotes adipose tissue browning and reduces obesity and insulin resistance. Nat Med 21(2): 159-165. 10.1038/nm.3760
- Wang H, Zhao Z, Zhou J, Guo Y, Wang G, Hao H, Xu X (2017). A novel intestinal-restricted FXR agonist. Bioorg Med Chem Lett 27(15): 3386-3390. 10.1016/j.bmcl.2017.06.003
- Lu Y, Zheng W, Lin S, Guo F, Zhu Y, Wei Y, Liu X, Jin S, Jin L, Li Y (2018). Identification of an Oleanane-Type Triterpene Hedragonic Acid as a Novel Farnesoid X Receptor Ligand with Liver Protective Effects and Anti-inflammatory Activity. Mol Pharmacol 93(2): 63-72. 10.1124/mol.117.109900
- Zhu C, Zuo Y, Wang R, Liang B, Yue X, Wen G, Shang N, Huang L, Chen Y, Du J, Bu X (2014). Discovery of potent cytotoxic ortho-aryl chalcones as new scaffold targeting tubulin and mitosis with affinity-based fluorescence. J Med Chem 57(15): 6364-6382. 10.1021/jm500024v
- Yang F, Tang X, Ding L, Zhou Y, Yang Q, Gong J, Wang G, Wang Z, Yang L (2016). Curcumin protects ANIT-induced cholestasis through signaling pathway of FXR-regulated bile acid and inflammation. Sci Rep 6: 33052. 10.1038/srep33052
- Polyak SJ, Ferenci P, Pawlotsky JM (2013). Hepatoprotective and antiviral functions of silymarin components in hepatitis C virus infection. Hepatology 57(3): 1262-1271. 10.1002/hep.26179
- Gu M, Zhao P, Huang J, Zhao Y, Wang Y, Li Y, Li Y, Fan S, Ma YM, Tong Q, Yang L, Ji G, Huang C (2016). Silymarin Ameliorates Metabolic Dysfunction Associated with Diet-Induced Obesity via Activation of Farnesyl X Receptor. Front Pharmacol 7: 345. 10.3389/fphar.2016.00345
- Nakanishi S, Toki S, Saitoh Y, Tsukuda E, Kawahara K, Ando K, Matsuda Y (1995). Isolation of myosin light chain kinase inhibitors from microorganisms: dehydroaltenusin, altenusin, atrovenetinone, and cyclooctasulfur. Biosci Biotechnol Biochem 59(7): 1333-1335. 10.1271/bbb.59.1333
- Zheng Z, Zhao Z, Li S, Lu X, Jiang M, Lin J, An Y, Xie Y, Xu M, Shen W, Guo GL, Huang Y, Li S, Zhang X, Xie W (2017). Altenusin, a Nonsteroidal Microbial Metabolite, Attenuates Nonalcoholic Fatty Liver Disease by Activating the Farnesoid X Receptor. Mol Pharmacol 92(4): 425-436. 10.1124/mol.117.108829
- Guo Z (2016). Artemisinin anti-malarial drugs in China. Acta Pharm Sin B 6(2): 115-124. 10.1016/j.apsb.2016.01.008
- Xu W, Lu C, Yao L, Zhang F, Shao J, Zheng S (2017). Dihydroartemisinin protects against alcoholic liver injury through alleviating hepatocyte steatosis in a farnesoid X receptor-dependent manner. Toxicol Appl Pharmacol 315: 23-34. 10.1016/j.taap.2016.12.001
- Duboc H, Tache Y, Hofmann AF (2014). The bile acid TGR5 membrane receptor: from basic research to clinical application. Dig Liver Dis 46(4): 302-312. 10.1016/j.dld.2013.10.021
- Ullmer C, Alvarez Sanchez R, Sprecher U, Raab S, Mattei P, Dehmlow H, Sewing S, Iglesias A, Beauchamp J, Conde-Knape K (2013). Systemic bile acid sensing by G protein-coupled bile acid receptor 1 (GPBAR1) promotes PYY and GLP-1 release. Br J Pharmacol 169(3): 671-684. 10.1111/bph.12158
- Watanabe M, Houten SM, Mataki C, Christoffolete MA, Kim BW, Sato H, Messaddeq N, Harney JW, Ezaki O, Kodama T, Schoonjans K, Bianco AC, Auwerx J (2006). Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature 439(7075): 484-489. 10.1038/nature04330
- Eggink H, Soeters M, Pols T (2014). TGR5 ligands as potential therapeutics in inflammatory diseases. Int J Interferon, Cytokine Mediator Res 6(1): 27-38. 10.2147/ijicmr.S40102
- Chen X, Xu H, Ding L, Lou G, Liu Y, Yao Y, Chen L, Huang W, Fu X (2015). Identification of miR-26a as a target gene of bile acid receptor GPBAR-1/TGR5. PLoS One 10(6): e0131294. 10.1371/journal.pone.0131294
- Fu X, Dong B, Tian Y, Lefebvre P, Meng Z, Wang X, Pattou F, Han W, Wang X, Lou F, Jove R, Staels B, Moore DD, Huang W (2015). MicroRNA-26a regulates insulin sensitivity and metabolism of glucose and lipids. J Clin Invest 125(6): 2497-2509. 10.1172/JCI75438
- Guo C, Chen WD, Wang YD (2016). TGR5, Not Only a Metabolic Regulator. Front Physiol 7: 646. 10.3389/fphys.2016.00646
- Pellicciari R, Gioiello A, Macchiarulo A, Thomas C, Rosatelli E, Natalini B, Sardella R, Pruzanski M, Roda A, Pastorini E, Schoonjans K, Auwerx J (2009). Discovery of 6alpha-ethyl-23(S)-methylcholic acid (S-EMCA, INT-777) as a potent and selective agonist for the TGR5 receptor, a novel target for diabesity. J Med Chem 52(24): 7958-7961. 10.1021/jm901390p
- Thomas C, Gioiello A, Noriega L, Strehle A, Oury J, Rizzo G, Macchiarulo A, Yamamoto H, Mataki C, Pruzanski M, Pellicciari R, Auwerx J, Schoonjans K (2009). TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab 10(3): 167-177. 10.1016/j.cmet.2009.08.001
- Kumar DP, Asgharpour A, Mirshahi F, Park SH, Liu S, Imai Y, Nadler JL, Grider JR, Murthy KS, Sanyal AJ (2016). Activation of Transmembrane Bile Acid Receptor TGR5 Modulates Pancreatic Islet alpha Cells to Promote Glucose Homeostasis. J Biol Chem 291(13): 6626-6640. 10.1074/jbc.M115.699504
- Festa C, Renga B, D'Amore C, Sepe V, Finamore C, De Marino S, Carino A, Cipriani S, Monti MC, Zampella A, Fiorucci S (2014). Exploitation of cholane scaffold for the discovery of potent and selective farnesoid X receptor (FXR) and G-protein coupled bile acid receptor 1 (GP-BAR1) ligands. J Med Chem 57(20): 8477-8495. 10.1021/jm501273r
- Carino A, Cipriani S, Marchiano S, Biagioli M, Santorelli C, Donini A, Zampella A, Monti MC, Fiorucci S (2017). BAR502, a dual FXR and GPBAR1 agonist, promotes browning of white adipose tissue and reverses liver steatosis and fibrosis. Sci Rep 7: 42801. 10.1038/srep42801
- Sato H, Genet C, Strehle A, Thomas C, Lobstein A, Wagner A, Mioskowski C, Auwerx J, Saladin R (2007). Anti-hyperglycemic activity of a TGR5 agonist isolated from Olea europaea. Biochem Biophys Res Commun 362(4): 793-798. 10.1016/j.bbrc.2007.06.130
- Kumar DP, Rajagopal S, Mahavadi S, Mirshahi F, Grider JR, Murthy KS, Sanyal AJ (2012). Activation of transmembrane bile acid receptor TGR5 stimulates insulin secretion in pancreatic beta cells. Biochem Biophys Res Commun 427(3): 600-605. 10.1016/j.bbrc.2012.09.104
- Genet C, Strehle A, Schmidt C, Boudjelal G, Lobstein A, Schoonjans K, Souchet M, Auwerx J, Saladin R, Wagner A (2010). Structure-Activity Relationship Study of Betulinic Acid, A Novel and Selective TGR5 Agonist, and Its Synthetic Derivatives: Potential Impact in Diabetes. J Med Chem 53(1): 178-190. 10.1021/jm900872z
- de Melo CL, Queiroz MG, Arruda Filho AC, Rodrigues AM, de Sousa DF, Almeida JG, Pessoa OD, Silveira ER, Menezes DB, Melo TS, Santos FA, Rao VS (2009). Betulinic acid, a natural pentacyclic triterpenoid, prevents abdominal fat accumulation in mice fed a high-fat diet. J Agric Food Chem 57(19): 8776-8781. 10.1021/jf900768w
- Szewczyk JR, Laudeman CP, Evans KA, Li YH, Dock ST, Chen Z (2007). Use of sulfonamide derivatives in the treatment of disorders of the metabolism and the nervous system. WO2007/127505-A2
- Hodge RJ, Lin J, Vasist Johnson LS, Gould EP, Bowers GD, Nunez DJ, Team SBP (2013). Safety, Pharmacokinetics, and Pharmacodynamic Effects of a Selective TGR5 Agonist, SB-756050, in Type 2 Diabetes. Clin Pharmacol Drug Dev 2(3): 213-222. 10.1002/cpdd.34
- Zambad S, Tuli D, Matur A, Ghalsasi S, Chaudhary A, Deshpande S, Gupta R, Chauthaiwale V, Dutt C (2013). TRC210258, a novel TGR5 agonist, reduces glycemic and dyslipidemic cardiovascular risk in animal models of diabesity. Diabetes, Metab Syndr Obes: Targets Ther 7: 1-14. 10.2147/dmso.S50209
- Evans KA, Budzik BW, Ross SA, Wisnoski DD, Jin J, Rivero RA, Vimal M, Szewczyk GR, Jayawickreme C, Moncol DL, Rimele TJ, Armour SL, Weaver SP, Griffin RJ, Tadepalli SM, Jeune MR, Shearer TW, Chen ZB, Chen L, Anderson DL, Becherer JD, De Los Frailes M, Colilla FJ (2009). Discovery of 3-aryl-4-isoxazolecarboxamides as TGR5 receptor agonists. J Med Chem 52(24): 7962-7965. 10.1021/jm901434t
- Budzik BW, Evans KA, Wisnoski DD, Jin J, Rivero RA, Szewczyk GR, Jayawickreme C, Moncol DL, Yu H (2010). Synthesis and structure-activity relationships of a series of 3-aryl-4-isoxazolecarboxamides as a new class of TGR5 agonists. Bioorg Med Chem Lett 20(4): 1363-1367. 10.1016/j.bmcl.2010.01.003
- Futatsugi K, Bahnck KB, Brenner MB, Buxton J, Chin JE, Coffey SB, Dubins J, Flynn D, Gautreau D, Guzman-Perez A, Hadcock JR, Hepworth D, Herr M, Hinchey T, Janssen AM, Jennings SM, Jiao W, Lavergne SY, Li B, Li M, Munchhof MJ, Orr STM, Piotrowski DW, Roush NS, Sammons M, Stevens BD, Storer G, Wang J, Warmus JS, Wei L, et al. (2013). Optimization of triazole-based TGR5 agonists towards orally available agents. MedChemComm 4(1): 205-210. 10.1039/c2md20174g
- Bollu V, Boren BC, Dalgard JE, Flatt BT, Haq N, Hudson S, Mohan R, Morrissey M, Pratt B, Wang T (2010). Triazole and imidazole derivatives for use as tgr5 agonists in the treatment of diabetes and obesity. WO2010/093845-A1
- Agarwal S, Patil A, Aware U, Deshmukh P, Darji B, Sasane S, Sairam KV, Priyadarsiny P, Giri P, Patel H, Giri S, Jain M, Desai RC (2016). Discovery of a Potent and Orally Efficacious TGR5 Receptor Agonist. ACS Med Chem Lett 7(1): 51-55. 10.1021/acsmedchemlett.5b00323
- Agarwal S, Sasane S, Kumar J, Darji B, Bhayani H, Soman S, Kulkarni N, Jain M (2019). Novel 2-mercapto imidazole and triazole derivatives as potent TGR5 receptor agonists. Medicine in Drug Discovery 1: 100002. 10.1016/j.medidd.2019.100002
- Keitel V, Cupisti K, Ullmer C, Knoefel WT, Kubitz R, Haussinger D (2009). The membrane-bound bile acid receptor TGR5 is localized in the epithelium of human gallbladders. Hepatology 50(3): 861-870. 10.1002/hep.23032
- Alemi F, Kwon E, Poole DP, Lieu T, Lyo V, Cattaruzza F, Cevikbas F, Steinhoff M, Nassini R, Materazzi S, Guerrero-Alba R, Valdez-Morales E, Cottrell GS, Schoonjans K, Geppetti P, Vanner SJ, Bunnett NW, Corvera CU (2013). The TGR5 receptor mediates bile acid-induced itch and analgesia. J Clin Invest 123(4): 1513-1530. 10.1172/JCI64551
- Lasalle M, Hoguet V, Hennuyer N, Leroux F, Piveteau C, Belloy L, Lestavel S, Vallez E, Dorchies E, Duplan I, Sevin E, Culot M, Gosselet F, Boulahjar R, Herledan A, Staels B, Deprez B, Tailleux A, Charton J (2017). Topical Intestinal Aminoimidazole Agonists of G-Protein-Coupled Bile Acid Receptor 1 Promote Glucagon Like Peptide-1 Secretion and Improve Glucose Tolerance. J Med Chem 60(10): 4185-4211. 10.1021/acs.jmedchem.6b01873
- Duan H, Ning M, Chen X, Zou Q, Zhang L, Feng Y, Zhang L, Leng Y, Shen J (2012). Design, synthesis, and antidiabetic activity of 4-phenoxynicotinamide and 4-phenoxypyrimidine-5-carboxamide derivatives as potent and orally efficacious TGR5 agonists. J Med Chem 55(23): 10475-10489. 10.1021/jm301071h
- Duan H, Ning M, Zou Q, Ye Y, Feng Y, Zhang L, Leng Y, Shen J (2015). Discovery of Intestinal Targeted TGR5 Agonists for the Treatment of Type 2 Diabetes. J Med Chem 58(8): 3315-3328. 10.1021/jm500829b
- Hansen M, Sonne DP, Knop FK (2014). Bile acid sequestrants: glucose-lowering mechanisms and efficacy in type 2 diabetes. Curr Diab Rep 14(5): 482. 10.1007/s11892-014-0482-4
- Chen T, Reich NW, Bell N, Finn PD, Rodriguez D, Kohler J, Kozuka K, He L, Spencer AG, Charmot D, Navre M, Carreras CW, Koo-McCoy S, Tabora J, Caldwell JS, Jacobs JW, Lewis JG (2018). Design of Gut-Restricted Thiazolidine Agonists of G Protein-Coupled Bile Acid Receptor 1 (GPBAR1, TGR5). J Med Chem 61(17): 7589-7613. 10.1021/acs.jmedchem.8b00308
- Wang XX, Wang D, Luo Y, Myakala K, Dobrinskikh E, Rosenberg AZ, Levi J, Kopp JB, Field A, Hill A, Lucia S, Qiu L, Jiang T, Peng Y, Orlicky D, Garcia G, Herman-Edelstein M, D'Agati V, Henriksen K, Adorini L, Pruzanski M, Xie C, Krausz KW, Gonzalez FJ, Ranjit S, Dvornikov A, Gratton E, Levi M (2018). FXR/TGR5 Dual Agonist Prevents Progression of Nephropathy in Diabetes and Obesity. J Am Soc Nephrol 29(1): 118-137. 10.1681/ASN.2017020222
- Hu YB, Liu XY, Zhan W (2018). Farnesoid X receptor agonist INT-767 attenuates liver steatosis and inflammation in rat model of nonalcoholic steatohepatitis. Drug Des Devel Ther 12: 2213-2221. 10.2147/DDDT.S170518
- Miyazaki-Anzai S, Masuda M, Levi M, Keenan AL, Miyazaki M (2014). Dual activation of the bile acid nuclear receptor FXR and G-protein-coupled receptor TGR5 protects mice against atherosclerosis. PLoS One 9(9): e108270. 10.1371/journal.pone.0108270
- Li Z, Huang J, Wang F, Li W, Wu X, Zhao C, Zhao J, Wei H, Wu Z, Qian M, Sun P, He L, Jin Y, Tang J, Qiu W, Siwko S, Liu M, Luo J, Xiao J (2019). Dual Targeting of Bile Acid Receptor-1 (TGR5) and Farnesoid X Receptor (FXR) Prevents Estrogen-Dependent Bone Loss in Mice. J Bone Miner Res 34(4): 765-776. 10.1002/jbmr.3652
- Rizzo G, Passeri D, De Franco F, Ciaccioli G, Donadio L, Rizzo G, Orlandi S, Sadeghpour B, Wang XX, Jiang T, Levi M, Pruzanski M, Adorini L (2010). Functional characterization of the semisynthetic bile acid derivative INT-767, a dual farnesoid X receptor and TGR5 agonist. Mol Pharmacol 78(4): 617-630. 10.1124/mol.110.064501
- Cipriani S, Renga B, D'Amore C, Simonetti M, De Tursi AA, Carino A, Monti MC, Sepe V, Zampella A, Fiorucci S (2015). Impaired Itching Perception in Murine Models of Cholestasis Is Supported by Dysregulation of GPBAR1 Signaling. PLoS One 10(7): e0129866. 10.1371/journal.pone.0129866
- Xu J, Li Z, Luo J, Yang F, Liu T, Liu M, Qiu WW, Tang J (2012). Synthesis and biological evaluation of heterocyclic ring-fused betulinic acid derivatives as novel inhibitors of osteoclast differentiation and bone resorption. J Med Chem 55(7): 3122-3134. 10.1021/jm201540h
- Ma SY, Ning MM, Zou QA, Feng Y, Ye YL, Shen JH, Leng Y (2016). OL3, a novel low-absorbed TGR5 agonist with reduced side effects, lowered blood glucose via dual actions on TGR5 activation and DPP-4 inhibition. Acta Pharmacol Sin 37(10): 1359-1369. 10.1038/aps.2016.27
- Brunton S (2014). GLP-1 receptor agonists vs. DPP-4 inhibitors for type 2 diabetes: is one approach more successful or preferable than the other? Int J Clin Pract 68(5): 557-567. 10.1111/ijcp.12361
- Li T, Chiang JY (2009). Regulation of bile acid and cholesterol metabolism by PPARs. PPAR Res 2009: 501739. 10.1155/2009/501739
- Jahan A, Chiang JY (2005). Cytokine regulation of human sterol 12alpha-hydroxylase (CYP8B1) gene. Am J Physiol Gastrointest Liver Physiol 288(4): G685-695. 10.1152/ajpgi.00207.2004
- Pathak P, Chiang JYL (2019). Sterol 12alpha-Hydroxylase Aggravates Dyslipidemia by Activating the Ceramide/mTORC1/SREBP-1C Pathway via FGF21 and FGF15. Gene Expr 19(3): 161-173. 10.3727/105221619X15529371970455
- Slatis K, Gafvels M, Kannisto K, Ovchinnikova O, Paulsson-Berne G, Parini P, Jiang ZY, Eggertsen G (2010). Abolished synthesis of cholic acid reduces atherosclerotic development in apolipoprotein E knockout mice. J Lipid Res 51(11): 3289-3298. 10.1194/jlr.M009308
- Kaur A, Patankar JV, de Haan W, Ruddle P, Wijesekara N, Groen AK, Verchere CB, Singaraja RR, Hayden MR (2015). Loss of Cyp8b1 improves glucose homeostasis by increasing GLP-1. Diabetes 64(4): 1168-1179. 10.2337/db14-0716
- Chevre R, Trigueros-Motos L, Castano D, Chua T, Corliano M, Patankar JV, Sng L, Sim L, Juin TL, Carissimo G, Ng LFP, Yi CNJ, Eliathamby CC, Groen AK, Hayden MR, Singaraja RR (2018). Therapeutic modulation of the bile acid pool by Cyp8b1 knockdown protects against nonalcoholic fatty liver disease in mice. FASEB J 32(7): 3792-3802. 10.1096/fj.201701084RR
- Straniero S, Laskar A, Savva C, Hardfeldt J, Angelin B, Rudling M (2020). Of mice and men: murine bile acids explain species differences in the regulation of bile acid and cholesterol metabolism. J Lipid Res 61(4): 480-491. 10.1194/jlr.RA119000307
- Guo GL, Chiang JYL (2020). Is CYP2C70 the key to new mouse models to understand bile acids in humans? J Lipid Res 61(3): 269-271. 10.1194/jlr.C120000621
- Honda A, Miyazaki T, Iwamoto J, Hirayama T, Morishita Y, Monma T, Ueda H, Mizuno S, Sugiyama F, Takahashi S, Ikegami T (2020). Regulation of bile acid metabolism in mouse models with hydrophobic bile acid composition. J Lipid Res 61(1): 54-69. 10.1194/jlr.RA119000395
- Takahashi S, Fukami T, Masuo Y, Brocker CN, Xie C, Krausz KW, Wolf CR, Henderson CJ, Gonzalez FJ (2016). Cyp2c70 is responsible for the species difference in bile acid metabolism between mice and humans. J Lipid Res 57(12): 2130-2137. 10.1194/jlr.M071183
- Li-Hawkins J, Gåfvels M, Olin M, Lund EG, Andersson U, Schuster G, Björkhem I, Russell DW, Eggertsen G (2002). Cholic acid mediates negative feedback regulation of bile acid synthesis in mice. J Clin Invest 110(8): 1191-1200. 10.1172/jci0216309
- Wang DQ, Tazuma S, Cohen DE, Carey MC (2003). Feeding natural hydrophilic bile acids inhibits intestinal cholesterol absorption: studies in the gallstone-susceptible mouse. Am J Physiol-Gastroint Liver Physiol 285(3): G494-G502. 10.1152/ajpgi.00156.2003
- Franchini PLA, Tietjen IW, Hayden MR, Singaraja RR, Radomski CCA, Percival MD, Eyers MS, Dean RA, Xie Z (2014). Cyp8b1 and uses thereof in therapeutic and diagnostic methods. WO2014/018375-A1
- Caldwell JP, Caplen MA, Cumming JN, Dykstra KD, Hruza A, Lankin C, Li D, Liu H, McCracken A, McKittrick B (2018). Compounds useful for altering the levels of bile acids for the treatment of diabetes and cardiometabolic disease. WO2018/034917-A1
- Fan L, Joseph JF, Durairaj P, Parr MK, Bureik M (2019). Conversion of chenodeoxycholic acid to cholic acid by human CYP8B1. Biol Chem 400(5): 625-628. 10.1515/hsz-2018-0379
- Tremaroli V, Backhed F (2012). Functional interactions between the gut microbiota and host metabolism. Nature 489(7415): 242-249. 10.1038/nature11552
- Foley MH, O'Flaherty S, Barrangou R, Theriot CM (2019). Bile salt hydrolases: Gatekeepers of bile acid metabolism and host-microbiome crosstalk in the gastrointestinal tract. PLoS Pathog 15(3): e1007581. 10.1371/journal.ppat.1007581
- Chand D, Avinash VS, Yadav Y, Pundle AV, Suresh CG, Ramasamy S (2017). Molecular features of bile salt hydrolases and relevance in human health. Biochim Biophys Acta Gen Subj 1861(1 Pt A): 2981-2991. 10.1016/j.bbagen.2016.09.024
- Horackova S, Plockova M, Demnerova K (2018). Importance of microbial defence systems to bile salts and mechanisms of serum cholesterol reduction. Biotechnol Adv 36(3): 682-690. 10.1016/j.biotechadv.2017.12.005
- Inagaki T, Moschetta A, Lee YK, Peng L, Zhao G, Downes M, Yu RT, Shelton JM, Richardson JA, Repa JJ, Mangelsdorf DJ, Kliewer SA (2006). Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci U S A 103(10): 3920-3925. 10.1073/pnas.0509592103
- Liu R, Hong J, Xu X, Feng Q, Zhang D, Gu Y, Shi J, Zhao S, Liu W, Wang X, Xia H, Liu Z, Cui B, Liang P, Xi L, Jin J, Ying X, Wang X, Zhao X, Li W, Jia H, Lan Z, Li F, Wang R, Sun Y, Yang M, Shen Y, Jie Z, Li J, Chen X, et al. (2017). Gut microbiome and serum metabolome alterations in obesity and after weight-loss intervention. Nat Med 23(7): 859-868. 10.1038/nm.4358
- Dong Z, Lee BH (2018). Bile salt hydrolases: Structure and function, substrate preference, and inhibitor development. Protein Sci 27(10): 1742-1754. 10.1002/pro.3484
- Begley M, Hill C, Gahan CG (2006). Bile salt hydrolase activity in probiotics. Appl Environ Microbiol 72(3): 1729-1738. 10.1128/AEM.72.3.1729-1738.2006
- Korpela K, Salonen A, Virta LJ, Kekkonen RA, Forslund K, Bork P, de Vos WM (2016). Intestinal microbiome is related to lifetime antibiotic use in Finnish pre-school children. Nat Commun 7: 10410. 10.1038/ncomms10410
- Geng W, Long SL, Chang YJ, Saxton AM, Joyce SA, Lin J (2020). Evaluation of bile salt hydrolase inhibitor efficacy for modulating host bile profile and physiology using a chicken model system. Sci Rep 10(1): 4941. 10.1038/s41598-020-61723-7
- Dahiya DK, Renuka, Puniya M, Shandilya UK, Dhewa T, Kumar N, Kumar S, Puniya AK, Shukla P (2017). Gut Microbiota Modulation and Its Relationship with Obesity Using Prebiotic Fibers and Probiotics: A Review. Front Microbiol 8: 563. 10.3389/fmicb.2017.00563
- Yao L, Seaton SC, Ndousse-Fetter S, Adhikari AA, DiBenedetto N, Mina AI, Banks AS, Bry L, Devlin AS (2018). A selective gut bacterial bile salt hydrolase alters host metabolism. eLife 7: e37182. 10.7554/eLife.37182
- Wang Z, Zeng X, Mo Y, Smith K, Guo Y, Lin J (2012). Identification and characterization of a bile salt hydrolase from Lactobacillus salivarius for development of novel alternatives to antibiotic growth promoters. Appl Environ Microbiol 78(24): 8795-8802. 10.1128/AEM.02519-12
- Smith K, Zeng X, Lin J (2014). Discovery of bile salt hydrolase inhibitors using an efficient high-throughput screening system. PLoS One 9(1): e85344. 10.1371/journal.pone.0085344
- Adhikari AA, Seegar TCM, Ficarro SB, McCurry MD, Ramachandran D, Yao L, Chaudhari SN, Ndousse-Fetter S, Banks AS, Marto JA, Blacklow SC, Devlin AS (2020). Development of a covalent inhibitor of gut bacterial bile salt hydrolases. Nat Chem Biol 16(3): 318-326. 10.1038/s41589-020-0467-3
- Song Z, Cai Y, Lao X, Wang X, Lin X, Cui Y, Kalavagunta PK, Liao J, Jin L, Shang J, Li J (2019). Taxonomic profiling and populational patterns of bacterial bile salt hydrolase (BSH) genes based on worldwide human gut microbiome. Microbiome 7(1): 9. 10.1186/s40168-019-0628-3
–
ACKNOWLEDGMENTS
We apologize that we could not in-clude all relevant publications in this review due to the space limitation. This work was supported by the Nation-al Natural Science Foundation of China (21877015) to YW and National Institute of Diabetes and Digestive and Kidney Diseases (R01DK124627) to WH.
COPYRIGHT
© 2020

Improving glucose and lipids metabolism: drug development based on bile acid related targets by Korenkova et al. is licensed under a Creative Commons Attribution 4.0 International License.


