Research Articles:
Cell Stress, Vol. 7, No. 9, pp. 69 - 89; doi: 10.15698/cst2023.09.289
Chromatin assembly factor-1 preserves genome stability in ctf4∆ cells by promoting sister chromatid cohesion
1 Marseille Cancer Research Centre (CRCM), U1068 INSERM, UMR7258 CNRS, UM105 Aix Marseille Univ, Institut Paoli-Calmettes, Marseille, France. Ligue Nationale Contre le Cancer (Labeled Equip).
Keywords: CAF-1, Ctf4, replication, chromatin assembly, sister chromatid cohesion.
Abbreviatons:
CAF-1 – chromatin assembly factor-1,
H3K56ac – Histone H3 lysine 56 acetylation,
SCC – sister chromatid cohesion,
UV – ultraviolet,
CPT – camptothecin,
HU – hydroxyurea,
MMS – methyl-methanesulfonate, WT – wild-type,
HR – homologous recombination,
γH2AX – phosphorylation of histone H2A on serine 129,
Polα – DNA-polymerase α,
ts – thermosensitive.
Received originally: 23/02/2023 Received in revised form: 31/07/2023
Accepted: 01/08/2023
Published: 14/08/2023
Correspondence:
Yves Corda, Marseille Cancer Research Centre, U1068 INSERM, UMR7258 CNRS, UM105 Aix Marseille Univ, Institut Paoli-Calmettes, 27, Blvd Leï Roure, BP30059, 13273 Marseille Cedex 9, France; yves.corda@inserm.fr
Conflict of interest statement: The authors declare that they have no conflict of interest.
Please cite this article as: Nagham Ghaddar, Pierre Luciano, Vincent Géli and Yves Corda (2023). Chromatin assembly factor-1 preserves genome stability in ctf4∆ cells by promoting sister chromatid cohesion. Cell Stress 7(9): 69-89. doi: 10.15698/cst2023.09.289
Abstract
Chromatin assembly and the establishment of sister chromatid cohesion are intimately connected to the progression of DNA replication forks. Here we examined the genetic interaction between the heterotrimeric chromatin assembly factor-1 (CAF-1), a central component of chromatin assembly during replication, and the core replisome component Ctf4. We find that CAF-1 deficient cells as well as cells affected in newly-synthesized H3-H4 histones deposition during DNA replication exhibit a severe negative growth with ctf4∆ mutant. We dissected the role of CAF-1 in the maintenance of genome stability in ctf4∆ yeast cells. In the absence of CTF4, CAF-1 is essential for viability in cells experiencing replication problems, in cells lacking functional S-phase checkpoint or functional spindle checkpoint, and in cells lacking DNA repair pathways involving homologous recombination. We present evidence that CAF-1 affects cohesin association to chromatin in a DNA-damage-dependent manner and is essential to maintain cohesion in the absence of CTF4. We also show that Eco1-catalyzed Smc3 acetylation is reduced in absence of CAF-1. Furthermore, we describe genetic interactions between CAF-1 and essential genes involved in cohesin loading, cohesin stabilization, and cohesin component indicating that CAF-1 is crucial for viability when sister chromatid cohesion is affected. Finally, our data indicate that the CAF-1-dependent pathway required for cohesion is functionally distinct from the Rtt101-Mms1-Mms22 pathway which functions in replicated chromatin assembly. Collectively, our results suggest that the deposition by CAF-1 of newly-synthesized H3-H4 histones during DNA replication creates a chromatin environment that favors sister chromatid cohesion and maintains genome integrity.
INTRODUCTION
Nucleosome assembly during DNA replication is tightly coupled to ongoing DNA synthesis. Chromatin assembly factor-1 (CAF-1) is a conserved histone chaperone, essential for cell survival in multicellular organisms, that plays a key role in replication-dependent nucleosome assembly [1] [2] [3] [4] [5] and preserves genome stability [6] [7]. In budding yeast, CAF-1 consists of three subunits called Cac1, Cac2, and Cac3, which differ in their ability to bind H3-H4. Deletion of any CAF-1 subunits is viable but leads to multiple defects including replisome dysfunction and DNA damage sensitivity [3] [7] [8] [9] [10] [11] [12] [13]. The ability of CAF-1 to deposit H3-H4 onto replicating DNA depends on its physical interaction with PCNA, a processivity factor for DNA polymerases, which is localized at the sites of DNA synthesis during replication and repair [14] [15] [16] [17] [18].
–
Lysine 56 of histone H3 is transiently acetylated during S phase of the cell cycle and after DNA damage and is rapidly de-acetylated, by the action of the sirtuins Hst3 and Hst4, when cells enter the transition between G2 and M phases and after DNA repair [19] [20]. Histone H3 lysine 56 acetylation (H3K56ac) is mediated by the histone acetyl-transferase Rtt109 and the histone chaperone Asf1 [21] [22] [23] [24] leading to H3 and H4 ubiquitination by the Rtt101-Mms1-Mms22 E3 ligase complex [25]. H3-H4 ubiquitination promotes histones H3-H4 deposition at the fork proximity, coordinates nucleosome formation, and facilitates the stable progression of the replication fork [25] [26] [27]. Besides H3K56ac function in replication-coupled chromatin assembly, H3K56ac is also required for transcription, DNA repair-coupled chromatin assembly, inactivation of the DNA damage checkpoint, and meiosis [20] [28] [29] [30] [31] [32] [33] [34] [35]. H3K56ac together with H3K121,122 ubiquitylation mediated by Rtt101-Mms1-Mms22 promote sister chromatid cohesion (SCC), establishing a potential functional connection between histone deposition and cohesin activity [28] [36] [37].
–
Cohesion holds the two copies of the sister chromatids together from the moment of duplication to the onset of anaphase, subsequently, ensuring accurate chromosome segregation during mitosis [38] [39]. Sister chromatid cohesion is mediated at many points along the sister chromatids by the cohesin ring complex. In Saccharomyces cerevisiae, the cohesin ring complex consists of Smc1, Smc3, Scc1/Mcd1, and Scc3 subunits and is loaded onto the chromosomes by the Scc2-Scc4 deposition complex, in the G1/S phase, at broad nucleosome-free regions [40] [41] [42]. In addition, the cohesion ring complex preferentially accumulates at centromeres and between convergent transcribed genes [43] [44]. Scc2-Scc4 determines cohesins localization across the genome [41] [45] [46] [47] [48] [49] and is necessary for maintaining stable cohesion-DNA association during G1 [50]. Scc2-Scc4 directly interacts with the kinetochore protein Ctf19 at CEN loci. This interaction is dependent on Ctf19 phosphorylation by DDK, an important event for centromeric cohesion [51]. Subsequently, cohesins are converted to a tethering competent state through the action of the essential replication fork-associated acetyltransferase Eco1 that acetylates Smc3 at lysine 112 and lysine 113 [52] [53] [54] [55] leading to cohesion establishment during S phase [56] [57]. Recent studies revealed that Eco1-dependent Smc3 acetylation is promoted by transient DNA structures that form during Okazaki fragment maturation [58]. Cohesion generation can also be induced in G2/M when a DSB is present [45] [55] [59]. In anaphase, after the formation of the mitotic spindle, the Scc1 subunit is cleaved by separase, allowing sister chromatids segregation [60]. It is well established that PCNA interacts and recruits Eco1 to replication forks, where the levels of PCNA correlate with cohesion establishment [61]. Indeed, protein complexes such as Ctf18-RFC and Elg1-RFC, which influence PCNA loading and unloading, affect cohesion [62] [63] [64] [65]. Recently it has been shown that several factors associated with replication forks (Chl1, MCM, Bre1, and the ubiquitin ligase complex Rtt101-Mms1-Mms22) also recruit Eco1 and/or promote Eco1-dependent cohesion establishment during DNA replication [37] [39] [66] [67] [68]. In addition to Eco1 and the components of the cohesin ring complex, which are all required for viability, genetic analyses have identified an important number of replication proteins, functioning in S-phase, that mediate cohesion establishment. Two genetically distinct pathways, that involve multiple replication fork-associated proteins being non-essential for cell viability, contribute to cohesion establishment at the replication forks in yeast [69]. The first pathway is involved with Scc2 in the de novo loading of the nucleoplasmic cohesin pool at the site of DNA synthesis. It includes the S-phase checkpoint protein Mrc1, a core component of the replisome progressing complex required for normal replication fork progression [70] [71] [72], and the Ctf18-Ctf8-Dcc1 complex which forms an alternative replication factor C complex with Rfc2-Rfc5 (Ctf18-RFC) [73] [74]. The second pathway, independent of Scc2, is involved in conversion of the preloaded cohesin rings on the DNA template into a cohesive form. This pathway is composed of the replisome components Csm3, Tof1, Chl1, and Ctf4 [74]. Csm3 and Tof1 form the replication checkpoint complex with Mrc1 [75] [141]. Chl1 helicase controls replication fork progression [76], and physically engages with cohesin during cohesion establishment [66] [77]. Ctf4, which was identified in budding yeast as a chromosome transmission fidelity factor, is required for the maintenance of genome stability and SCC [56] [78] [79].
–
Ctf4 is a core component of the replisome progression complex [56] that forms a hub connecting replication forks to an important number of proteins [77] [80] [81]. During normal replication, Ctf4 recruits and stabilizes DNA polymerase-alpha at the replication forks and coordinates DNA unwinding and synthesis [82] [83] [84]. Furthermore, Ctf4 forms an axis with Mcm2 and Pol alpha to facilitate the transfer of parental H3-H4 to lagging strands [85]. Among various partners, Ctf4 interacts and recruits Chl1 to the replisome to coordinate replication fork progression and cohesion establishment [77]. Ctf4 also interacts with Mms22 to recruit the Rtt101-Mms1-Mms22 E3 ubiquitin ligase complex to the replisome during S-phase [86]. This interaction is important to maintain genome stability in presence of replicative stress through the H3K56ac-dependent CAF-1-independent pathway [82] [84] [87] [88] [89] [90] [91] [92] [93]. The Mms22/Ctf4 interaction also contributes to cohesion establishment by promoting Eco1 recruitment and stabilization at replication forks, and it has been proposed that Rtt101-Mms1-Mms22 E3 ubiquitin ligase acts through Ctf4 to coordinate replication coupled sister chromatid cohesion and H3K56ac-dependent nucleosome assembly [37].
–
We previously reported that during replicative stress, replisome function is modulated by H3K56ac and that Ctf4 is harmful upon DNA damage in the absence of the functional DNA repair/tolerance branch of the CAF-1-independent H3K56ac pathway [92]. In this study, we present pieces of evidence that H3K56ac and Rtt101-Mms1-Mms22 E3 ubiquitin ligase are not required for growth in the absence of Ctf4 contrary to CAF-1 and replication-coupled chromatin assembly. We report that the loss of CAF-1 function increases the cohesion defect observed in ctf4Δ cells, affects the Eco1-dependent Smc3 acetylation required for cohesion establishment, and provokes cell death in yeast cells affected in the major SCC establishment pathways. Taken together, our experiments are consistent with a model in which the nucleosome assembly function of CAF-1 is required to create an adequate structural environment required for sister chromatid cohesion establishment at DNA replication forks.
RESULTS
CAF-1 exhibits a synthetic genetic interaction with the replisome component Ctf4
We have previously observed that the replication function of Ctf4 is strongly deleterious for yeast cells experiencing constitutive replicative damages in the absence of a functional H3K56ac-dependent pathway [92]. Having discovered that the effects observed are a direct consequence of the CAF-1-independent H3K56ac role in repairing/tolerating replicative DNA damage, we now investigate the consequences associated with CTF4 inactivation in yeast cells defective in H3K56ac-dependent nucleosome assembly. H3K56ac facilitates replication-coupled chromatin assembly through a function that is dependent on CAF-1 and Rtt106 [25] [26] [27]. This pathway coordinates nucleosome assembly and stability of advancing replication forks but is not required for H3K56ac-mediated protection against replicative DNA-damaging agents by DNA repair/tolerance mechanisms [27] [94]. We carried out tetrad analysis after sporulating diploid heterozygous for cac1Δ rtt106Δ ctf4Δ. Dissection of meiotic tetrads shows that cac1Δ rtt106Δ ctf4Δ segregants grow at a very slow rate (Figure 1A). Interestingly, the growth of cac1Δ ctf4Δ double mutant was also considerably reduced and seemed slightly better than that of the triple mutant cac1Δ rtt106Δ ctf4Δ, suggesting that deletion of RTT106 slightly exacerbates the growth defect of cac1Δ ctf4Δ cells. Finally, the growth difference between rtt106Δ ctf4Δ and ctf4Δ is much smaller than that between cac1Δ ctf4Δ and ctf4Δ, which means that RTT106 was less required than CAC1 in the absence of CTF4 (Figure 1A). Taking into account that Cac1 and Rtt106 coordinate to deposit newly synthesized histone H3-H4 onto replicated DNA during S phase and DNA repair [95] [96], and based on the fact that Cac1 plays a most major role in this mechanism during DNA synthesis, these results suggest that the function of CAC1 important for ctf4Δ cells growth is related to its chromatin assembly function. This function requires all CAF-1 subunits; Cac1, Cac2, and Cac3. Because CAF-1 subunits seem to have distinct functions in addition to their common nucleosome assembly function during DNA synthesis [97] [98] [99] [100] we analyzed the growth of cac2Δ ctf4Δ and cac3Δ ctf4Δ cells by monitoring the meiotic progeny of the diploid strains heterozygous for CTF4 and CAC2, and CTF4 and CAC3, deletions. We found that both cac2Δ and cac3Δ exhibited a strong negative interaction with ctf4Δ (Figure S1A). Moreover, we observed that cac3Δ ctf4Δ mutant was less affected in growth than cac2Δ ctf4Δ and cac1Δ ctf4Δ mutants (Figure S1A and Figure S2A, B, C). This observation could be explained by the fact that Cac3 depletion has a minor effect on nucleosome formation compared to cac1Δ and cac2Δ mutants [100]. Taken together these results strongly suggest that the nucleosome assembly function of CAF-1 during DNA synthesis is crucial in the absence of CTF4.
–
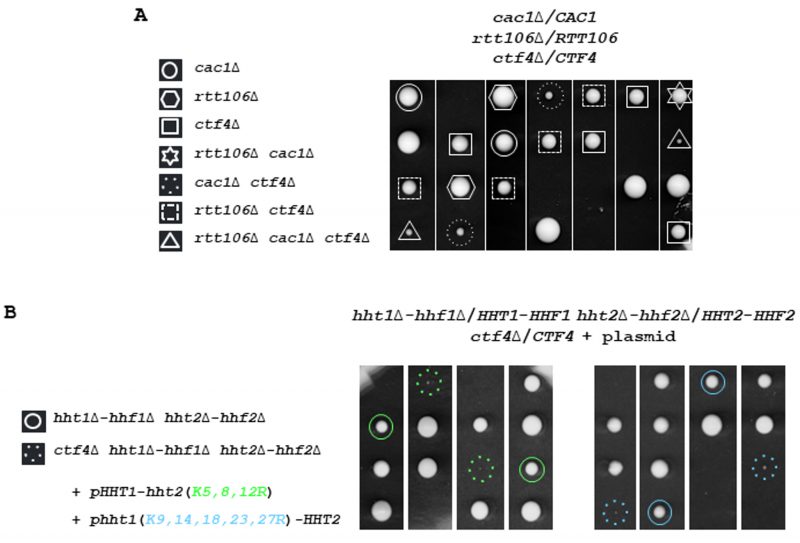 |
FIGURE 1: CTF4 inactivation results in cell lethality in different genetic contexts affecting chromatin assembly. (A) Defective Cac1/Rtt106-dependent chromatin assembly affects growth in absence of CTF4. Tetrads from rtt106Δ/RTT106 cac1Δ/CAC1 ctf4Δ/CTF4 diploid strain were dissected. In this and subsequent figures, the spores from a given tetrad are in vertical line in a YPD plate. Fifty tetrads were dissected. Five representative tetrads are shown after 3 days at 30°. (B) Mutations at histone lysine residues implicated in nucleosome assembly strongly affect growth of ctf4Δ cells. One hundred tetrads from diploids for hht1Δ-hhf1Δ/HHT1-HHF1 hht2Δ-hhf2Δ/HHT2-HHF2 ctf4Δ/CTF4 expressing either HHT2 and hhf2(K5,8,12R), or HHF1 and hht1(K9,14,18,23,27R) from a centromeric plasmid were dissected and analyzed for the presence of auxotrophic markers. The circle indicates spore expressing H4K5,8,12R (green), or H3K9,14,18,23,27R (blue) as the sole source of H4 or H3 histones, respectively. The dashed circle indicates ctf4Δ spore expressing H4K5,8,12R (green), or H3K9,14,18,23,27R (blue) as the sole source of H4 or H3 histones, respectively. |
Mutations affecting DNA-replication coupled nucleosome assembly exhibit a synthetic interaction with ctf4Δ
The HIR complex (formed by Hir1, Hir2, Hir3, and Hpc2) promotes replication-independent chromatin assembly [101]. This complex is important for normal growth and silencing in the absence of CAF-1, indicating functional overlap between HIR and CAF-1 complexes [102]. We found that HIR1 was dispensable for the growth of ctf4Δ cells (Figures S1B and S2J), suggesting that the replication-independent chromatin assembly function is not important for ctf4Δ cells. We next conducted an extensive genetic analysis to assess the importance of genes encoding histone chaperones and histone acetyltransferases in ctf4Δ mutant cells (Figure S2). We first confirm that RTT106 inactivation affected ctf4Δ cell growth less severely than CAC1 inactivation (Figure S2E). Among many candidates tested we found that the Spt16 subunit of the heterodimeric FACT complex (Spt16-Pob3), which binds histone H3-H4 and Cac2 subunit of CAF-1, and functions in DNA replication-coupled nucleosome assembly [103], exhibited negative genetic interactions with ctf4Δ (Figure S2H). In addition, we show that rfa1-A88P mutation, which exhibits attenuated nucleosome assembly on nascent chromatin [104] was lethal with ctf4Δ (Figure S2K). Finally, we observed that the lysine acetyltransferase Gcn5, which regulates the interaction between H3-H4 and CAF-1 to promote the deposition of newly-synthesized histones [105] was also important for the normal growth of ctf4Δ cells (Figure S2M). We noticed that asf1Δ as well as rtt109Δ only slightly affected ctf4Δ cells (Figure S2D and L). H3 and H4 N-terminal tail acetylation serve as an important regulator of nucleosome assembly [26] [105] [106] [107]. We therefore, determine the importance of different H3-H4 histone modifications involved in nucleosome assembly, in ctf4Δ cells.
–
We next crossed a strain expressing H3K9,14,18,23,27R or H4K5,8,12R from a centromeric plasmid as the sole source of histone H3 or H4 with the ctf4Δ strain and analyzed the spores after diploids sporulation. We observed that in the absence of CTF4, the mutation at H3 or H4 lysine residues, implicated in nucleosome assembly, became deleterious (Figure 1B). We further analyzed the consequences of the absence of Nap1, which promotes H2A-H2B tetramer assembly in nucleosomes, in ctf4Δ cells. We found that nap1Δ did not negatively affect the growth of ctf4Δ cells (Figure S1C). Altogether, these genetic analyses indicate that defects in chromatin assembly during replication related to H3-H4 histones are deleterious in absence of CTF4. These results further support the notion that CAF-1 has a crucial role in a process linked to the Ctf4 function.
–
Cac1 is important for genome integrity in absence of CTF4
To eliminate the possibility that cac1Δ ctf4Δ growth defects originate from meiotic events, we deleted CAC1 in the ctf4Δ strain background by gene targeting. We first confirmed the slow growth phenotype, observed for cac1Δ ctf4Δ, during the segregation analyses and observed that this phenotype is amplified at both 25°C and 35°C (Figure 2A). We next examined the sensitivity of cac1Δ ctf4Δ mutant to both ultraviolet (UV) light and chronic exposure to DNA-damaging agents. We found that the cac1Δ ctf4Δ double mutant was more sensitive to UV, camptothecin (CPT), hydroxyurea (HU), and methyl-methanesulfonate (MMS) compared to every single mutant (Figure 2B). Then, we assessed in ctf4Δ mutant, the importance of Cac1 in the absence of Rrm3 which facilitates the progression of replication forks through non-histone DNA-protein complexes [108] [109]. We found that Cac1 was crucial for rrm3Δ ctf4Δ cells (Figure S3). These results indicate that replication stress and replication-induced DNA damage are lethal for cac1Δ ctf4Δ mutant. We next evaluated if the absence of Cac1 in ctf4Δ cells cause a synthetic interaction with mutations affecting the S-phase checkpoint pathway. We found that the absence of Mec1 or Rad53 kinases, that activate both branches of the S-phase checkpoint pathway [110], strongly affect the viability of cac1Δ ctf4Δ cells (Figure 2C).
–
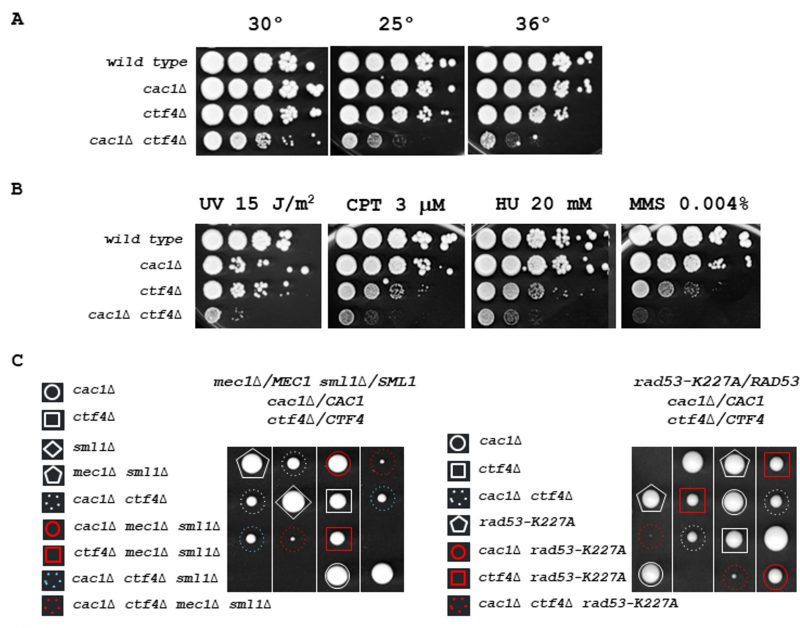 |
FIGURE 2: CAC1 is important for growth in the absence of CTF4. (A) cac1Δ ctf4Δ growth is affected at various temperatures. Tenfold serial dilutions of wild-type, cac1Δ, ctf4Δ, and cac1Δ ctf4Δ cells were spotted onto YPD plates and incubated at 30° (left), 25° (middle), or 36° (right) for 3 days. (B) cac1Δ ctf4Δ growth is affected in presence of DNA damage. Tenfold serial dilutions of wild-type, cac1Δ, ctf4Δ, and cac1Δ ctf4Δ cells were assayed on normal growth media (YPD), after UV irradiation or not, and on media containing the indicated DNA-damaging agents, camptothecin (CPT), hydroxyurea (HU), and methyl-methanesulfonate (MMS). (C) The S-phase checkpoint is required for cac1Δ ctf4Δ mutant viability. The diploid strains mec1Δ/MEC1 sml1Δ/SML1 cac1Δ/CAC1 ctf4Δ/CTF4 (left) and rad53-K227A/RAD53 cac1Δ/CAC1 ctf4Δ/CTF4 (right) were sporulated and one hundred tetrads were dissected on YPD plates and incubated at 30° for 5 days. Four representative tetrads are shown for each dissection. mec1Δ sml1Δ and rad53-K227A mutations are lethal in cac1Δ ctf4Δ cells. |
To evaluate if homologous recombination (HR) was important for cac1Δ ctf4Δ mutant, we investigated whether cac1Δ ctf4Δ leads to an increase in spontaneous Rad52 foci, which reflect HR proteins recruitment into repair foci [111] [112]. We found that cac1Δ ctf4Δ cells exhibited a higher frequency of Rad52-YFP foci compared to cac1Δ and ctf4Δ single mutants (Figure 3A, left). We also found that cac1Δ ctf4Δ exhibited an abnormally high frequency of spontaneous Rfa1 foci compared to that in cac1Δ and ctf4Δ single mutants (Figure 3A, right). These results suggest that the absence of Cac1 spontaneously creates chromosome breaks or ssDNA gaps during replication in ctf4Δ cells and that these damages, are repaired by HR.
–
In S. cerevisiae, phosphorylation of histone H2A on serine 129 (γH2AX) is tightly associated to DNA damage, and it has been shown that DNA double-strand breaks levels can be obtained by measuring levels of γH2AX [113] [114]. To determine if the absence of Cac1 increased DNA damage in ctf4Δ cells, we analyzed by Western blot γH2AX levels, after a nocodazole-imposed mitotic arrest, (Figure 3B). In agreement with previously published results showing that cac1Δ mutant had only a weak requirement for replication or DNA-damage checkpoint proteins [99], we found a slight increase of H2A phosphorylation in absence of Cac1 compared to wild-type (WT) cells. In contrast, we observed a more consistent increase over WT cells in cac1Δ ctf4Δ mutant, indicating that the absence of Cac1 induces DNA damages in ctf4Δ cells.
–
Having found that Cac1 was required for the viability of ctf4Δ mutant in absence of Rrm3 helicase (Figure S3), which helps replication fork traverse protein-DNA complexes [108] and assists fork progression across TERs [109], we sought to evaluate whether loss of CAF-1 function affected genome stability in ctf4Δ cells. For this end, we used the natural Escherichia coli Tus/Ter barrier system known to induce an unidirectionally and site-specific replication fork stalling in S. cerevisiae. Interestingly, this system, which reflects the natural protein-bound DNA barriers arising in yeast cells, represents one of the most physiological replicative stresses that yeast cells could encounter [115] [116] [117]. The Tus/Ter barrier system, composed by a 21-bp DNA sequence (Ter) which is bound by the Tus terminator protein, is coupled with the genetic URA3 gene reporter located immediately upstream the Tus/Ter barrier (Figure 3C, top), allowing us to quantify mutagenic outcomes as previously described [118] (Ghaddar et al 2023 Nat Commun, in press). In correlation with our previous data showing an increased Rad52 and Rfa1 foci formation and an increased level of γH2AX in cac1Δ ctf4Δ double mutant, we observed an increase in mutation rate in cac1Δ ctf4Δ cells compared to cac1Δ and ctf4Δ cells expressing Tus protein (Figure 3C, bottom).
–
Finally, we found that the elimination of Mad2 spindle checkpoint, which causes impairment of microtubule-kinetochore attachment and incomplete sister chromatid cohesion, negatively affects the growth of cac1Δ ctf4Δ cells (Figure S4). Overall, these results reflect the requirement of both DNA damage and spindle checkpoint for the viability of cac1Δ ctf4Δ cells and that HR is required for cell growth in the absence of both CAC1 and CTF4. They show that loss of CAF-1 function causes DNA damages and leads to spontaneous mutation in ctf4Δ cells, revealing that CAF-1 prevents DNA damage formation and maintains genome stability.
–
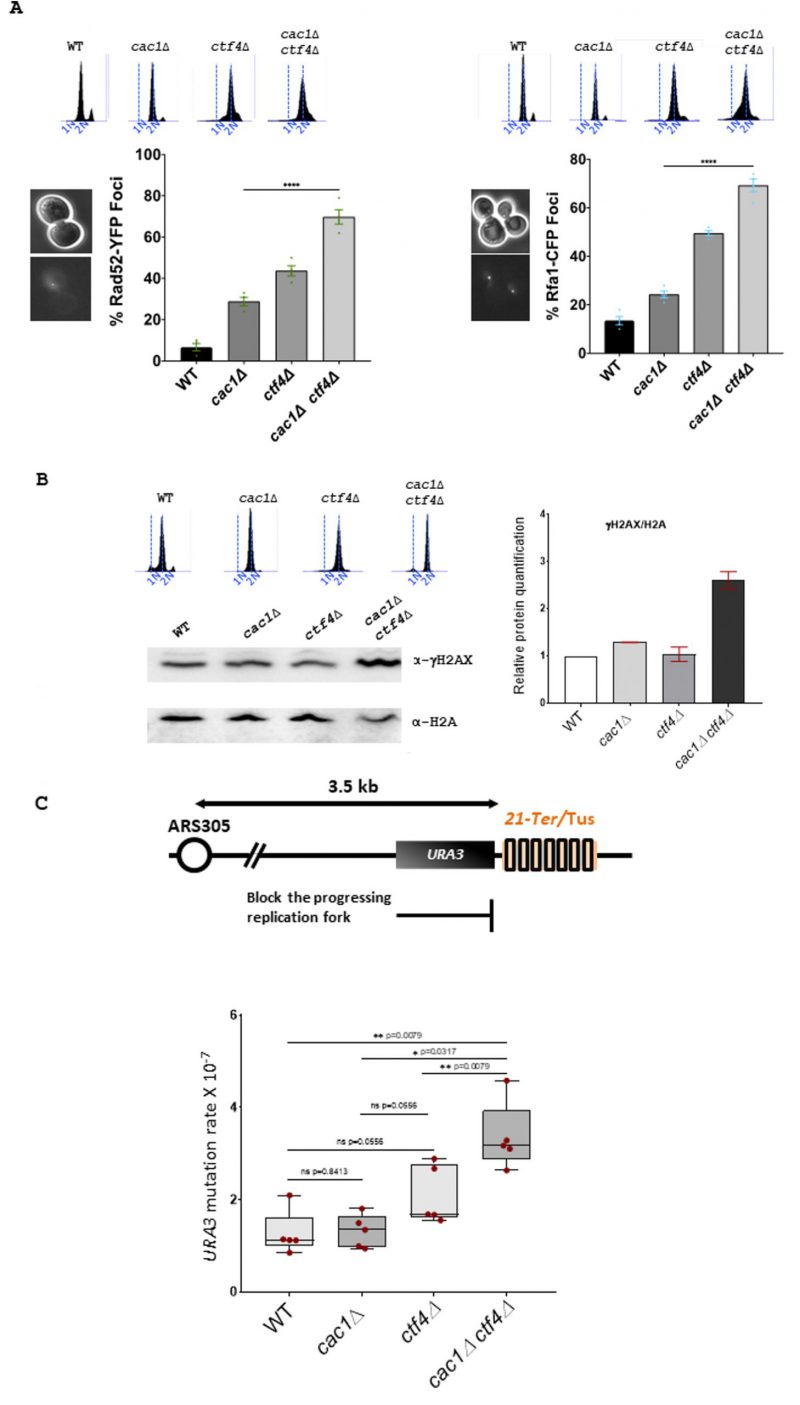 | FIGURE 3: Genomic integrity is affected in cac1Δ ctf4Δ cells. (A) Left, Rad52 foci are increased in cac1Δ ctf4Δ cells. Wild-type, cac1Δ, ctf4Δ, and cac1Δ ctf4Δ cells encoding Rad52-YFP were analyzed with fluorescence microscopy. Right, Rfa1 foci are increased in cac1Δ ctf4Δ cells. Wild-type, cac1Δ, ctf4Δ, and cac1Δ ctf4Δ cells encoding Rfa1-CFP were analyzed with fluorescence microscopy. Numbers indicate the percentage of cells that contained Rad52-YFP (left) or Rfa1-CFP foci (right). DNA replication was monitored by FACS analysis of DNA content. At least 200 cells were analyzed for each strain from three independent experiments. Statistical significance was measured using the two-tailed Mann-Whitney test. (B) γH2AX is increased in the absence of Cac1 in ctf4Δ cells. Left, Western blot was used to detect phosphorylation of H2A serine 129 (γH2AX). Right, histogram shows for WT, cac1Δ, ctf4Δ, and cac1Δ ctf4Δ, γH2AX/H2A ratios calculated based on Western blots signal intensities. DNA replication was monitored by FACS analysis of DNA content. The experiment has been done in duplicate. (C) CAF-1 inactivation increases mutation rate in ctf4Δ cells at the Tus/Ter barrier. Top, Schematic representation of the unidirectional and site-specific Tus-Ter replication fork barrier. Ter sequence is integrated 3.5 kb downstream ARS305 on Chromosome III where Tus protein (colored rectangles) binds specifically to Ter sequence, causing replication fork pausing. Upstream to Tus/Ter replication fork barrier is the URA3 reporter gene, which permits the positive selection for ura3 mutations in presence of 5-FOA to measure mutation rate. Bottom, exponentially growing cells expressing Tus protein were plated for 3 days at 30° on YPGal plates and plated out on 5-FOA to select for ura3 mutation. Box-and-whisker plots, representing the upper and lower quartile with the median, show the mutation rate in WT, cac1Δ, ctf4Δ, and cac1Δ ctf4Δ cells. Statistical analyses were done on n=5 independent experiments using two-tailed Mann-Whitney test; *p<0.05; ** p< 0.005; **** p<0.0001; ns, not significant. |
Dissecting the importance of Cac1 in functions mediated by Ctf4
Ctf4 performs different functions in DNA metabolism. To explore the significance of the negative genetic interaction between CAC1 and CTF4, we sought to investigate which function of Ctf4 is responsible for this negative interaction. To determine if the growth defect observed in cac1Δ ctf4Δ mutant is a direct effect of uncoupling between helicase and DNA-polymerase α (Polα) [82] [83] [119], we analyzed the consequences of inactivating CAC1 in the thermosensitive (ts) mutant cdc17-1 encoding the catalytic subunit of Polα. We found that cac1Δ did not affect the viability of cdc17-1 cells (Figure S5A), which indicates that the growth defect of cac1Δ ctf4Δ was not related to the ctf4Δ cell's inability to incorporate Polα into the replisome. This result shows that replication fork architecture defects, due to uncoupling arising in ctf4Δ mutant, are not responsible for the synthetic fitness defects observed in cac1Δ ctf4Δ cells. Moreover, the loss of Ctf4 affects DNA damage tolerance function due to faulty MCM-uncoupled Polα/Primase activity [93]. Thus, this result also indicates that the severe growth defect observed in cac1Δ ctf4Δ was not a consequence of defective DNA damage tolerance. We next searched for synthetic growth defects caused by combining cac1Δ mutation with the mcm2-3A mutation known to affect the Ctf4-Mcm2-Polα-dependent transfer of (H3-H4) parental histones [85] [120]. We found that affecting the parental histones transfer onto the lagging strand did not cause growth defects in absence of CAF-1 function (Figure S5B).
–
Another major function assigned to Ctf4 is associated with its role in cohesion establishment. Genetic analyses have defined two pathways for cohesion establishment at the replication fork, one containing CTF4, CHL1, CSM3, TOF1, and the second containing MRC1, CTF18, CTF8, DDC1 [69]. We addressed if any of these non-essential replisome proteins, previously implicated in cohesion establishment, were required for growth in cac1Δ cells. We found that only CTF4 inactivation became deleterious in absence of CAC1 (Figure S5C-J).
–
These data suggest that the important growth defect observed in the ctf4Δ cac1Δ mutant was unrelated to the Ctf4 functions mentioned above.
–
CAC1 is required to sustain the viability of cells affected in essential cohesion pathways
Because Ctf4 is involved in multiple cohesion establishment pathways, its absence negatively influences sister chromatid cohesion in different ways [69] [77] [93] [121]. Among the non-essential proteins associated with replisomes involved in the two parallel pathways for cohesion establishment at the replication fork [69], ctf4Δ mutant is the only one that causes lethality in the absence of the S-phase acetyltransferase ECO1 [121], a protein that locks sister chromatid entrapment by acetylating Smc3 both during S phase and in response to DNA damage [54] [55] [123] [124]. Therefore, we wondered whether affecting cohesion, in a more severe way than deleting non-essential cohesion genes, might reveal the importance of CAC1 for the growth of cells affected in cohesion. We first investigated the relationship between CAC1 and the essential cohesion genes ECO1. Since the inactivation of RAD61/WPL1, which counteracts cohesion-establishing reaction, suppresses eco1Δ lethality [54] [55], we deleted one allele of ECO1 and one allele of RAD61 in a cac1Δ/CAC1 diploid strain. We found that cac1Δ rad61Δ cells grew normally and that cac1Δ cells exhibited a synthetic sick phenotype with eco1Δ rad61Δ mutations at 30°C (Figure 4A, left). RAD61 inactivation impacts cohesion, chromatin structure, and intra-chromosomal loops organized by cohesins [125] [126] [127]. Thus, we also analyzed the consequence on cac1Δ cells of the temperature-sensitive eco1-1 mutation which confers severe cohesion defects at 37°C, and to a lesser extent, at lower temperatures [53]. We found that the eco1-1 mutant was strongly affected by CAC1 inactivation (Figure 4A, right), which confirmed that Cac1 was required for efficient growth in eco1 mutants. These data suggest that Cac1 could act, at least in part, in a cohesion pathway parallel to the Smc3ac function.
–
To further test whether in the absence of CAC1 cohesion defects are deleterious, we combined cac1Δ with mutations in genes that affect the cohesin loader (Scc2-Scc4) and the cohesin ring complex (Scc1). We first analyzed the consequences of CAC1 deletion in scc2-4 temperature-sensitive mutant defective in cohesion establishment. Whilst at permissive temperature (25°C), scc2-4 cac1Δ cells were indistinguishable from scc2-4 and cac1Δ, the double mutant cells were strongly affected at the semi-permissive temperature of 30°C (Figure 4B). We next focused on Scc1, which is involved in both establishment of cohesion in S phase and the maintenance of cohesion in G2/M. Using the temperature-sensitive scc1-73 allele [128], we found that impairment of cohesin function strongly affects cell viability in the absence of any of CAF-1 subunits, Cac1, Cac2, or Cac3 (Figure 4C, and Figure S6A). Interestingly, we observed a slight difference in the severity of cac1Δ, cac2Δ, and cac3Δ mutant phenotypes, with the cac3Δ mutant showing a weaker phenotype (Figure S6A). Again, this could be explained by previous observations showing that CAC3 inactivation has a minor effect on nucleosome formation compared to CAC1 or CAC2 inactivation [100]. The similar weaker phenotype observed for cac3Δ compared to cac1Δ and cac2Δ mutants in both scc1-73 (Figure S6A) and ctf4Δ cells (Figure 1 and Figure S2), suggests that the requirement for Cac1 in ctf4Δ cells is related to important cohesion defects linked to the absence of Ctf4. cac1-F233L and cac1-F233A,F234G mutations were previously shown to alter Cac1 binding to PCNA as well as Cac1 DNA-replication-linked nucleosome assembly function [17]. To investigate the importance of Cac1 recruitment to the replication forks through its interaction with PCNA, we determined the impact of cac1-F233L and cac1-F233A, F234G mutations in scc1-73 cells. We found that both mutations caused growth defects for scc1-73 cells at permissive temperature (Figure S6B). These data indicate that the PCNA-dependent CAF-1 recruitment to chromatin is required when cohesin function is affected. These results confirm that CAF-1 complex is required for cell viability when cohesion is affected. On the other hand, CAF-1 and Rtt106 function in a coordinated manner in nucleosome assembly [107]. Henceforth, we tested whether RTT106 deletion affects the growth of the scc1-73 mutant. A significant growth defect was observed in rtt106Δ scc1-73 cells at the semi-permissive temperature compared to each rtt106Δ and scc1-73 single mutant (Figure S6C). In agreement with the fact that RTT106 exerts a minor role compared to CAF-1 in new histones deposition during DNA synthesis [26], we found that the growth defect of the rtt106Δ scc1-73 mutant was much weaker than that of the cac1Δ scc1-73 mutant. Furthermore, cac1Δ rtt106Δ scc1-73 triple mutant exhibited a more dramatic effect compared to the rtt106Δ scc1-73 and cac1Δ scc1-73 double mutants (Figure S6C). Taken together these results indicate that CAF-1, and more generally nucleosome assembly during replication, is crucial in maintaining genome stability when SCC is compromised. These results, in association with our previous findings showing that cac1Δ ctf4Δ cells are more affected than rtt106Δ ctf4Δ cells and that RTT106 inactivation amplified the growth defect of cac1Δ ctf4Δ cells (Figure 1A), strongly suggest that the requirement for CAF-1 in ctf4Δ mutant was related to important cohesion defects linked to the absence of CTF4.
–
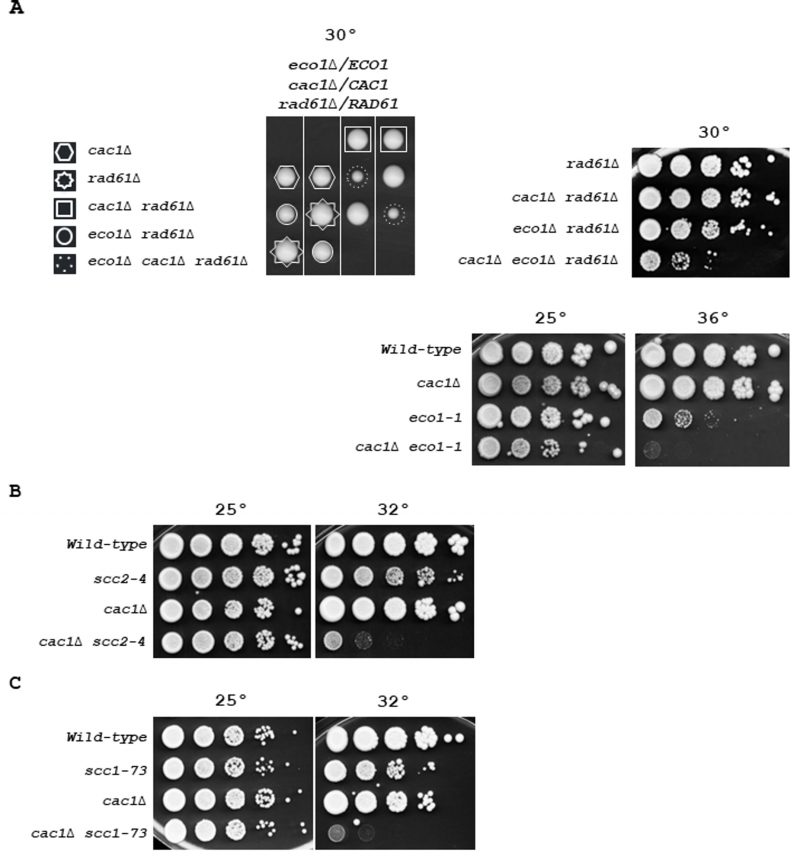 | FIGURE 4: Essential sister chromatid cohesion genes are required for the viability of CAF-1 deficient cells. (A) Genetic interaction of CAC1 with ECO1. Left, fifty tetrads from eco1Δ/ECO1 cac1Δ/CAC1 rad61Δ/RAD61 diploid strain were dissected. Tetrads were grown at 30° for 5 days. Dashed circles indicate eco1Δ cac1Δ rad61Δ mutant. Right, top, tenfold serial dilutions of indicated genotypes were spotted onto YPD plates and incubated at 30° for 3 days. Right, down, tenfold serial dilutions of wild-type, cac1Δ, eco1-1, and eco1-1 cac1Δ cells were spotted onto YPD plates and incubated at 25° (left), or 36°C (right) for 3 days. (B) Genetic interaction of CAC1 with SCC2. Tenfold serial dilutions of exponentially growing cells were spotted onto YPD plates and incubated at 25° (left), or 32° (right) for 3 days. (C) Genetic interaction of CAC1 with SCC1. Tenfold serial dilutions of exponentially growing cells were spotted onto YPD plates and grown at 25° (left), or 32° (right) for 3 days. |
Ctf4 interacts with Mms22 [82] [84] [90], an adaptor protein of the Rtt101-Mms1 E3 ubiquitin ligase complex to tether the Rtt101-Mms1-Mms22 E3 ubiquitin ligase to active replisome during S-phase [86]. Previous studies showed that Rtt101-Mms1-Mms22 inactivation is epistatic with ctf4Δ regarding cohesion defects [37]. We first assessed the importance of the ubiquitin ligase in ctf4Δ cells and found that its absence did not significantly affect growth (Figure S7). We next addressed the consequences of deleting RTT101, MMS1, or MMS22, in cac1Δ, scc1-73, and cac1Δ scc1-73 cells. We found that at 30°C, the growth of cac1Δ rtt101Δ, cac1Δ mms1Δ, and cac1Δ mms22 double mutants was not strongly affected compared to one of the rtt101Δ, mms1Δ, and mm22Δ single mutants (Figure 5A, B, C, left). These results strongly suggest that sister chromatid cohesion established through the interaction between Rtt101-Mms1-Mms22 E3 ubiquitin ligase and Ctf4 has little or no involvement in the growth defect observed for cac1Δ ctf4Δ cells. Moreover, we found that impairment of cohesin function dramatically affected cac1Δ viability compared to rtt101Δ or mms1Δ (Figure 5A, B, left). We observed a higher impact for MMS22 inactivation compared to RTT101 or MMS1 inactivation. This may be explained by the multiple roles of Mms22 in response to DNA damage [31] [92] [96] and/or by an Mms22 role in promoting cohesion through its direct interaction with Eco1 [37] [39]. Interestingly, at the permissive temperature for the scc1-73 mutant (25°C, right), we observed that scc1-73 cac1Δ rtt101Δ, scc1-73 cac1Δ mms1Δ, and scc1-73 cac1Δ mms22Δ cells are more affected than each of the double mutants, suggesting that CAF-1 could act in parallel with the Scc1 and Rtt101-Mms1-Mms22 cohesion pathways. Taken together these genetic analyses indicate that the growth defect of cac1Δ ctf4Δ cells is not the consequence of cohesion defects caused by a deficiency in the Rtt101-Mms1-Mms22-Ctf4 replication-coupled sister chromatid cohesion pathway, but rely on another pathway independent of the Rtt101-Mms1-Mms22 E3 ubiquitin ligase complex.
–
 | FIGURE 5: Cac1 functions in a different pathway from that of Rtt101-Mms1-Mms22 in cohesion. Genetic interactions among RTT101, MMS1, MMS22, SCC1, and CAC1. (A) Effect associated with RTT101 inactivation on viability of cac1Δ, scc1-73, and cac1Δ scc1-73 cells. (B) Effect associated with MMS1 inactivation on viability of cac1Δ, scc1-73, and cac1Δ scc1-73 cells cells. (C) Effect associated with MMS22 inactivation on the viability of cac1Δ, scc1-73, and cac1Δ scc1-73 cells. Genetic interactions were assessed by spotting a tenfold series dilution of cells of the indicated genotype onto YPD. Cells were grown at 30° for 3 days (left), or at 25°C (right) for 4 days. |
Cac1 is required for sister chromatid cohesion in ctf4Δ cells
In contrast to ctf4Δ cells, which present important cohesion defects [129], cac1Δ cells exhibit almost none or only moderate cohesion defects according to previous studies [37] [121]. To address whether the synthetic growth defect of cac1Δ ctf4Δ cells could be due to an additive effect on sister chromatid cohesion, we compared cohesion in metaphase-arrested cac1Δ and ctf4Δ cells to cohesion in cac1Δ ctf4Δ double mutant. To evaluate sister chromatid cohesion, we used a haploid strain containing Lac operator tandem repeats integrated at a site near the centromere of chromosome III and expressing a GFP-Lac repressor fusion protein. We found that ctf4Δ cells exhibited cohesion defects and that cohesion was not significantly affected by the absence of CAF-1, suggesting that CAF-1 is not important for cohesion in a wild-type context. However, cohesion defects in the cac1Δ ctf4Δ double mutant were significantly more severe than that in ctf4Δ single mutant (Figure 6). While we do not know the reason behind this increased cohesion defect, this result together with our genetic analyses, strongly suggests that, when cohesion is severely affected, CAF-1 exerts an important role required for cohesion maintenance and cell viability.
–
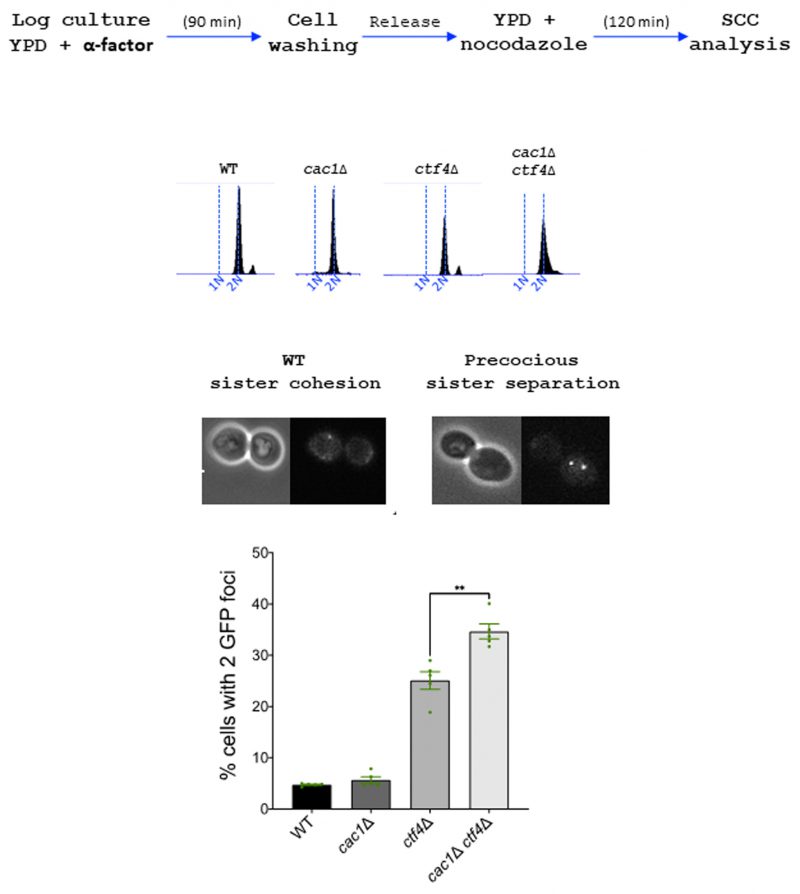 | FIGURE 6: Cac1 is important to maintain cohesion in ctf4Δ cells. Sister chromatid cohesion was analyzed by monitoring the tagged centromere of chromosome III. Top, experimental design. DNA replication was monitored by FACS analysis of DNA content. Over 100 cells were counted for each experiment. The results represent the average of five independent experiments. Statistical significance was measured using Two Tailed Mann-Whitney test. |
Cohesin association to chromatin is increased in the absence of Cac1 in WT and ctf4Δ cells
We next investigated if the level of Scc1, at known cohesin binding sites on chromatin, was affected in the absence of CAF-1 function by performing ChIP experiments in WT and cac1Δ cells. To our surprise, we found that deletion of CAC1 markedly increased cohesin levels both at centromeres and chromosome arms (Figure 7A). Cohesin enrichment is enhanced genome-wide in response to DSB induction and this enrichment at undamaged sites globally tight-ens sister chromatid cohesion [124] [130] [131]. Taking into account that CAF-1 plays multiple roles in maintaining genome stability [3] [6] [7] [11] [13] [96] [98] [122] and that the absence of Cac1 in both wild-type and ctf4Δ cells increased Rad52 foci (Figure 3A, left), Rfa1 foci (Figure 3A, right) [7], and γH2AX (Figure 3B), a possible explanation could be that DNA-damage-induced cohesion establishment was the source of the high cohesin level arising in absence of Cac1. Because Chk1, which mediates the DNA damage response in parallel with RAD53, is a key component of the damage-induced cohesion establishment pathway required for the generation of damage-induced cohesion [134], we analyzed cohesin levels in absence of Chk1 in WT and cac1Δ cells. We observed that the cohesin levels were indistinguishable between chk1Δ and wild-type cells and found that deletion of CHK1 in cac1Δ mutant reduced the association of Scc1 at both centromeres (CEN3, CEN9) and chromosome arms (PAO1) (Figure 7B). This indicates that DNA damage-induced cohesion establishment is the source of the increased Scc1 level observed in absence of CAC1.
–
Cohesin association with centromeres, promoters, DSBs, and stalled replication forks depends on the Scc2-Scc4 complex [41] [45] [135] [136]. To determine whether the Scc2-Scc4 complex was involved in the higher level of cohesins observed in cac1Δ cells, we used the scc2-4 thermosensitive mutant and compared the level of Scc1 at centromeres and chromosome arms in wild-type, scc2-4, cac1Δ and cac1Δ scc2-4 cells. To this end, we synchronized cells by α-factor pheromone block, released them into S-phase at 25°C, and subsequently cultivate them at the restrictive temperature for the scc2-4 mutation in the presence of nocodazole to arrest cells in G2/M. As expected, at restrictive temperatures, cohesin levels were strongly decreased in scc2-4 mutant compared to wild-type cells (Figure 7C). Interestingly, our chromatin immunoprecipitation experiments revealed similar cohesin levels both at centromeres and chromosome arms of scc2-4 and scc2-4 cac1Δ cells, revealing that the increased level of cohesin association observed in cac1Δ cells depends on the cohesin loader Scc2 (Figure 7C).
–
We further compared the level of Scc1 on chromatin in WT, cac1Δ, ctf4Δ, and cac1Δ ctf4Δ cells. As expected we found that Scc1 binding at chromatin was greatly reduced in ctf4Δ cells [77] [121]. Compared to cac1Δ cells, cac1Δ ctf4Δ cells exhibited a slight reduction in Scc1 binding, which interestingly, remained much higher than in ctf4Δ mutant and WT (Figure 7D). The fact that Scc1 occupancy is higher in cac1Δ ctf4Δ double mutant compared to ctf4Δ single mutant while the double mutant exhibits a stronger cohesion defect, suggests that cohesins are not fully functional in cac1Δ ctf4Δ cells, possibly because they are not able to reach an efficient cohesive state when ctf4Δ mutation is combined with CAC1 gene deletion.
–
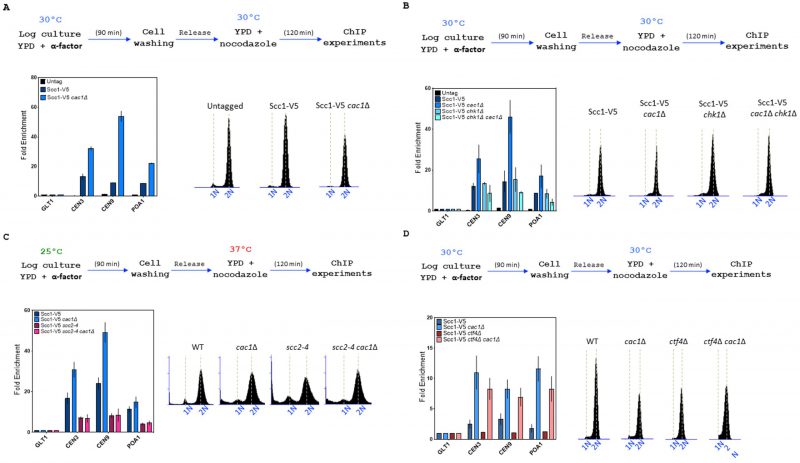 |
FIGURE 7: CAC1 inactivation increases cohesin level at chromatin in WT and ctf4Δ cells. Top, experimental design. In all experiments, DNA replication was monitored by FACS analysis of DNA content. (A) CAC1 inactivation increases cohesin level at centromere and chromosome arm. Cells of the indicated genotype were synchronized in G1 and were released into nocodazole imposed mitotic arrest for 120 minutes. Scc1 levels at two centromeres (CEN3 and CEN9), a chromosome arm cohesin binding site (POA1), and a negative control-binding site (GLT1) were measured by ChIP, followed by real-time qPCR. SEM shown represents four independent experiments. (B) The increased cohesin level at chromatin observed in absence of CAC1 is due to DNA damage. ChIP-qPCR analyses of Scc1 level at centromere and chromosome arm in WT, cac1Δ, chk1Δ, and chk1Δ cac1Δ. Same experimental conditions as in (A). (C) ChIP-qPCR analyses of Scc1 level at centromere and chromosome arm in WT, scc2-4, cac1Δ, and cac1Δ scc2-4. (D) CAC1 inactivation increases cohesin level at centromere and chromosome arm in absence of CTF4. |
Cac1 is required for the efficient acetylation of Smc3
Cohesion establishment mainly depends on Eco1 which acetylates the Smc3 cohesin subunit at lysine 112 (K112) and 113 (K113) both during S phase and independently of DNA replication [52] [53] [54] [137] [138]. To further understand the genetic relationship between CAC1 and CTF4 related to cohesion, we investigate whether CAF-1 contributed to Smc3ac in WT and ctf4Δ cells. We analyzed Smc3ac after a nocodazole-imposed mitotic arrest by Western blot using a validated antibody. As expected from previous data [121], we observed that the Smc3-K112,113 Eco1-dependent acetylation is strongly diminished in ctf4Δ cells. Our quantitative Western blotting of Smc3-K112,113 acetylation (Smc3-K112,113ac) showed that cac1Δ mutation did not amplify the Smc3-K112,113ac defect observed in absence of Ctf4 but interestingly, revealed that CAF-1 deficient cells displayed partial loss of Smc3-K112,113ac (Figure 8). Because CAC1 is required for SCC in ctf4Δ mutant (Figure 6), this suggests that CAF-1 acts, at least in part, in a pathway parallel to Smc3ac to promote cohesion.
–
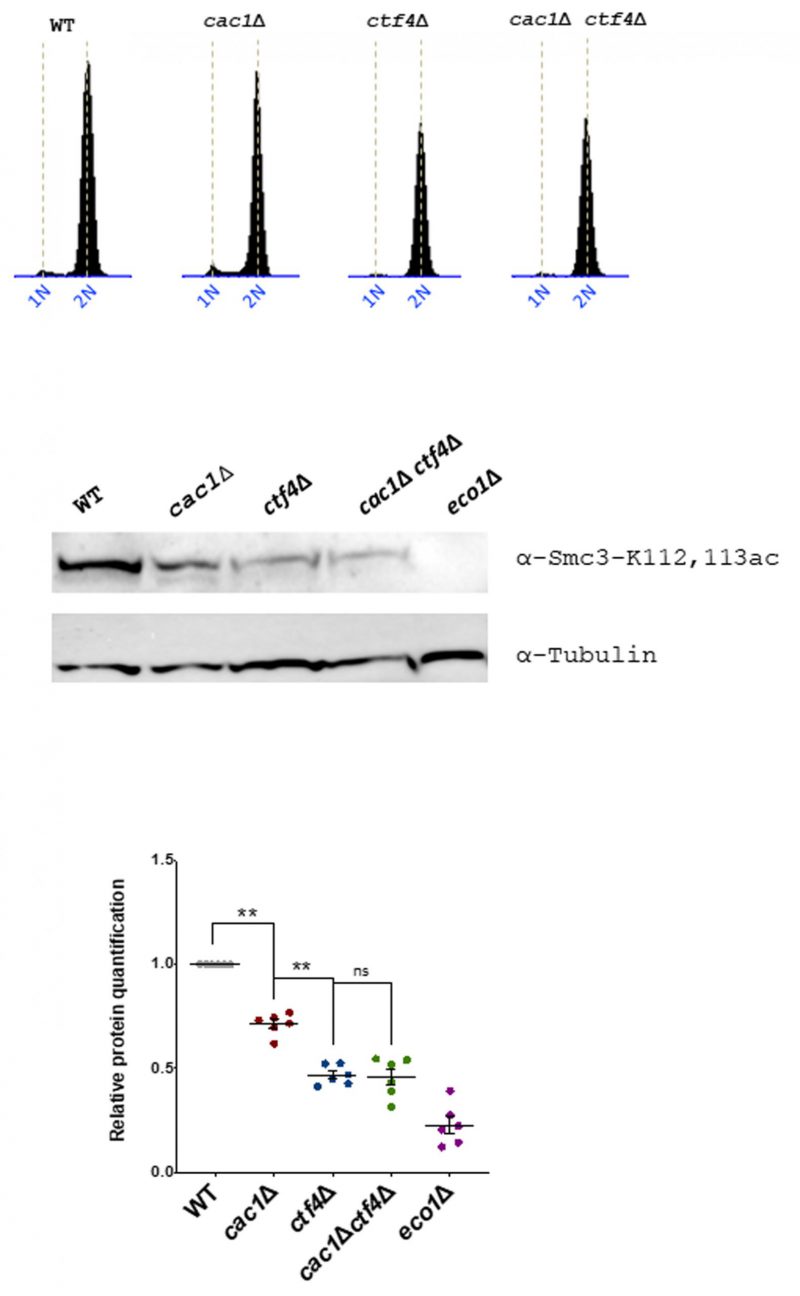 | FIGURE 8: Eco1-catalyzed Smc3 acetylation is reduced in the absence of Cac1. Cells of the indicated genotypes were synchronized in G1 using α-factor and released into nocodazole-imposed mitotic arrest at 25°. Top, FACS analysis of the DNA content was used to monitor cell cycle progression. Middle, acetylated Smc3 was immunoblotted in the cell lysate by an antibody specific to Smc3-K112,113Ac (gift from Adele Marston, The Wellcome Centre for Cell Biology, Edinburgh, UK). Down, quantitative Western blotting analyses of Smc3-K112,113 acetylation. The results represented the average of six independent experiments. Statistical significance was measured using Two Tailed Mann-Whitney test. |
DISCUSSION
H3K56 acetylation is a histone mark required for genome stability maintenance during replication stress and for chromatin assembly during replication (reviewed in [139]). In a previous study, we found that replisome function is modulated during replicative stress by H3K56ac through an interaction between Rtt101-Mms1-Mms22 and Ctf4. We revealed that Ctf4 became deleterious under replication stress in the absence of the DNA repair/tolerance branch of the H3K56ac pathway [92] but left unanswered the importance of Ctf4 when the nucleosome assembly branch of the H3K56ac pathway is affected during replication. Here we show that mutants lacking any subunit of the chromatin assembly factor CAF-1 (Cac1, Cac2, Cac3) exhibit synthetic sickness in absence of CTF4 gene, suggesting that Cac1 and Ctf4 jointly participate in an essential process of the cells. To uncover this essential process, we performed genetic analyses. We observed that ctf4Δ is deleterious when combined with mutations affecting various histone chaperones known to be involved in replication-coupled chromatin assembly, and with mutations at H3 or H4 histone lysine residues implicated in nucleosome assembly during replication. On the contrary, inactivation of the HIR complex, which is involved in replication-independent nucleosome assembly, as well as deletion of NAP1, encoding for a histone chaperone involved in H2A and H2B histones deposition, does not induce a growth defect in ctf4Δ cells. These data highlight the crucial role of replication-coupled chromatin assembly in absence of CTF4. We further show that asf1Δ and rtt109Δ deletions caused only a modest synthetic sickness with ctf4Δ (compared to cac1Δ, cac2Δ or cac3Δ) even though they abrogate the H3K56ac pathways. Along the same line, the ctf4Δ mutant is only slightly affected by the absence of the Rtt101-Mms1-Mms22 E3 ubiquitin ligase complex which functions with Asf1 and Rtt109 in the H3K56ac pathways [25] [92]. We infer that this difference reflects the much more important role exerted by CAF-1 in chromatin assembly during replication. This assumption is reinforced by previous data showing that mutating the Rtt101-Mms1-Mms22 complex affects the association of H3-H4 with the histone chaperones Asf1 and Rtt106 but does not alter the binding of H3-H4 to CAF-1 [25], suggesting that H3 ubiquitination promotes the transfer of H3-H4 from Asf1 to Rtt106 but not to CAF-1. Moreover, nucleosome assembly proceeds in an ordered manner in the absence of Asf1, Rtt109, and H3K56ac, but appears to be severely disrupted upon deletion of CAF-1 [140]. Thus, we propose that the H3K56ac-dependent DNA repair/tolerance mechanisms (that do not require CAF-1), as well as the H3K56ac-chromatin assembly function of Rtt101-Mms1-Mms22, are not important for the growth of ctf4Δ mutant contrary to CAF-1-dependent replication-coupled nucleosome assembly function whose mutation leads to synthetic sickness in ctf4Δ cells. Taken together, our data strongly suggest that the synthetic sickness observed for CAF-1 deficient cells in absence of CTF4 is a direct consequence of nucleosome assembly defects arising during replication.
–
We show that the combined absence of CAC1 and CTF4 is lethal for cells experiencing exogenous DNA damage and that Rrm3, whose function is to assist fork progression across pausing sites, is essential for the viability of cac1Δ ctf4Δ cells. In addition, we show that both the S-phase and the spindle-assembly checkpoints are required for cac1Δ ctf4Δ viability. We also report a much higher frequency of spontaneously arising Rfa1 and Rad52 foci, an increased level of γH2AX, as well as an increased level of spontaneous mutation rate in cac1Δ ctf4Δ cells compared to every single mutant and wild-type cells. Therefore, we conclude that the absence of CAF-1 function causes an important genetic instability in ctf4Δ cells leading to the emergence of toxic DNA structures and/or DSB during replication, that needs to be repaired by homologous recombination.
–
Ctf4 has been implicated in multiple chromosomal functions [77] [81] [82] [84] [85] [92] [93] [119] [141] [142]. Our genetic analysis conducted to uncover which function of Ctf4 is crucial in absence of CAC1 reveals that the functions of Ctf4, in maintaining normal replisome architecture, in promoting the coordination of leading and lagging strands during replication, and in DNA damage tolerance, are not responsible for the sickness observed for cac1Δ ctf4Δ cells. In addition, contrary to DNA-replication coupled nucleosome assembly, the transfer of parental histones to lagging strands during replication promoted by Ctf4 is not required for the growth of cac1Δ ctf4Δ cells. These findings suggest that the problem arising in cac1Δ ctf4Δ mutant lies in the deposition, during replication fork progression, of H3-H4 histones synthesized de novo rather than in recycling parental histones.
–
Ctf4 is also required for cohesion, which is established at the time of replication. Two parallel pathways for cohesion establishment at the replication fork, involving non-essential genes encoding for proteins associated with replisomes, one containing CSM3, TOF1, CTF4, and CHL1, and the second containing MRC1, CTF18, CTF8, and DCC1, have been defined [69]. Our genetic analyses revealed that, among the different genes involved in these parallel pathways, CTF4 is the only gene required for the growth of cac1Δ mutant. We also found that the E3 ubiquitin ligase complex Rtt101-Mms1-Mms22, known to interact and function with Ctf4 in sister chromatid cohesion [37], is not required for the growth of cac1Δ. At first sight, these results suggest that the defective process leading to cac1Δ ctf4Δ sickness is unrelated to Ctf4 function in cohesion. Ctf4 physically interacts and recruits Chl1 to the replisome which in turn interacts with cohesin to promote cohesion [77]. Interestingly, unlike CHL1 deletion, and unlike deletion of genes encoding for the other establishment factors, CTF4 deletion is lethal in eco1Δ (rad61Δ/wpl1Δ) mutant cells [121] that are strongly affected in Smc3-K112,113ac and as a consequence severely defective for sister chromatid cohesion [55]. This suggests that Ctf4 also acts, at least in part, in a pathway parallel to Smc3ac and independently of the Chl1 pathway. Thus, we reasoned that Ctf4 function in SCC is more important than the one exercised by each of the other proteins encoded by the non-essential genes involved in SCC. In that case, the deficient cohesion function of ctf4Δ mutant could be responsible for the sickness observed during the combined absence of CAC1 and CTF4. Indeed, cell fractionation experiments have suggested that Ctf4 helps the chromatin recruitment of Ctf18-RFC [56], a complex involved in the de novo loading of cohesin onto nascent DNAs through a CTF4-independent pathway required for normal cohesion and cell viability in the absence of Ctf4 [74]. Moreover, Ctf4 also recruits the Rtt101-Mms1-Mms22 complex to the replisome through its interaction with Mms22, a protein that promotes Eco1 recruitment at the DNA replication fork and subsequently cohesion [37]. Thus, because Ctf4 plays a major role in cohesion establishment through multiple pathways, we propose that cac1Δ ctf4Δ sickness is a direct consequence of a role for CAF-1 function that is revealed when cohesion is affected. Such hypothesis is strongly reinforced by our findings revealing that CAC1 inactivation increases the cohesion defect of ctf4Δ cells, and by the strong negative synthetic genetic interactions detected between caf-1 mutants and mutations affecting essential cohesion genes (Figure 9A, B). Our findings show that mutations abolishing H3K56ac (asf1Δ and rtt109Δ) or Rtt101-Mms1-Mms22 E3 complex function (rtt101Δ, mms1Δ, or mms22Δ) do not significantly affect the growth of ctf4Δ cells contrary to CAF-1 mutations and mutations at genes playing a key role in DNA-replication coupled nucleosome assembly. We thus assume that CAF-1 function in cohesion is independent of the H3K56ac-Rtt101-Mms1-Mms22-Ctf4-dependent pathway previously described by Zhang and colleagues [37]. This pathway is known to protect against replicative damage by DNA repair/tolerance mechanisms, in a CAF-1-independent manner [27] [92]. Moreover, based on co-precipitation analyses showing that the Rtt101-Mms1-Mms22-dependent H3 ubiquitination affects H3-H4 association with Asf1 and Rtt106 but not with CAF-1 [25], our data showing that CAF-1 inactivation affects scc1-73 cohesin mutant viability much more strongly than Rtt101-Mms1 E3 ubiquitin ligase inactivation, led us to propose that the chromatin function of CAF-1 required for cell viability in presence of SCC defects is independent of H3K56ac and Rtt101-Mms1-Mms22.
–
We further report that deleting CAC1 increases the level of Scc1 on chromatin. Consistent with the observation that cac1Δ cells exhibit higher levels of Rfa1 spontaneous foci, of Rad52 spontaneous foci, and of γH2AX which recruits the cohesin loader at DNA double-strand breaks [143], we found that the increased Scc1 level observed in the cac1Δ mutant requires an intact DNA damage response. Thus, it is possible that the increased Scc1 level, induced in response to DNA damage observed in cac1Δ cells, is sufficient to maintain a wild-type level of cohesion. This may explain, in agreement with previous work [121], the absence of SCC defect that we observed in CAF-1 deficient cells. Indeed, we observed that deleting CAC1 leads to a WT level of Scc1 both at centromeres and chromosome arms in the absence of the cohesin loader complex Scc2-Scc4. Because both cohesin loading and cohesin translocation on chromatin depend mainly on Scc2 and Scc4 proteins [144] [145] it is possible that the abnormal level of cohesin observed in the absence of Cac1 is the consequence of an abnormal cohesin redistribution in response to DNA damage. Nucleosomes inhibit cohesin loading [42] and it has been shown that fewer nucleosomes are deposited on replicated DNA in CAF-1 deficient cells [146]. Thus, it is possible that the temporal delay in nucleosome assembly and the increased inter-nucleosome spacing in nascent chromatin arising in CAF-1 mutants [147] [148] alter chromatin structure and cause replicative DNA damages that facilitates cohesin loading.
–
We further found that deleting CAC1 in ctf4Δ mutant also increased Scc1 level on chromatin. How could CAF-1 inactivation in ctf4Δ cells simultaneously increase cohesin levels on chromatin and cohesion defects? Ctf4 is essential for converting cohesin associated with un-replicated DNA into functional cohesive structures [74] and to recruit Mms22 to the replisome [86], a protein that in turn, can recruits Eco1 at the DNA replication fork [37] [39]. A simple explanation could be that, although the level of cohesin increases in cac1Δ ctf4Δ mutant compared to ctf4Δ mutant, the level of functional cohesins is reduced in cac1Δ ctf4Δ mutant due to CAF-1 inactivation.
–
What could be the function of CAF-1 in cohesion? We do not know whether the observed effects are only the consequence of nucleosome formation defects during replication or if CAF-1 plays a direct role in SCC. So far, no physical interaction between CAF-1 and any cohesin establishment/maintenance factors nor cohesin proteins has been identified. However, it has been shown that the over-expression of CAF-1 subunits can suppress the non-viability of temperature sensitivity of eco1 mutant at restrictive temperatures [37], and we have shown that CAF-1 inactivation negatively affects Smc3-K112,113ac, indicating that CAF-1 can influence cohesin acetylation at the replication fork during S phase, and suggesting that CAF-1 could function directly in cohesion establishment. CAF-1 and Eco1 both interact with PCNA through a PIP-box, and this interaction is crucial for Eco1 to promote cohesion establishment as well as for CAF-1 to sustain the viability of cohesin mutants. One possibility is that CAF-1 facilitates the recruitment and/or the stabilization of the acetyltransferase Eco1 at the replication fork. It was recently nicely shown that two transient DNA structures that form during Okazaki fragment maturation promotes cohesin acetylation to stabilize newly established sister chromatid cohesion [58]. Okazaki fragments processing and nucleosome assembly are interlinked [140] [147]. Depletion of CAF-1, but not the absence of H3K56ac, completely ablates the nucleosome-sized periodicity of Okazaki fragments, and generates fewer but longer Okazaki fragments [140]. It is possible that the lower nucleosome density and the longer Okazaki fragments generated in the absence of CAF-1 affect the surrounding and/or the access of proteins required for cohesion, leading to a defective Smc3ac and to cohesion defects.
–
Our genetic pieces of evidence argue that the CAF-1-related defects in SCC are additive with those arising from improper loading of cohesin (scc2-4 mutant), from improper stabilization of cohesin (eco1 and scc1-73 mutants), and improper activation of cohesin (ctf4Δ mutant). This favors the idea that CAF-1 is involved in SCC through a cohesin-independent pathway that is additive with the cohesin-mediated pairing pathways. CAF-1 could participate in the maintenance of cohesion through its capacity to create a chromatin structure that maintains sister chromatids in proximity when cohesion is affected. If so, this chromatin structure mediated by CAF-1 should be established during fork progression because we have shown that the interaction between CAF-1 and PCNA, which recruits CAF-1 at replication forks, is crucial for the viability of scc1-73 mutant in the presence of cohesion defects. Disruption of the interaction between PCNA and CAF-1 causes silencing defects [14] [16]. CAF-1 contributes to the maintenance of silencing independently of Asf1 (and H3K56ac) at the transcriptionally silent HML loci [149] and is important to ensure the inheritance of the appropriate epigenetic state [12] [13]. CAF-1-mediated chromatin structure may help the recruitment of a specific factor not directly involved in cohesion, but that favors cohesion. Cac1 directly interacts with the heterochromatin protein Sir1 which interacts with the origin recognition complex ORC [150] [151] and contributes to silencing. Interestingly, ORC mutants do not exhibit any cohesion defects by themselves but genetic analyses revealed an interaction between the ORC genes and SCC genes [152] [153] [154]. ORC is involved in Smc3ac [153], and similarly to caf-1 mutations, orc mutations are additive with eco1-1 and cohesin defects [152] [153]. Thus, it may be that CAF-1 similarly to ORC maintains a global chromatin structure that is not important for cohesion establishment and maintenance but is required to maintain sister chromatids in proximity in the presence of important cohesion defects (Figure 9C).
–
In summary, we have demonstrated that CAF-1 (but not H3K56ac) is crucial to guard genome stability in the absence of the replisomal protein Ctf4. We show that yeast lacking both CAC1 and CTF4 present an increased mutation rate, and require the S-phase and the spindle checkpoint pathways as well as HR to survive, revealing the presence of important damages. Furthermore, our detailed genetic analyses demonstrate that CAF-1 is required for cell growth in the presence of SCC defects and highlight the major role played by Ctf4 in cohesion. We also point out that the absence of CAF-1 increases the level of cohesin on chromatin and reduces cohesin acetylation. This work reveals novel roles for CAF1 related to its nucleosome assembly function, in the maintenance of genome stability.
–
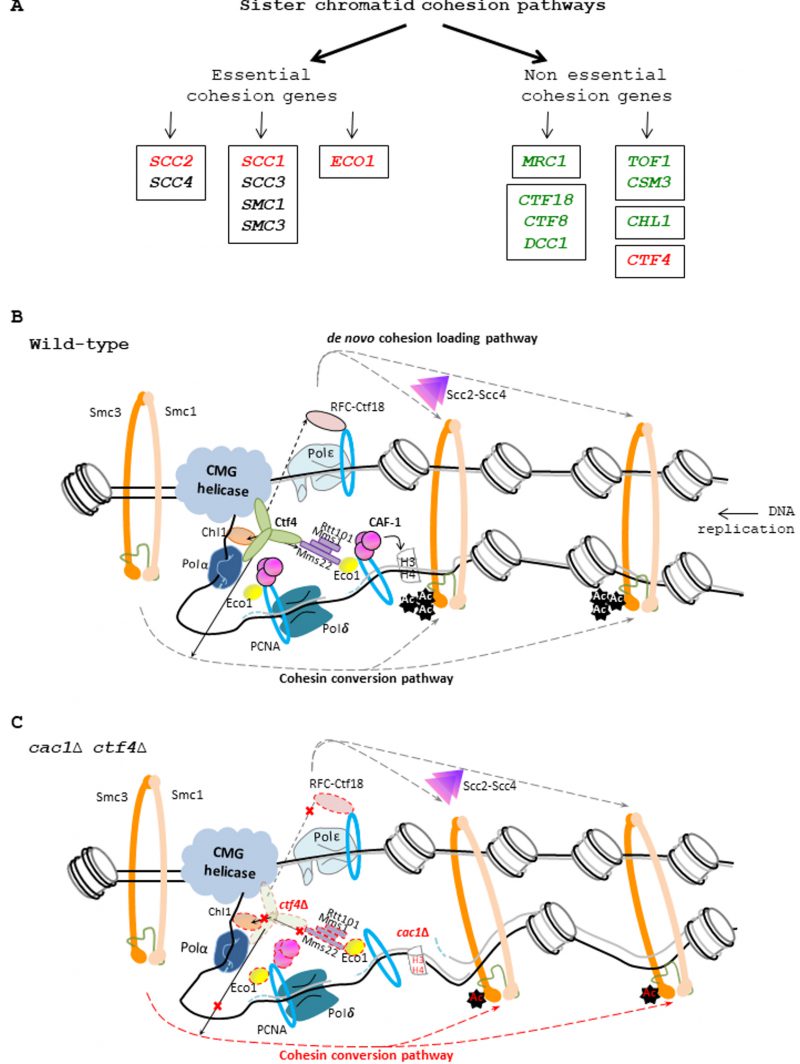 |
FIGURE 9: CAF-1 and the replisomal protein Ctf4 are important for cohesion. (A) Genetic interactions between CAF-1 complex and genes involved in sister chromatid cohesion. Left, essential cohesion genes. Right, non-essential cohesion genes encoding replisome-associated proteins that are essential for efficient Smc3 acetylation by Eco1, and for cohesion establishment [69] [121]. The Mrc1, CTF18-RFC pathway is involved in de novo loading of cohesins onto nascent DNAs [74]. The Tof1/Csm3, Chl1, Ctf4 pathway is involved in conversion of chromosome associated cohesins into cohesive structure during S phase [74]. Genes enclosed encode protein to form a complex. Genes in green are not required for growth in absence of CAF-1 function. Genes in red are required for growth in absence of CAF-1 function. For essential genes, we used thermosensitive mutants to analyze the genetic interactions with CAF-1 mutants. Genes in black: Genetic interaction with CAF-1 mutants non-determined. (B) Illustration of the role played by Ctf4 in the SCC establishment. Ctf4 links DNA replication with sister chromatid cohesion establishment by recruiting Chl1 helicase to the replisome where it directly interacts with cohesins and assists Eco1, which acetylates Smc3, in replication coupled-cohesin establishment [38] [77]. Ctf4 interacts and tethers the Rtt101-Mms1-Mms22 E3 ubiquitin ligase to the replisome which in turn recruits and/or promotes Eco1-dependent sister chromatid cohesion [37] [86]. Ctf4 can also help the chromatin recruitment of Ctf18-RFC [56] a complex that, loads and unloads PCNA, could act as a binding platform for recruiting Eco1, and is involved with the cohesin loader Scc2-Scc4 in the de novo loading of cohesins onto nascent DNA [74]. Finally, Ctf4 is also required for the conversion of cohesins rings preloaded onto the DNA template into a cohesive form [74]. (C) Proposed model explaining the importance of CAF-1 in ctf4Δ cells. In absence of Ctf4, multiple cohesion establishment pathways are affected (red cross) leading to major defects in SCC establishment. In absence of CAF-1 fewer nucleosomes are deposited on replicated DNA, generating fewer but longer Okazaki fragments [140], leading to increased inter-nucleosome spacing in nascent chromatin and inappropriate epigenetic states [13] [148]. In these conditions, the nucleosome assembly function of CAF-1 is required to maintain SCC and efficient cell growth in yeast affected in cohesion. In ctf4Δ cac1Δ cells, the altered chromatin structure arising in absence of CAF-1 function increases the SCC defects induced by the absence of Ctf4, leading to severe growth defects and genomic instability. |
MATERIALS AND METHODS
Strain construction
All strains used in this study are presented in Table S1. To obtain gene deletions we amplified by PCR a disruption cassette containing the appropriate marker, as described previously [155].
–
Spore viability and growth
Diploid strains were sporulated at 25°C during 3 days on solid sporulation media and treated with 3 μl of 1 mg/ml zymolyase 20T (Seikagaku Biobusiness, Japan) during 10 minutes in water before tetrads were dissected on rich media (YPD plate). We used a MSN400 micromanipulator from SINGER Instruments. Viable colonies were scored 3 days or 5 days later. An average of 50 tetrads were dissected. The number of tetrads analyzed is denoted in the figure legends. Quantification of the spore growth was done by image analysis of area of growth using Image J.
–
Fluorescence microscopy
Microscopy analyses were carried out in liquid media supplemented in adenine using a Nikon Eclipse Ti microscope with a 100x objective. We used a Neo sCMOS camera (Andor) to collect the images. The exposure time was DIC: 500 ms; CFP: 75 ms, YFP: 75 ms, and GFP 75 ms. We used ImageJ to analyze the images on 2D-maximum projections from 11-Z-stacks spaced 0.5 μ each. All the cells analyzed were prepared by growing the cells at 30°C in YPD media supplemented in adenine.
–
Mutation rate analysis
Yeast cells were grown overnight in liquid YPD medium at 30°C. Cells were then diluted to 0.2 OD600nm in 1 ml of water. Tenfold serial dilutions were done in water and each dilution was plated on YPGal plates to induced TUS expression and obtain separated colonies after 3 days growth at 30°C. YPGal plates were then replica plated twice onto 5-FOA plates to confirm the 5-FOA resistant phenotype of the growing colonies. Mutation rates were measured by fluctuation analysis [156] [157]. Statistical analyses were done on n=5 independent experiments using two-tailed Mann-Whitney test.
–
Chromatin Immunoprecipitation (ChIP)
We used the following steps to prepare chromatin samples: We first crosslinked the cells for 15 minutes with formaldehyde (1%) and used glycine (125 mM) to quench the reaction for 5 minutes. Cells were then lysed in 50 mM HEPES-KOH [pH7.5], 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate (FA lysis buffer) supplemented with cocktail of protease inhibitors by vortexing cells with glass beads (6 cycles × 20 s, with cooling between the cycles). After a centrifugation to remove debris, we then used a Bioruptor Pico sonicator to share the chromatin to around 200 bp. Insoluble materials were removed by centrifugation for 10 minutes (14,000 rpm, 4 °C). We mixed 500 μg of chromatin and the recommended amount of anti-V5 and conducted the immunoprecipitation overnight. We next added 25 μl of FA lysis buffer containing Protein G-Sepharose beads (3 hours incubation at 4 °C). Protein G-Sepharose beads were next washed successively once with FA buffer and twice with a FA buffer containing 500 mM NaCl. We next washed the protein G-Sepharose beads two times using a wash buffer composed of 10 mM Tris-HCl [pH 8.0], 0.25 M LiCl, 1 mM EDTA, 0.5% NP-40, 0.5% Na-Deoxycholate and one time with a TE buffer (10 mM Tris-HCl [pH8.0], 1 mM EDTA). The precipitated materials were eluted after incubation at 65 °C (10 minutes) in a buffer containing 50 mM Tris-HCl [pH7.5], 10 mM EDTA and 1% SDS. The decrosslinking step was performed overnight at 65 °C. We used the Invisorb Fragment CleanUp Kit to purify the DNA fragments.
–
Quantitative PCR analysis (qPCR)
All the qPCR experiments realized for individual gene analysis were conducted with the BioRad CFX384 qPCR machine using the following parameters: Five minutes at 95°C followed by forty cycles (15 s at 95 °C + 15 s at 50 °C + 40 s at 72 °C), followed by ten minutes at 95°C. The oligonucleotides used for the qPCR reactions are listed in Table S2.
–
Western blotting
Cells were grown in YPD and blocked in G2 as described previously. 10 ml cultures were collected and crosslinked with 1% formaldehyde for 10 minutes followed by quenching using 1.2M glycine. Pellets were resuspended in 5% TCA (5ml) and left on ice for 10 minutes, the pellets were snap frozen in liquid nitrogen and resuspended in 1 ml acetone at room temperature. The pellets are left to dry for at least 3 hours, and then resuspended in 100μl lysis buffer (50 mM Tris pH7.5, 1 mM EDTA pH7.5) 2.75 μl; 1M DTT, 20 μl 50x protease inhibitors). Glass beads were added to break cells in fast prep 3x45secs. 50 μl of 3xSDS sample buffer was added to the lysate followed by immediate heating at 95°C for 5 min, cooled and centrifuged before loading onto SDS-PAGE gels (8–10%). PAGE was carried out using a Bio-Rad Mini Trans-Blot System (Bio-Rad) in SDS running buffer (25 mM Tris, 190 mM glycine, 0.01% SDS). SDS-PAGE gels were transferred onto nitrocellulose membrane (0.45 μM, Amersham-GE Healthcare, Amersham, UK) in transfer buffer (25 mM Tris, 1.5% glycine, 0.02% SDS, 20% EtOH) in a Bio-Rad Mini Trans-Blot system. Membranes were blocked in 5% milk in PBS with 0.05% Tween20 (PBST) for at least 1 hr at room temperature before incubating in primary antibody in 2% milk/PBST overnight at 4°C. Membranes were washed in PBST three times for 15 min, incubated in secondary antibody in 2% milk/PBST for overnight at 4°C, and washed in PBST three times. Signals were detected with Amersham ECL detection reagents (RPN2105; Cytiva) and images were directly acquired with a ChemiDoc MP Imaging System (Bio-Rad). Primary antibodies used were rabbit anti-Smc3-K112,113Ac (a kind gift from Pr Adele Marston), mouse anti-V5 (Invitrogen), and rat anti-tubulin (Santa-Cruz). The ECL signals were quantified using Image Lab 6.0 (Bio-Rad). The quantification of the relative levels of proteins were calculated by normalizing the ratio signals of Smc3K112,113ac to tubulin. The statistical analyses were performed using one-tailed Mann-Whitney test.
REFERENCES
- Almouzni G, and Méchali M (1988). Assembly of spaced chromatin promoted by DNA synthesis in extracts from Xenopus eggs. EMBO J. 7(3): 665–672. 10.1002/j.1460-2075.1988.tb02861.x
- Kaufman PD, Kobayashi R, Kessler N, and Stillman B (1995). The p150 and p60 subunits of chromatin assembly factor I: a molecular link between newly synthesized histones and DNA replication. Cell. 81(7): 1105–1114. 10.1016/s0092-8674(05)80015-7
- Kaufman PD, Kobayashi R, and Stillman B (1997). Ultraviolet radiation sensitivity and reduction of telomeric silencing in Saccharomyces cerevisiae cells lacking chromatin assembly factor-I. Genes Dev. 11(3): 345–357. 10.1101/gad.11.3.345
- Verreault A, Kaufman PD, Kobayashi R, and Stillman B (1996). Nucleosome assembly by a complex of CAF-1 and acetylated histones H3/H4. Cell. 87(1): 95–104. 10.1016/s0092-8674(00)81326-4
- Tyler JK, Adams CR, Chen SR, Kobayashi R, Kamakaka RT, and Kadonaga JT (1999). The RCAF complex mediates chromatin assembly during DNA replication and repair. Nature. 402(6761): 555–560. 10.1038/990147
- Chan JE, and Kolodner RD (2011). A Genetic and Structural Study of Genome Rearrangements Mediated by High Copy Repeat Ty1 Elements. PLoS Genet. 7(5): e1002089. 10.1371/journal.pgen.1002089
- Tsirkas I, Dovrat D, Lei Y, Kalyva A, Lotysh D, Li Q, and Aharoni A (2021). Cac1 WHD and PIP domains have distinct roles in replisome progression and genomic stability. Curr Genet. 67(1): 129–139. 10.1007/s00294-020-01113-8
- Monson EK, de Bruin D, and Zakian VA (1997). The yeast Cac1 protein is required for the stable inheritance of transcriptionally repressed chromatin at telomeres. Proc Natl Acad Sci U S A. 94(24): 13081–13086. 10.1073/pnas.94.24.13081
- Enomoto S, and Berman J (1998). Chromatin assembly factor I contributes to the maintenance, but not the re-establishment, of silencing at the yeast silent mating loci. Genes Dev. 12(2): 219–232. 10.1101/gad.12.2.219
- Smith JS, Caputo E, and Boeke JD (1999). A Genetic Screen for Ribosomal DNA Silencing Defects Identifies Multiple DNA Replication and Chromatin-Modulating Factors. Mol Cell Biol. 19(4): 3184–3197. 10.1128/MCB.19.4.3184
- Linger J, and Tyler JK (2005). The yeast histone chaperone chromatin assembly factor 1 protects against double-strand DNA-damaging agents. Genetics. 171(4): 1513–1522. 10.1534/genetics.105.043000
- Young TJ, Cui Y, Irudayaraj J, and Kirchmaier AL (2019). Modulation of Gene Silencing by Cdc7p via H4 K16 Acetylation and Phosphorylation of Chromatin Assembly Factor CAF-1 in Saccharomyces cerevisiae. Genetics. 211(4): 1219–1237. 10.1534/genetics.118.301858
- Young TJ, Cui Y, Pfeffer C, Hobbs E, Liu W, Irudayaraj J, and Kirchmaier AL (2020). CAF-1 and Rtt101p function within the replication-coupled chromatin assembly network to promote H4 K16ac, preventing ectopic silencing. PLOS Genet. 16(12): e1009226. 10.1371/journal.pgen.1009226
- Shibahara K, and Stillman B (1999). Replication-Dependent Marking of DNA by PCNA Facilitates CAF-1-Coupled Inheritance of Chromatin. Cell. 96(4): 575–585. 10.1016/S0092-8674(00)80661-3
- Moggs JG, Grandi P, Quivy J-P, Jónsson ZO, Hübscher U, Becker PB, and Almouzni G (2000). A CAF-1–PCNA-Mediated Chromatin Assembly Pathway Triggered by Sensing DNA Damage. Mol Cell Biol. 20(4): 1206–1218. 10.1128/MCB.20.4.1206-1218.2000
- Zhang Z, Shibahara K, and Stillman B (2000). PCNA connects DNA replication to epigenetic inheritance in yeast. Nature. 408(6809): 221–225. 10.1038/35041601
- Krawitz DC, Kama T, and Kaufman PD (2002). Chromatin assembly factor I mutants defective for PCNA binding require Asf1/Hir proteins for silencing. Mol Cell Biol. 22(2): 614–625. 10.1128/MCB.22.2.614-625.2002
- Ben-Shahar TR, Castillo AG, Osborne MJ, Borden KLB, Kornblatt J, and Verreault A (2009). Two Fundamentally Distinct PCNA Interaction Peptides Contribute to Chromatin Assembly Factor 1 Function. Mol Cell Biol. 29(24): 6353–6365. 10.1128/MCB.01051-09
- Masumoto H, Hawke D, Kobayashi R, and Verreault A (2005). A role for cell-cycle-regulated histone H3 lysine 56 acetylation in the DNA damage response. Nature. 436(7048): 294–298. 10.1038/nature03714
- Recht J, Tsubota T, Tanny JC, Diaz RL, Berger JM, Zhang X, Garcia BA, Shabanowitz J, Burlingame AL, Hunt DF, Kaufman PD, and Allis CD (2006). Histone chaperone Asf1 is required for histone H3 lysine 56 acetylation, a modification associated with S phase in mitosis and meiosis. Proc Natl Acad Sci U S A. 103(18): 6988–6993. 10.1073/pnas.0601676103
- Driscoll R, Hudson A, and Jackson SP (2007). Yeast Rtt109 Promotes Genome Stability by Acetylating Histone H3 on Lysine 56. Science. 315(5812): 649–652. 10.1126/science.1135862
- Han J, Zhou H, Horazdovsky B, Zhang K, Xu R-M, and Zhang Z (2007). Rtt109 acetylates histone H3 lysyne 56 and functions in DNA replication. Science. 315(5812): 653–655. 10.1126/science.1133234
- Han J, Zhou H, Li Z, Xu R-M, and Zhang Z (2007). Acetylation of lysine 56 of histone H3 catalyzed by RTT109 and regulated by ASF1 is required for replisome integrity. J Biol Chem. 282(39): 28587–28596. 10.1074/jbc.M702496200
- Tsubota T, Berndsen CE, Erkmann JA, Smith CL, Yang L, Freitas MA, Denu JM, and Kaufman PD (2007). Histone H3-K56 acetylation is catalyzed by histone chaperone-dependent complexes. Mol Cell. 25(5): 703–712. 10.1016/j.molcel.2007.02.006
- Han J, Zhang H, Zhang H, Wang Z, Zhou H, and Zhang Z (2013). A Cul4 E3 Ubiquitin Ligase Regulates Histone Hand-Off during Nucleosome Assembly. Cell. 155(4): 817–829. 10.1016/j.cell.2013.10.014
- Li Q, Zhou H, Wurtele H, Davies B, Horazdovsky B, Verreault A, and Zhang Z (2008). Acetylation of histone H3 lysine 56 regulates replication-coupled nucleosome assembly. Cell. 134(2): 244–255. 10.1016/j.cell.2008.06.018
- Clemente-Ruiz M, González-Prieto R, and Prado F (2011). Histone H3K56 Acetylation, CAF1, and Rtt106 Coordinate Nucleosome Assembly and Stability of Advancing Replication Forks. PLoS Genet. 7(11): e1002376. 10.1371/journal.pgen.1002376
- Thaminy S, Newcomb B, Kim J, Gatbonton T, Foss E, Simon J, and Bedalov A (2007). Hst3 is regulated by Mec1-dependent proteolysis and controls the S phase checkpoint and sister chromatid cohesion by deacetylating histone H3 at lysine 56. J Biol Chem. 282(52): 37805–37814. 10.1074/jbc.M706384200
- Chen C-C, Carson JJ, Feser J, Tamburini B, Zabaronick S, Linger J, and Tyler JK (2008). Acetylated Lysine 56 on Histone H3 Drives Chromatin Assembly after Repair and Signals for the Completion of Repair. Cell. 134(2): 231–243. 10.1016/j.cell.2008.06.035
- Endo H, Kawashima S, Sato L, Lai MS, Enomoto T, Seki M, and Horikoshi M (2010). Chromatin dynamics mediated by histone modifiers and histone chaperones in postreplicative recombination: H3-K56-Ac-mediated recombination. Genes Cells. 15(9): 945–958. 10.1111/j.1365-2443.2010.01435.x
- Wurtele H, Kaiser GS, Bacal J, St-Hilaire E, Lee E-H, Tsao S, Dorn J, Maddox P, Lisby M, Pasero P, and Verreault A (2012). Histone H3 Lysine 56 Acetylation and the Response to DNA Replication Fork Damage. Mol Cell Biol. 32(1): 154–172. 10.1128/MCB.05415-11
- Tanaka A, Tanizawa H, Sriswasdi S, Iwasaki O, Chatterjee AG, Speicher DW, Levin HL, Noguchi E, and Noma K (2012). Epigenetic Regulation of Condensin-Mediated Genome Organization during the Cell Cycle and upon DNA Damage through Histone H3 Lysine 56 Acetylation. Mol Cell. 48(4): 532–546. 10.1016/j.molcel.2012.09.011
- Haber JE, Braberg H, Wu Q, Alexander R, Haase J, Ryan C, Lipkin-Moore Z, Franks-Skiba KE, Johnson T, Shales M, Lenstra TL, Holstege FCP, Johnson JR, Bloom K, and Krogan NJ (2013). Systematic Triple-Mutant Analysis Uncovers Functional Connectivity between Pathways Involved in Chromosome Regulation. Cell Rep. 3(6): 2168–2178. 10.1016/j.celrep.2013.05.007
- Muñoz-Galván S, Jimeno S, Rothstein R, and Aguilera A (2013). Histone H3K56 Acetylation, Rad52, and Non-DNA Repair Factors Control Double-Strand Break Repair Choice with the Sister Chromatid. PLoS Genet. 9(1): e1003237. 10.1371/journal.pgen.1003237
- Karányi Z, Hornyák L, and Székvölgyi L (2020). Histone H3 Lysine 56 Acetylation Is Required for Formation of Normal Levels of Meiotic DNA Breaks in S. cerevisiae. Front Cell Dev Biol. 7: 364. 10.3389/fcell.2019.00364
- Prado F, and Maya D (2017). Regulation of Replication Fork Advance and Stability by Nucleosome Assembly. Genes. 8(2): 49. 10.3390/genes8020049
- Zhang J, Shi D, Li X, Ding L, Tang J, Liu C, Shirahige K, Cao Q, and Lou H (2017). Rtt101-Mms1-Mms22 coordinates replication-coupled sister chromatid cohesion and nucleosome assembly. EMBO Rep. 18(8): 1294–1305. 10.15252/embr.201643807
- Choudhary K, and Kupiec M (2022). The cohesin complex of yeasts: sister chromatid cohesion and beyond. FEMS Microbiol Rev. fuac045. 10.1093/femsre/fuac045
- Zuilkoski CM, and Skibbens RV (2022). Integrating Sister Chromatid Cohesion Establishment to DNA Replication. Genes. 13(4): 625. 10.3390/genes13040625
- Nasmyth K, and Haering CH (2009). Cohesin: its roles and mechanisms. Annu Rev Genet. 43: 525–558. 10.1146/annurev-genet-102108-134233
- Ciosk R, Shirayama M, Shevchenko A, Tanaka T, Toth A, Shevchenko A, and Nasmyth K (2000). Cohesin's binding to chromosomes depends on a separate complex consisting of Scc2 and Scc4 proteins. Mol Cell. 5(2): 243–254. 10.1016/s1097-2765(00)80420-7
- Muñoz S, Minamino M, Casas-Delucchi CS, Patel H, and Uhlmann F (2019). A Role for Chromatin Remodeling in Cohesin Loading onto Chromosomes. Mol Cell. 74(4): 664-673.e5. 10.1016/j.molcel.2019.02.027
- Lengronne A, Katou Y, Mori S, Yokobayashi S, Kelly GP, Itoh T, Watanabe Y, Shirahige K, and Uhlmann F (2004). Cohesin relocation from sites of chromosomal loading to places of convergent transcription. Nature. 430(6999): 573–578. 10.1038/nature02742
- Tanaka H, Kubota Y, Tsujimura T, Kumano M, Masai H, and Takisawa H (2009). Replisome progression complex links DNA replication to sister chromatid cohesion in Xenopus egg extracts. Genes Cells Devoted Mol Cell Mech. 14(8): 949–963. 10.1111/j.1365-2443.2009.01322.x
- Ström L, Lindroos HB, Shirahige K, and Sjögren C (2004). Postreplicative Recruitment of Cohesin to Double-Strand Breaks Is Required for DNA Repair. Mol Cell. 16(6): 1003–1015. 10.1016/j.molcel.2004.11.026
- Ünal E, Arbel-Eden A, Sattler U, Shroff R, Lichten M, Haber JE, and Koshland D (2004). DNA Damage Response Pathway Uses Histone Modification to Assemble a Double-Strand Break-Specific Cohesin Domain. Mol Cell. 16(6): 991–1002. 10.1016/j.molcel.2004.11.027
- Gruber S, Arumugam P, Katou Y, Kuglitsch D, Helmhart W, Shirahige K, and Nasmyth K (2006). Evidence that Loading of Cohesin Onto Chromosomes Involves Opening of Its SMC Hinge. Cell. 127(3): 523–537. 10.1016/j.cell.2006.08.048
- Hinshaw SM, Makrantoni V, Kerr A, Marston AL, and Harrison SC (2015). Structural evidence for Scc4-dependent localization of cohesin loading. eLife. 4: e06057. 10.7554/eLife.06057
- Murayama Y, and Uhlmann F (2015). DNA Entry into and Exit out of the Cohesin Ring by an Interlocking Gate Mechanism. Cell. 163(7): 1628–1640. 10.1016/j.cell.2015.11.030
- Srinivasan M, Petela NJ, Scheinost JC, Collier J, Voulgaris M, B Roig M, Beckouët F, Hu B, and Nasmyth KA (2019). Scc2 counteracts a Wapl-independent mechanism that releases cohesin from chromosomes during G1. eLife. 8: e44736. 10.7554/eLife.44736
- Hinshaw SM, Makrantoni V, Harrison SC, and Marston AL (2017). The Kinetochore Receptor for the Cohesin Loading Complex. Cell. 171(1): 72-84.e13. 10.1016/j.cell.2017.08.017
- Skibbens RV, Corson LB, Koshland D, and Hieter P (1999). Ctf7p is essential for sister chromatid cohesion and links mitotic chromosome structure to the DNA replication machinery. Genes Dev. 13(3): 307–319. 10.1101/gad.13.3.307
- Tóth A, Ciosk R, Uhlmann F, Galova M, Schleiffer A, and Nasmyth K (1999). Yeast cohesin complex requires a conserved protein, Eco1p(Ctf7), to establish cohesion between sister chromatids during DNA replication. Genes Dev. 13(3): 320–333. 10.1101/gad.13.3.320
- Rolef Ben-Shahar T, Heeger S, Lehane C, East P, Flynn H, Skehel M, and Uhlmann F (2008). Eco1-dependent cohesin acetylation during establishment of sister chromatid cohesion. Science. 321(5888): 563–566. 10.1126/science.1157774
- Ünal E, Heidinger-Pauli JM, Kim W, Guacci V, Onn I, Gygi SP, and Koshland DE (2008). A Molecular Determinant for the Establishment of Sister Chromatid Cohesion. Science. 321(5888): 566–569. 10.1126/science.1157880
- Lengronne A, McIntyre J, Katou Y, Kanoh Y, Hopfner K-P, Shirahige K, and Uhlmann F (2006). Establishment of sister chromatid cohesion at the S. cerevisiae replication fork. Mol Cell. 23(6): 787–799. 10.1016/j.molcel.2006.08.018
- Moldovan G-L, Pfander B, and Jentsch S (2006). PCNA controls establishment of sister chromatid cohesion during S phase. Mol Cell. 23(5): 723–732. 10.1016/j.molcel.2006.07.007
- Minamino M, Bouchoux C, Canal B, Diffley JFX, and Uhlmann F (2023). A replication fork determinant for the establishment of sister chromatid cohesion. Cell. S0092-8674(22)01587–2. 10.1016/j.cell.2022.12.044
- Hou W, Li Y, Zhang J, Xia Y, Wang X, Chen H, and Lou H (2022). Cohesin in DNA damage response and double-strand break repair. Crit Rev Biochem Mol Biol. 57(3): 333–350. 10.1080/10409238.2022.2027336
- Uhlmann F, Lottspeich F, and Nasmyth K (1999). Sister-chromatid separation at anaphase onset is promoted by cleavage of the cohesin subunit Scc1. Nature. 400(6739): 37–42. 10.1038/21831
- Liu HW, Bouchoux C, Panarotto M, Kakui Y, Patel H, and Uhlmann F (2020). Division of Labor between PCNA Loaders in DNA Replication and Sister Chromatid Cohesion Establishment. Mol Cell. 78(4): 725-738.e4. 10.1016/j.molcel.2020.03.017
- Bermudez VP, Maniwa Y, Tappin I, Ozato K, Yokomori K, and Hurwitz J (2003). The alternative Ctf18-Dcc1-Ctf8-replication factor C complex required for sister chromatid cohesion loads proliferating cell nuclear antigen onto DNA. Proc Natl Acad Sci. 100(18): 10237–10242. 10.1073/pnas.1434308100
- Bylund GO, and Burgers PMJ (2005). Replication Protein A-Directed Unloading of PCNA by the Ctf18 Cohesion Establishment Complex. Mol Cell Biol. 25(13): 5445–5455. 10.1128/MCB.25.13.5445-5455.2005
- Maradeo ME, and Skibbens RV (2009). The Elg1-RFC clamp-loading complex performs a role in sister chromatid cohesion. PloS One. 4(3): e4707. 10.1371/journal.pone.0004707
- Parnas O, Zipin-Roitman A, Mazor Y, Liefshitz B, Ben-Aroya S, and Kupiec M (2009). The ELG1 clamp loader plays a role in sister chromatid cohesion. PloS One. 4(5): e5497. 10.1371/journal.pone.0005497
- Skibbens RV (2004). Chl1p, a DNA Helicase-Like Protein in Budding Yeast, Functions in Sister-Chromatid Cohesion. Genetics. 166(1): 33–42. 10.1534/genetics.166.1.33
- Zhang W, Yeung CHL, Wu L, and Yuen KWY (2017). E3 ubiquitin ligase Bre1 couples sister chromatid cohesion establishment to DNA replication in Saccharomyces cerevisiae. eLife. 6: e28231. 10.7554/eLife.28231
- Ivanov MP, Ladurner R, Poser I, Beveridge R, Rampler E, Hudecz O, Novatchkova M, Hériché J, Wutz G, Lelij P, Kreidl E, Hutchins JR, Axelsson-Ekker H, Ellenberg J, Hyman AA, Mechtler K, and Peters J (2018). The replicative helicase MCM recruits cohesin acetyltransferase ESCO2 to mediate centromeric sister chromatid cohesion. EMBO J. 37(15). 10.15252/embj.201797150
- Xu H, Boone C, and Brown GW (2007). Genetic dissection of parallel sister-chromatid cohesion pathways. Genetics. 176(3): 1417–1429. 10.1534/genetics.107.072876
- Xu H, Boone C, and Klein HL (2004). Mrc1 Is Required for Sister Chromatid Cohesion To Aid in Recombination Repair of Spontaneous Damage. Mol Cell Biol. 24(16): 7082–7090. 10.1128/MCB.24.16.7082-7090.2004
- Calzada A, Hodgson B, Kanemaki M, Bueno A, and Labib K (2005). Molecular anatomy and regulation of a stable replisome at a paused eukaryotic DNA replication fork. Genes Dev. 19(16): 1905–1919. 10.1101/gad.337205
- Szyjka SJ, Viggiani CJ, and Aparicio OM (2005). Mrc1 is required for normal progression of replication forks throughout chromatin in S. cerevisiae. Mol Cell. 19(5): 691–697. 10.1016/j.molcel.2005.06.037
- Naiki T, Kondo T, Nakada D, Matsumoto K, and Sugimoto K (2001). Chl12 (Ctf18) Forms a Novel Replication Factor C-Related Complex and Functions Redundantly with Rad24 in the DNA Replication Checkpoint Pathway. Mol Cell Biol. 21(17): 5838–5845. 10.1128/MCB.21.17.5838-5845.2001
- Srinivasan M, Fumasoni M, Petela NJ, Murray A, and Nasmyth KA (2020). Cohesion is established during DNA replication utilising chromosome associated cohesin rings as well as those loaded de novo onto nascent DNAs. eLife. 9: e56611. 10.7554/eLife.56611
- Bando M, Katou Y, Komata M, Tanaka H, Itoh T, Sutani T, and Shirahige K (2009). Csm3, Tof1, and Mrc1 Form a Heterotrimeric Mediator Complex That Associates with DNA Replication Forks. J Biol Chem. 284(49): 34355–34365. 10.1074/jbc.M109.065730
- Batté A, van der Horst SC, Tittel-Elmer M, Sun SM, Sharma S, van Leeuwen J, Chabes A, and van Attikum H (2022). Chl1 helicase controls replication fork progression by regulating dNTP pools. Life Sci Alliance. 5(4): e202101153. 10.26508/lsa.202101153
- Samora CP, Saksouk J, Goswami P, Wade BO, Singleton MR, Bates PA, Lengronne A, Costa A, and Uhlmann F (2016). Ctf4 Links DNA Replication with Sister Chromatid Cohesion Establishment by Recruiting the Chl1 Helicase to the Replisome. Mol Cell. 63(3): 371–384. 10.1016/j.molcel.2016.05.036
- Spencer F, Gerring SL, Connelly C, and Hieter P (1990). Mitotic chromosome transmission fidelity mutants in Saccharomyces cerevisiae. Genetics. 124(2): 237–249. 10.1093/genetics/124.2.237
- Gambus A, Jones RC, Sanchez-Diaz A, Kanemaki M, van Deursen F, Edmondson RD, and Labib K (2006). GINS maintains association of Cdc45 with MCM in replisome progression complexes at eukaryotic DNA replication forks. Nat Cell Biol. 8(4): 358–366. 10.1038/ncb1382
- Yuan Z, Georgescu R, Santos R de LA, Zhang D, Bai L, Yao NY, Zhao G, O'Donnell ME, and Li H (2019). Ctf4 organizes sister replisomes and Pol α into a replication factory. eLife. 8: e47405. 10.7554/eLife.47405
- Villa F, Simon AC, Ortiz Bazan MA, Kilkenny ML, Wirthensohn D, Wightman M, Matak-Vinkovíc D, Pellegrini L, and Labib K (2016). Ctf4 Is a Hub in the Eukaryotic Replisome that Links Multiple CIP-Box Proteins to the CMG Helicase. Mol Cell. 63(3): 385–396. 10.1016/j.molcel.2016.06.009
- Gambus A, van Deursen F, Polychronopoulos D, Foltman M, Jones RC, Edmondson RD, Calzada A, and Labib K (2009). A key role for Ctf4 in coupling the MCM2-7 helicase to DNA polymerase α within the eukaryotic replisome. EMBO J. 28(19): 2992–3004. 10.1038/emboj.2009.226
- Tanaka H, Katou Y, Yagura M, Saitoh K, Itoh T, Araki H, Bando M, and Shirahige K (2009). Ctf4 coordinates the progression of helicase and DNA polymerase α. Genes Cells. 14(7): 807–820. 10.1111/j.1365-2443.2009.01310.x
- Mimura S, Yamaguchi T, Ishii S, Noro E, Katsura T, Obuse C, and Kamura T (2010). Cul8/Rtt101 forms a variety of protein complexes that regulate DNA damage response and transcriptional silencing. J Biol Chem. 285(13): 9858–9867. 10.1074/jbc.M109.082107
- Gan H, Serra-Cardona A, Hua X, Zhou H, Labib K, Yu C, and Zhang Z (2018). The Mcm2-Ctf4-Polα Axis Facilitates Parental Histone H3-H4 Transfer to Lagging Strands. Mol Cell. 72(1): 140-151.e3. 10.1016/j.molcel.2018.09.001
- Buser R, Kellner V, Melnik A, Wilson-Zbinden C, Schellhaas R, Kastner L, Piwko W, Dees M, Picotti P, Maric M, Labib K, Luke B, and Peter M (2016). The Replisome-Coupled E3 Ubiquitin Ligase Rtt101Mms22 Counteracts Mrc1 Function to Tolerate Genotoxic Stress. PLOS Genet. 12(2): e1005843. 10.1371/journal.pgen.1005843
- Luke B, Versini G, Jaquenoud M, Zaidi IW, Kurz T, Pintard L, Pasero P, and Peter M (2006). The Cullin Rtt101p Promotes Replication Fork Progression through Damaged DNA and Natural Pause Sites. Curr Biol. 16(8): 786–792. 10.1016/j.cub.2006.02.071
- Duro E, Vaisica JA, Brown GW, and Rouse J (2008). Budding yeast Mms22 and Mms1 regulate homologous recombination induced by replisome blockage. DNA Repair. 7(5): 811–818. 10.1016/j.dnarep.2008.01.007
- Zaidi IW, Rabut G, Poveda A, Scheel H, Malmström J, Ulrich H, Hofmann K, Pasero P, Peter M, and Luke B (2008). Rtt101 and Mms1 in budding yeast form a CUL4(DDB1)-like ubiquitin ligase that promotes replication through damaged DNA. EMBO Rep. 9(10): 1034–1040. 10.1038/embor.2008.155
- Mimura S, Komata M, Kishi T, Shirahige K, and Kamura T (2009). SCF(Dia2) regulates DNA replication forks during S-phase in budding yeast. EMBO J. 28(23): 3693–3705. 10.1038/emboj.2009.320
- Vaisica JA, Baryshnikova A, Costanzo M, Boone C, and Brown GW (2011). Mms1 and Mms22 stabilize the replisome during replication stress. Mol Biol Cell. 22(13): 2396–2408. 10.1091/mbc.E10-10-0848
- Luciano P, Dehé P-M, Audebert S, Géli V, and Corda Y (2015). Replisome function during replicative stress is modulated by histone h3 lysine 56 acetylation through Ctf4. Genetics. 199(4): 1047–1063. 10.1534/genetics.114.173856
- Fumasoni M, Zwicky K, Vanoli F, Lopes M, and Branzei D (2015). Error-Free DNA Damage Tolerance and Sister Chromatid Proximity during DNA Replication Rely on the Polα/Primase/Ctf4 Complex. Mol Cell. 57(5): 812–823. 10.1016/j.molcel.2014.12.038
- Prado F, and Clemente-Ruiz M (2012). Nucleosome assembly and genome integrity: The fork is the link. Bioarchitecture. 2(1): 6–10. 10.4161/bioa.19737
- Ransom M, Dennehey BK, and Tyler JK (2010). Chaperoning histones during DNA replication and repair. Cell. 140(2): 183–195. 10.1016/j.cell.2010.01.004
- Diao L-T, Chen C-C, Dennehey B, Pal S, Wang P, Shen Z-J, Deem A, and Tyler JK (2017). Delineation of the role of chromatin assembly and the Rtt101Mms1 E3 ubiquitin ligase in DNA damage checkpoint recovery in budding yeast. PLOS ONE. 12(7): e0180556. 10.1371/journal.pone.0180556
- Johnston SD, Enomoto S, Schneper L, McClellan MC, Twu F, Montgomery ND, Haney SA, Broach JR, and Berman J (2001). CAC3(MSI1) suppression of RAS2(G19V) is independent of chromatin assembly factor I and mediated by NPR1. Mol Cell Biol. 21(5): 1784–1794. 10.1128/MCB.21.5.1784-1794.2001
- Harkness TAA, Arnason TG, Legrand C, Pisclevich MG, Davies GF, and Turner EL (2005). Contribution of CAF-I to Anaphase-Promoting-Complex-Mediated Mitotic Chromatin Assembly in Saccharomyces cerevisiae. Eukaryot Cell. 4(4): 673–684. 10.1128/EC.4.4.673-684.2005
- Kats ES, Albuquerque CP, Zhou H, and Kolodner RD (2006). Checkpoint functions are required for normal S-phase progression in Saccharomyces cerevisiae RCAF- and CAF-I-defective mutants. Proc Natl Acad Sci. 103(10): 3710–3715. 10.1073/pnas.0511102103
- Mattiroli F, Gu Y, Balsbaugh JL, Ahn NG, and Luger K (2017). The Cac2 subunit is essential for productive histone binding and nucleosome assembly in CAF-1. Sci Rep. 7: 46274. 10.1038/srep46274
- Green EM, Antczak AJ, Bailey AO, Franco AA, Wu KJ, Yates JR, and Kaufman PD (2005). Replication-Independent Histone Deposition by the HIR Complex and Asf1. Curr Biol. 15(22): 2044–2049. 10.1016/j.cub.2005.10.053
- Sharp JA, Franco AA, Osley MA, and Kaufman PD (2002). Chromatin assembly factor I and Hir proteins contribute to building functional kinetochores in S. cerevisiae. Genes Dev. 16(1): 85–100. 10.1101/gad.925302
- Yang J, Zhang X, Feng J, Leng H, Li S, Xiao J, Liu S, Xu Z, Xu J, Li D, Wang Z, Wang J, and Li Q (2016). The Histone Chaperone FACT Contributes to DNA Replication-Coupled Nucleosome Assembly. Cell Rep. 14(5): 1128–1141. 10.1016/j.celrep.2015.12.096
- Liu S, Xu Z, Leng H, Zheng P, Yang J, Chen K, Feng J, and Li Q (2017). RPA binds histone H3-H4 and functions in DNA replication-coupled nucleosome assembly. Science. 355(6323): 415–420. 10.1126/science.aah4712
- Burgess RJ, Zhou H, Han J, and Zhang Z (2010). A Role for Gcn5 in Replication-Coupled Nucleosome Assembly. Mol Cell. 37(4): 469–480. 10.1016/j.molcel.2010.01.020
- Ai X, and Parthun MR (2004). The Nuclear Hat1p/Hat2p Complex. Mol Cell. 14(2): 195–205. 10.1016/S1097-2765(04)00184-4
- Burgess RJ, and Zhang Z (2013). Histone chaperones in nucleosome assembly and human disease. Nat Struct Mol Biol. 20(1): 14–22. 10.1038/nsmb.2461
- Azvolinsky A, Dunaway S, Torres JZ, Bessler JB, and Zakian VA (2006). The S. cerevisiae Rrm3p DNA helicase moves with the replication fork and affects replication of all yeast chromosomes. Genes Dev. 20(22): 3104–3116. 10.1101/gad.1478906
- Fachinetti D, Bermejo R, Cocito A, Minardi S, Katou Y, Kanoh Y, Shirahige K, Azvolinsky A, Zakian VA, and Foiani M (2010). Replication Termination at Eukaryotic Chromosomes Is Mediated by Top2 and Occurs at Genomic Loci Containing Pausing Elements. Mol Cell. 39(4): 595–605. 10.1016/j.molcel.2010.07.024
- Pardo B, Crabbé L, and Pasero P (2017). Signaling pathways of replication stress in yeast. FEMS Yeast Res. 17(2). 10.1093/femsyr/fow101
- Lisby M, Barlow JH, Burgess RC, and Rothstein R (2004). Choreography of the DNA Damage Response. Cell. 118(6): 699–713. 10.1016/j.cell.2004.08.015
- Thorpe PH, Alvaro D, Lisby M, and Rothstein R (2011). Bringing Rad52 foci into focus. J Cell Biol. 194(5): 665–667. 10.1083/jcb.201108095
- Downs JA, Lowndes NF, and Jackson SP (2000). A role for Saccharomyces cerevisiae histone H2A in DNA repair. Nature. 408(6815): 1001–1004. 10.1038/35050000
- Barroso SI, and Aguilera A (2021). Detection of DNA Double-Strand Breaks by γ-H2AX Immunodetection. In: Aguilera A, Carreira A, editors Homol. Recomb. Springer US, New York, NY. 10.1007/978-1-0716-0644-5_1
- Larsen NB, Hickson ID, and Mankouri HW (2014). Tus-Ter as a tool to study site-specific DNA replication perturbation in eukaryotes. Cell Cycle. 13(19): 2994–2998. 10.4161/15384101.2014.958912
- Larsen NB, Sass E, Suski C, Mankouri HW, and Hickson ID (2014). The Escherichia coli Tus–Ter replication fork barrier causes site-specific DNA replication perturbation in yeast. Nat Commun. 5(1): 3574. 10.1038/ncomms4574
- Willis NA, Chandramouly G, Huang B, Kwok A, Follonier C, Deng C, and Scully R (2014). BRCA1 controls homologous recombination at Tus/Ter-stalled mammalian replication forks. Nature. 510(7506): 556–559. 10.1038/nature13295
- Larsen NB, Liberti SE, Vogel I, Jørgensen SW, Hickson ID, and Mankouri HW (2017). Stalled replication forks generate a distinct mutational signature in yeast. Proc Natl Acad Sci. 114(36): 9665–9670. 10.1073/pnas.1706640114
- Simon AC, Zhou JC, Perera RL, van Deursen F, Evrin C, Ivanova ME, Kilkenny ML, Renault L, Kjaer S, Matak-Vinković D, Labib K, Costa A, and Pellegrini L (2014). A Ctf4 trimer couples the CMG helicase to DNA polymerase α in the eukaryotic replisome. Nature. 510(7504): 293–297. 10.1038/nature13234
- Foltman M, Evrin C, De Piccoli G, Jones RC, Edmondson RD, Katou Y, Nakato R, Shirahige K, and Labib K (2013). Eukaryotic Replisome Components Cooperate to Process Histones During Chromosome Replication. Cell Rep. 3(3): 892–904. 10.1016/j.celrep.2013.02.028
- Borges V, Smith DJ, Whitehouse I, and Uhlmann F (2013). An Eco1-independent sister chromatid cohesion establishment pathway in S. cerevisiae. Chromosoma. 122(1–2): 121–134. 10.1007/s00412-013-0396-y
- Rowlands H, Shaban K, Cheng A, Foster B, and Yankulov K (2019). Dysfunctional CAF-I reveals its role in cell cycle progression and differential regulation of gene silencing. Cell Cycle. 18(22): 3223–3236. 10.1080/15384101.2019.1673100
- Zhang J, Shi X, Li Y, Kim B-J, Jia J, Huang Z, Yang T, Fu X, Jung SY, Wang Y, Zhang P, Kim S-T, Pan X, and Qin J (2008). Acetylation of Smc3 by Eco1 Is Required for S Phase Sister Chromatid Cohesion in Both Human and Yeast. Mol Cell. 31(1): 143–151. 10.1016/j.molcel.2008.06.006
- Heidinger-Pauli JM, Ünal E, and Koshland D (2009). Distinct Targets of the Eco1 Acetyltransferase Modulate Cohesion in S Phase and in Response to DNA Damage. Mol Cell. 34(3): 311–321. 10.1016/j.molcel.2009.04.008
- Gandhi R, Gillespie PJ, and Hirano T (2006). Human Wapl Is a Cohesin-Binding Protein that Promotes Sister-Chromatid Resolution in Mitotic Prophase. Curr Biol. 16(24): 2406–2417. 10.1016/j.cub.2006.10.061
- Kueng S, Hegemann B, Peters BH, Lipp JJ, Schleiffer A, Mechtler K, and Peters J-M (2006). Wapl controls the dynamic association of cohesin with chromatin. Cell. 127(5): 955–967. 10.1016/j.cell.2006.09.040
- Barton RE, Massari LF, Robertson D, and Marston AL (2022). Eco1-dependent cohesin acetylation anchors chromatin loops and cohesion to define functional meiotic chromosome domains. eLife. 11: e74447. 10.7554/eLife.74447
- Michaelis C, Ciosk R, and Nasmyth K (1997). Cohesins: Chromosomal Proteins that Prevent Premature Separation of Sister Chromatids. Cell. 91(1): 35–45. 10.1016/S0092-8674(01)80007-6
- Hanna JS, Kroll ES, Lundblad V, and Spencer FA (2001). Saccharomyces cerevisiae CTF18 and CTF4 are required for sister chromatid cohesion. Mol Cell Biol. 21(9): 3144–3158. 10.1128/MCB.21.9.3144-3158.2001
- Unal E, Heidinger-Pauli JM, and Koshland D (2007). DNA double-strand breaks trigger genome-wide sister-chromatid cohesion through Eco1 (Ctf7). Science. 317(5835): 245–248. 10.1126/science.1140637
- Phipps J, and Dubrana K (2022). DNA Repair in Space and Time: Safeguarding the Genome with the Cohesin Complex. Genes. 13(2): 198. 10.3390/genes13020198
- Dorsett D, and Ström L (2012). The Ancient and Evolving Roles of Cohesin in Gene Expression and DNA Repair. Curr Biol. 22(7): R240–R250. 10.1016/j.cub.2012.02.046
- Wu N, and Yu H (2012). The Smc complexes in DNA damage response. Cell Biosci. 2: 5. 10.1186/2045-3701-2-5
- Heidinger-Pauli JM, Ünal E, Guacci V, and Koshland D (2008). The Kleisin Subunit of Cohesin Dictates Damage-Induced Cohesion. Mol Cell. 31(1): 47–56. 10.1016/j.molcel.2008.06.005
- Lopez-Serra L, Kelly G, Patel H, Stewart A, and Uhlmann F (2014). The Scc2-Scc4 complex acts in sister chromatid cohesion and transcriptional regulation by maintaining nucleosome-free regions. Nat Genet. 46(10): 1147–1151. 10.1038/ng.3080
- Delamarre A, Barthe A, de la Roche Saint-André C, Luciano P, Forey R, Padioleau I, Skrzypczak M, Ginalski K, Géli V, Pasero P, and Lengronne A (2020). MRX Increases Chromatin Accessibility at Stalled Replication Forks to Promote Nascent DNA Resection and Cohesin Loading. Mol Cell. 77(2): 395-410.e3. 10.1016/j.molcel.2019.10.029
- Beckouët F, Hu B, Roig MB, Sutani T, Komata M, Uluocak P, Katis VL, Shirahige K, and Nasmyth K (2010). An Smc3 Acetylation Cycle Is Essential for Establishment of Sister Chromatid Cohesion. Mol Cell. 39(5): 689–699. 10.1016/j.molcel.2010.08.008
- Lyons NA, and Morgan DO (2011). Cdk1-Dependent Destruction of Eco1 Prevents Cohesion Establishment after S Phase. Mol Cell. 42(3): 378–389. 10.1016/j.molcel.2011.03.023
- Gershon L, and Kupiec M (2021). The Amazing Acrobat: Yeast's Histone H3K56 Juggles Several Important Roles While Maintaining Perfect Balance. Genes. 12(3): 342. 10.3390/genes12030342
- Yadav T, and Whitehouse I (2016). Replication-Coupled Nucleosome Assembly and Positioning by ATP-Dependent Chromatin-Remodeling Enzymes. Cell Rep. 15(4): 715–723. 10.1016/j.celrep.2016.03.059
- Mayer ML, Pot I, Chang M, Xu H, Aneliunas V, Kwok T, Newitt R, Aebersold R, Boone C, Brown GW, and Hieter P (2004). Identification of Protein Complexes Required for Efficient Sister Chromatid Cohesion. Mol Biol Cell. 15(4): 1736–1745. 10.1091/mbc.e03-08-0619
- Sasaki M, and Kobayashi T (2017). Ctf4 Prevents Genome Rearrangements by Suppressing DNA Double-Strand Break Formation and Its End Resection at Arrested Replication Forks. Mol Cell. 66(4): 533-545.e5. 10.1016/j.molcel.2017.04.020
- Scherzer M, Giordano F, Ferran MS, and Ström L (2022). Recruitment of Scc2/4 to double-strand breaks depends on γH2A and DNA end resection. Life Sci Alliance. 5(5): e202101244. 10.26508/lsa.202101244
- Petela NJ, Gligoris TG, Metson J, Lee B-G, Voulgaris M, Hu B, Kikuchi S, Chapard C, Chen W, Rajendra E, Srinivisan M, Yu H, Löwe J, and Nasmyth KA (2018). Scc2 Is a Potent Activator of Cohesin's ATPase that Promotes Loading by Binding Scc1 without Pds5. Mol Cell. 70(6): 1134-1148.e7. 10.1016/j.molcel.2018.05.022
- Davidson IF, and Peters J-M (2021). Genome folding through loop extrusion by SMC complexes. Nat Rev Mol Cell Biol. 22(7): 445–464. 10.1038/s41580-021-00349-7
- Adkins MW, and Tyler JK (2004). The histone chaperone Asf1p mediates global chromatin disassembly in vivo. J Biol Chem. 279(50): 52069–52074. 10.1074/jbc.M406113200
- Smith DJ, and Whitehouse I (2012). Intrinsic coupling of lagging-strand synthesis to chromatin assembly. Nature. 483(7390): 434–438. 10.1038/nature10895
- Fennessy RT, and Owen-Hughes T (2016). Establishment of a promoter-based chromatin architecture on recently replicated DNA can accommodate variable inter-nucleosome spacing. Nucleic Acids Res. gkw331. 10.1093/nar/gkw331
- Janke R, King GA, Kupiec M, and Rine J (2018). Pivotal roles of PCNA loading and unloading in heterochromatin function. Proc Natl Acad Sci. 115(9). 10.1073/pnas.1721573115
- Sharp JA, Krawitz DC, Gardner KA, Fox CA, and Kaufman PD (2003). The budding yeast silencing protein Sir1 is a functional component of centromeric chromatin. Genes Dev. 17(19): 2356–2361. 10.1101/gad.1131103
- Özaydın B, and Rine J (2010). Expanded Roles of the Origin Recognition Complex in the Architecture and Function of Silenced Chromatin in Saccharomyces cerevisiae. Mol Cell Biol. 30(3): 626–639. 10.1128/MCB.00614-09
- Suter B, Tong A, Chang M, Yu L, Brown GW, Boone C, and Rine J (2004). The Origin Recognition Complex Links Replication, Sister Chromatid Cohesion and Transcriptional Silencing in Saccharomyces cerevisiae. Genetics. 167(2): 579–591. 10.1534/genetics.103.024851
- Shimada K, and Gasser SM (2007). The Origin Recognition Complex Functions in Sister-Chromatid Cohesion in Saccharomyces cerevisiae. Cell. 128(1): 85–99. 10.1016/j.cell.2006.11.045
- Popova VV, Brechalov AV, Georgieva SG, and Kopytova DV (2018). Nonreplicative functions of the origin recognition complex. Nucleus. 9(1): 460–473. 10.1080/19491034.2018.1516484
- Corda Y, Lee SE, Guillot S, Walther A, Sollier J, Arbel-Eden A, Haber JE, and Géli V (2005). Inactivation of Ku-mediated end joining suppresses mec1Delta lethality by depleting the ribonucleotide reductase inhibitor Sml1 through a pathway controlled by Tel1 kinase and the Mre11 complex. Mol Cell Biol. 25(23): 10652–10664. 10.1128/MCB.25.23.10652-10664.2005
- Drake JW (1991). A constant rate of spontaneous mutation in DNA-based microbes. Proc Natl Acad Sci. 88(16): 7160–7164. 10.1073/pnas.88.16.7160
- Lujan SA, Williams JS, Pursell ZF, Abdulovic-Cui AA, Clark AB, Nick McElhinny SA, and Kunkel TA (2012). Mismatch Repair Balances Leading and Lagging Strand DNA Replication Fidelity. PLoS Genet. 8(10): e1003016. 10.1371/journal.pgen.1003016
–
SUPPLEMENTAL INFORMATION
 Download Supplemental Information
Download Supplemental Information
ACKNOWLEDGMENTS
We gratefully acknowledge Adele Marston for anti-Smc3-K112,113Ac antibody. We thank Armelle Lengronne for providing PP1418 and PP1628 strains, Jennifer Gerton for providing eco1-1 mutant strain, Qing Li for rfa1-A88P mutant, Sue Biggins for SBY885 strain, and Zhiguo Zhang for ZGY1069 and ZGY1077 strains. We acknowledge Paul D. Kaufman for the pRS414-CAC1, the pRS414-cac1-F233L, and the pRS414-cac1-F233A, F234G plasmids. We thank Hocine Mankouri and Ian Hickson for providing the Tus/Ter barrier system. Thanks to Michel-Hervé Moimême for helpful discussions and permanent support. V.G. laboratory is supported by the "Ligue Na-tionale Contre le Cancer" (LNCC) (Equipe labellisée). N.G. is supported by “Bourse du Ministère de l’Enseignement Supérieur, et de la Recherche (MESR)” and by "Fondation pour la Recherche Médicale (FRM)".
COPYRIGHT
© 2023

Chromatin assembly factor-1 preserves genome stability in ctf4∆ cells by promoting sister chromatid cohesion by Ghaddar et al. is licensed under a Creative Commons Attribution 4.0 International License.


