Reviews:
Cell Stress, Vol. 7, No. 11, pp. 95 - 104; doi: 10.15698/cst2023.11.291
STING-driven activation of T cells: relevance for the adoptive cell therapy of cancer

1 Centre d’Étude des Pathologies Respiratoires, U1100, INSERM, Tours, France.
2 Faculté de Médecine, Université de Tours, Tours, France.
3 Brown Center for Immunotherapy, Indiana University Melvin and Bren Simon Comprehensive Cancer Center, Indiana University School of Medicine, Indianapolis, IN 46202, USA.
Keywords: Adoptive T cell therapy, STING, T cells, cancer, immunomodulation.
Abbreviatons:
ACT – adoptive cell therapy,
TME – tumor microenvironment,
STING – stimulator of interferon genes,
DC – dendritic cell,
ICI – immune checkpoint inhibitors,
TILs – tumor-infiltrating lymphocytes,
TCR – T cell receptor,
CAR-T – chimeric antigen receptor T cells,
MHC – major histocompatibility complex,
TLRs – Toll-like receptors,
NOD – nucleotide-binding oligomerization domain,
NLRs – NOD-like receptors,
RIG-I – Retinoic acid-inducible gene I,
RLRs – RIG-I-like receptors,
CLRs – C-type lectin receptors,
cGAS – cGAMP synthase,
IRF3 – interferon regulatory factor 3,
IFNs – type I interferons,
APCs – antigen-presenting cells.
Received originally: 20/06/2023 Received in revised form: 03/10/2023
Accepted: 05/10/2023
Published: 14/11/2023
Correspondence:
Lionel Apetoh, Brown Center for Immunotherapy, Indiana University Melvin and Bren Simon Comprehensive Cancer Center, Indiana University School of Medicine, Indianapolis, IN 46202, USA; lapetoh@iu.edu
Conflict of interest statement:
L.A. is a consultant for Brenus-Pharma. L.A. performed consultancy work for Roche, Merck, Bristol-Myers Squibb, and Orega Biotech and was a recipient of a research grant from Sanofi.
Please cite this article as: Fabian Richter, Christophe Paget and Lionel Apetoh (2023). STING-driven activation of T cells: relevance for the adoptive cell therapy of cancer. Cell Stress 7(11): 95-104. doi: 10.15698/cst2023.11.291
Abstract
Adoptive cell therapy (ACT) can successfully treat hematopoietic cancers but lacks efficacy against solid tumors. This is due to insufficient T cell infiltration, high tumor heterogeneity, frequent antigen loss with subsequent tumor escape, and the immunosuppressive tumor microenvironment (TME). Alternative methods to boost the anticancer efficacy of adoptively transferred cells are actively pursued. Among adjuvants that are utilized to stimulate anticancer immune responses, ligands of the stimulator of interferon genes (STING) pathway have received increasing attention. STING activation can trigger dendritic cell (DC) activation and endogenous immune responses, thereby preventing tumor escape. Activation of the STING pathway in the context of ACT was accordingly associated with improved T cell trafficking and persistence in the TME combined with the reduced presence of immunosuppressive cells. Recent findings also suggest cell-intrinsic effects of STING ligands on T cells. Activation of the STING signaling pathway was in this regard shown to enhance effector functions of CD4+ and CD8+ T cells, suggesting that the STING signaling could be exploited to harness T cell anticancer functions. In this review, we will discuss how the STING signaling can be used to enhance the anticancer efficacy of ACT.
THE RELEVANCE OF ADOPTIVE T CELL THERAPY IN CANCER
In 2018, Tasuku Honjo and James Allison were awarded the Nobel Prize for their research on immune checkpoint inhibitors (ICI). They found that inhibition of the suppressive molecules PD-1 and CTLA-4 enhanced the ability of the immune system to eliminate cancer cells [1]. Immune checkpoints are expressed on activated immune cells and serve to maintain self-tolerance and regulate immune responses. Tumor-induced engagement of these immune checkpoints is considered as a resistance mechanism that limits T cell activation [2]. Numerous anti-PD-1 and anti-CTLA-4 therapies were developed and showed unparalleled survival rates for patients with non-small cell lung cancer, renal cell carcinoma, and melanoma [3]. Although these treatments are now FDA-approved for many cancer types, a significant number of patients suffers from immune-related adverse effects or acquired resistance [4]. Especially in weakly immunogenic cancers, some patients cannot benefit from ICIs, highlighting the importance of further progress in this research area and the need for alternative therapeutic approaches [5]. One other treatment avenue with proven clinical success is adoptive cell therapy (ACT). In ACT, autologous immune cells are amplified ex-vivo, modified, and subsequently transferred back into the patient [6]. It has been successfully demonstrated that ICI and ACT can be combined [7][8], possibly providing additional benefits [9]. Some metastatic melanoma patients in whom ICI showed no therapeutic effect were successfully treated with ACT, suggesting that ACT can be used when ICIs fail [10][11][12][13].
–
Three main T cell-based approaches are currently considered for ACT, including the use of tumor-infiltrating lymphocytes (TILs), antigen-specific T cells equipped with a specific T cell receptor (TCR), and chimeric antigen receptor T cells (CAR-T) [14]. TCR-based ACT does not require an engineered construct but relies on the target antigen being presented via the major histocompatibility complex (MHC). CAR-T based ACT can target MHC independent antigens, as well as carbohydrates and glycolipids expressed on the cell surface of tumors [14]. CD8+ T cells can directly eliminate tumor cells due to their cytotoxic properties, which is why they have long been considered as the most suitable T cell subset for ACT. CD8+ T cell-mediated killing occurs through MHC-I-dependent recognition of tumor cells and subsequent granzyme- and/or FAS ligand-dependent elimination. This also requires cross-priming by DCs as well as co-stimulation by natural killer cells and/or CD4+ T cells derived cytokines [15]. However, the support of innate immunity to T cell adaptive immune responses is not always present in the TME. If CD8+ T cells cannot be primed and activated in the TME, CD8+ T cell-dependent tumor elimination fails [16]. The required co-stimulation to activate CD8+ T cells can also be provided by CD4+ T cells. Current strategies aim to exploit these properties of CD4+ T cells in ACT [17]. CD4+ T cells are also suitable for ACT because they support antigen presentation from DCs, T cell homing through the secretion of chemokines such as CXCL9-11, formation of CD8+ T cell memory, and direct tumor elimination by granzymes, perforin, TRAIL, or FasL (reviewed in [18]). A recent study examining long-persisting anti-CD19 CAR T cells demonstrated that 9 years after therapy, the long-term protection against CD19+ cells was mediated almost exclusively by cytotoxic CD4+ CAR T cells. This suggests that beyond the short-term tumor elimination mediated by CD8+ T cells, CD4+ T cells contribute to long-term remission [19].
–
Because it is now accepted that the immune system shapes the initiation and progression of cancer [20], increasing efforts are being made to design and exploit adjuvants that will boost anticancer immune responses. Immune adjuvants include agonists of pattern recognition receptors, such as Toll-like receptors (TLRs), nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), Retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs), C-type lectin receptors (CLRs) and cytosolic DNA sensors such as stimulator of interferon genes (STING) [21]. We will here discuss recent findings indicating that STING agonists enhance T cell anticancer responses and underscore their relevance in the context of ACT.
STING IN CANCER IMMUNOTHERAPY
The STING pathway was initially considered as a protective mechanism against intracellular pathogens through the detection of cytosolic double-stranded DNA. STING signaling is also induced following the detection of cytosolic self-DNA, which can originate from tumor cells due to genomic instability [22]. Double-stranded DNA in the cytosol is bound in a sequence-independent manner by the cGAMP synthase (cGAS), resulting in the formation of cGAMP, which acts as a second messenger and activates the STING protein in the ER. STING then undergoes conformational changes and translocates into the ER-Golgi intermediate compartment and Golgi compartment. Here, TANK-binding kinase 1 is recruited for further signal transduction and phosphorylates interferon regulatory factor 3 (IRF3). IRF3 translocates to the nucleus, where it leads to the synthesis of type I interferons (IFNs) and the activation of IFN-stimulated genes (reviewed in [22][23]). Increased type I IFN secretion subsequently induces tumor-specific priming of T cells by antigen-presenting cells (APCs). In tumor cells, STING induction can induce cell death and thus increases the amount of tumor antigens available for APCs. This enhances the activation of T cells, their infiltration into the tumor, thereby resulting in cancer cell elimination [24]. Treatment with STING pathway agonists accordingly promotes adaptive immune responses in the TME [25]. The resulting induction of IFN-γ-expressing CD8+ T cells mediates anticancer responses [26][27]. These properties support the use of STING agonists like 2′3′-cGAMP for cancer immunotherapy (Figure 1).
–
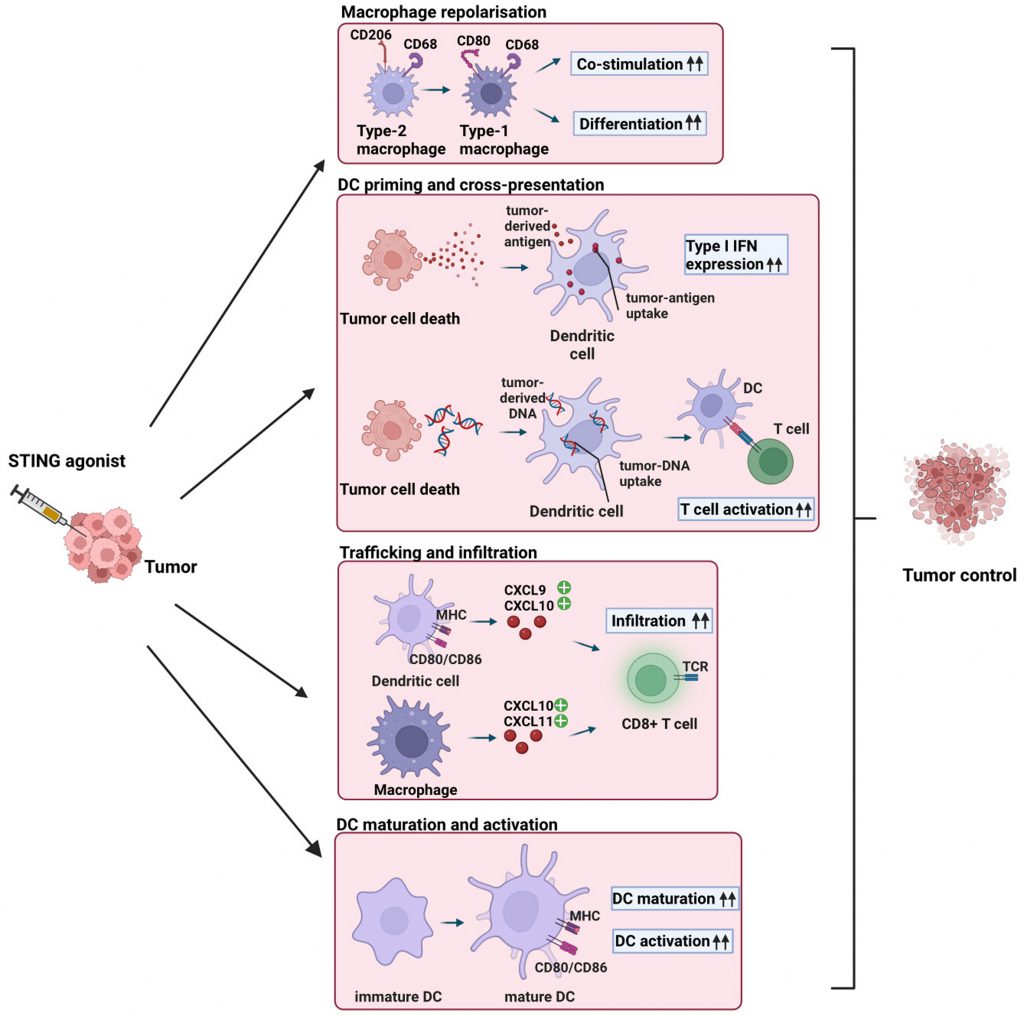 | FIGURE 1: STING agonist-induced effects on innate immune cells in the TME. Intratumoral injection of STING agonists induces repolarization of type-2 macrophages into type-1 macrophages, resulting in enhanced co-stimulation and differentiation of CD4+ and CD8+ T cells [39]. STING ligand-induced tumor cell death leads to the release of tumor-derived antigens and DNA, which are taken up by DCs, resulting in the release of type I IFNs and enhanced T cell activation [23][24][27]. Intratumoral injection of STING agonists results in the secretion of the chemokines CXCL9, CXCL10, and CXCL10, CXCL11, which are secreted by DCs and macrophages, respectively [37][41]. As a result, cytotoxic CD8+ T cells are recruited to the TME. STING agonists induces the secretion of type I IFN, leading to terminal differentiation of immature DCs and enhanced activation of DCs. Overall, intratumoral STING agonists injection leads to macrophage repolarization, DC cross-presentation, T cell trafficking, and DC maturation and activation, resulting in enhanced recruitment and activation of anti-tumor CD8+ T cells and improved tumor control. |
–
Because of STING agonist-mediated immune cell infiltration, combination therapies with ICIs were tested [28]. Promising preclinical results when combining ICIs and STING agonists were achieved in multiple cancer models, including melanoma and an HPV+ oral tumor [29][30][31][32]. However, these successes failed to translate into the clinic due to the pharmacokinetic properties of STING agonists that preclude effective drug delivery [33]. Furthermore, some tumors feature limited responses to STING ligands due to inhibitory mechanisms that include for instance p53 [34], ecto-nucleotide pyrophosphatases 1 (ENPP1) [35], Hypoxia-induced RNASEH2a upregulation [36], and TIM-3 [37]. To tackle some of these hurdles, a better bioavailability of STING agonists is needed. This could be achieved through more stable STING agonists or improved delivery systems (discussed in [33]).
STING-DRIVEN ACTIVATION OF INNATE IMMUNE CELLS FAVORS T CELL ACTIVATION
Two major uses for STING activation can be contemplated. The first is to deliver STING agonists directly into the TME of the host. This approach can trigger anticancer responses [38], thereby contributing to overall tumor elimination as discussed above. Macrophages and DCs are critical innate immune cells that affect CD8+ T cell-mediated tumor elimination. Type-2 macrophages can be repolarized into type-1 macrophages by STING activation, which can improve the antitumor response by enhancing the co-stimulation and differentiation of CD4+ and CD8+ T cells [39]. In DCs, cGAS-STING is required for antigen presentation and cross-priming of T cells [27]. DC-mediated cross-priming is followed by recruitment of cytotoxic T cells to the TME through the chemokines CXCL9 and CXCL10 [37][40].
–
Numerous studies (discussed in [24]) showed that disruption of the STING-axis led to compromised CD8+ T cell-mediated tumor elimination. STING activation in the TME therefore supports T cell functions. This was exploited using either CD8+ and CD4+ CAR T cells, as discussed further below.
–
Using a murine second-generation specific CD8+ CAR T cell model, the group of Sandra Hervas-Stubbs demonstrated that 2’3′-cGAMP treatment induced an endogenous T cell response, prevented antigen-loss variant outgrowth and led to an improved overall survival rate [42]. By using a bilateral B16-OVA tumor mouse model, the authors observed restrained tumor progression in the injected and opposite tumor, when the combination of 2’3′-cGAMP injection and antigen-specific CD8+ CAR-T cells was used. Most importantly, mice treated with the combination were the only ones to survive, while the control groups receiving standalone CAR-T cells or 2’3′-cGAMP died within the first 20-30 days.
–
Since the combined treatment enabled to control tumor progression in treated and untreated tumors and resulted in the survival of the mice, the authors concluded a strong synergistic effect of the STING agonist and the CAR T cells they used.
–
Subsequently, they investigated whether STING agonists could induce an immune response and thus could target the cancer systemically [42]. To verify this, the authors examined the presence of endogenous CD8+ T cells in treated and untreated tumors, lymph nodes, and peripheral blood three days after STING agonist administration. Furthermore, they analyzed the frequency of Ag-specific CD8+ T cells restricted to two immunodominant epitopes of B16-OVA tumors, OVA, which is a foreign antigen, and M8 is an endogenous one. Their analysis revealed a significantly increased number of M8-specific CD8+ T cells in the group receiving CAR T cells and administration of STING ligand in all tissues examined. The number of OVA-specific CD8+ T cells was generally lower, but significantly increased except in the contralateral lymph nodes. The increased presence of OVA+ or M8+ endogenous CD8+ T cells, particularly in the blood, lymph nodes, and untreated tumor suggests that the combination treatment induced an endogenous CD8+ T cell response. Overall, this may have led to the increased overall survival of the mice receiving the combined therapy. Notably, the administration of CAR T cells without STING ligand also resulted in high levels of OVA+ or M8+ endogenous CD8+ T cells in the untreated tumor. In line with the observations of Corrales et al. [40], the authors also found that cross-priming DCs are responsible for the ability of the combined therapy to eliminate tumors [42].
–
To next address the question of whether and to what extent the STING pathway was responsible for the observed antitumor responses, the authors used STING-deficient tumor-bearing mice or mouse STING-deficient CAR T cells. The authors found that the use of STING-deficient CAR T cells was associated with reduced antitumor effects and decreased overall survival of recipient mice but did not affect the frequency of endogenous tumor-specific T cells. In STING-deficient mice, no tumor growth delay and no increased overall survival was detected in response to the combined treatment, along with a complete lack of activated endogenous tumor-specific T cell response [42]. Therefore, the STING pathway in the host has to be fully functional for successful antitumor therapy. T cells possess an intrinsic tumor-eliminating potential, which can be triggered by STING agonist-mediated activation prior to transfer. This study by Sandra Hervas-Stubbs’s group extends previous findings obtained with other STING ligands. Indeed, DMXAA and synthetic cyclic dinucleotides (CDN) elicited a systemic antitumor response dependent on STING signaling in host cells [40]. Collectively, this suggests that intratumoral injection of STING ligands can be exploited in combination with CD8+ T cells to target cancer systemically.
–
Jonathan Serody’s group recently used mouse CD4+ T helper cells in combination with STING agonist to treat a locally advanced breast cancer model [43]. While the potential of CD4+ T cells for ACT is clear, the diversity of CD4+ T cell responses should be considered. CD4+ T cells are a highly heterogeneous group consisting of TH1, TH2, TH9, TH17, TFH, and TReg cell subsets. Each is characterized by a subset-specific cytokine profile and thereby fulfills different effector functions [44]. Although there is a large agreement that Tregs and TH1 cells respectively favor and restrict tumor progression, TH2, TH9 and TH17 cell functions in cancer are context-dependent [45]. TH17 cells can contrastingly affect cancer depending on their environment [46]. However, in the setting of adoptive cell therapy, IL-17-secreting T cells are considered beneficial, with reported antitumor activity in melanoma and lung cancer [47][48][49]. The effect of DMXAA and 2’3′-cGAMP in combination with murine Neu-specific TC/TH17 CAR T cells was investigated in the setting of breast cancer [43]. The authors have demonstrated that the combined treatment mediates antitumor control due to improved trafficking and persistence of CAR T cells in the TME [43]. TC/TH17 CAR T cells featured improved antitumor efficacy in mice receiving DMXAA, resulting in long-term control in some of the treated mice, while standalone treatments remained ineffective [43]. The infiltrating CAR-T cells were then analyzed by flow cytometry to determine the cause of the enhanced antitumor response. The authors found that some of the transferred TC/TH17 CAR-T cells had undergone a shift to TC/TH1 CAR-T cells and that DMXAA enhanced the accumulation of these TC/TH1 CAR-T cells in the TME. This phenotypic shift was accompanied by the upregulation of TC/TH1 signature transcription factors and cytokines (Tbx21 and IFN-γ). By using anti-IFN-γ mAb, they found that tumor elimination occurred in a TC/TH1 CAR-T cell mediated manner [43]. It was previously shown that TH17 cells possess some levels of plasticity enabling their transformation into IFN-γ-producing TH1-like cells [50]. This plasticity is necessary for their antitumor response in TH17 ACT [51]. However, the observation that activation of the STING signaling pathway further enhances this plasticity and the antitumor responses is of high interest for cancer immunotherapy. Despite the improved effect of the combined CAR T cell and STING agonist treatment, one question remains: Why do most tumors recur as IL-17-secreting T cells are known to be long-lived and self-renewing with superior persistence [52]? To address this, the authors have performed a single-cell transcriptome sequencing of CD45+ cells from the TME. The authors compared leukocytes on the day of best tumor control with their counterparts collected on the day of tumor recurrence. In DMXAA treated mice, they found a shift in the myeloid cells of the TME favoring M1-like macrophages. Interestingly, their depletion by means of liposome clodronate was associated with a complete loss of the DMXAA-induced therapeutic benefit. This shift was accompanied by a stronger M1 gene expression including nos2 and inhba, and a reduced expression of M2 associated genes like retnla, mrc1, folr2, and il10. Furthermore, M1-associated chemokines (cxcl9, cxcl10, and ccl5) were increasingly secreted to attract TC/TH1 T cells to the TME. In addition, they have found that while the combined therapy transiently relieves immunosuppression in the TME, this effect is short-lived and immunosuppressive myeloid cells were subsequently present in the TME. They also analyzed the TC/TH17 CAR T cells from the TME and discovered an increased expression of markers associated with dysfunction and apoptosis, providing evidence for their assumption that T cell dysfunction is the key limiting factor of the therapy. This suggested that both immunosuppression driven by myeloid cells and T cell dysfunction were responsible for the absence of complete responses to the combined therapy. To test this further, the combination of TC/TH17 CAR T cells and DMXAA was administered with anti-PD-1 and anti-GR-1 monoclonal antibodies twice a week, starting one day after the CAR T cell injection. This resulted in a marked increase in the ability of tumor-bearing mice to reject tumors. Importantly, only the combination of both antibodies was effective, indicating that both PD-L1 and myeloid cell-driven immunosuppression needed to be targeted to yield a therapeutic benefit. Next, the authors examined the effect of their TC/TH17 CAR T cells by comparing them with CAR T cells expanded with IL-7 and IL-15. The use of TC/TH17 CAR T cells resulted in an improved in vivo anticancer efficacy over the latter [43]. The authors linked this difference to the significantly higher proliferation rate of CD4+ CAR T cells in the spleen and the enhanced expansion of CD8+ CAR T cells with a central memory phenotype. Finally, the authors compared DMXAA with the other STING agonist 2’3′-cGAMP. Although the use of 2’3′-cGAMP resulted in significantly more TC/TH17 CAR T cells in the TME, there was only a minor difference in therapeutic efficacy [43]. Overall, this study suggests that STING ligands and CAR T cells have synergistic effects that can successfully fight breast cancer when combined.
T CELL-INTRINSIC STING ACTIVATION AND ITS RELEVANCE IN ACT
Direct administration of STING agonists faces obstacles such as difficulties with drug delivery and poor pharmacokinetics. It is further accompanied by reports of autoimmune events after excessive STING stimulation [33]. To circumvent this, cells can be directly activated with the STING agonist prior to transfer. This would prevent a STING-mediated detrimental inflammatory response in the host and minimize drug-induced adverse events. We will therefore next focus on the intrinsic effect of STING agonists in T cells and discuss how STING signaling affects their viability, cell proliferation, cytokine secretion, and antitumor functions (Figure 2).
–
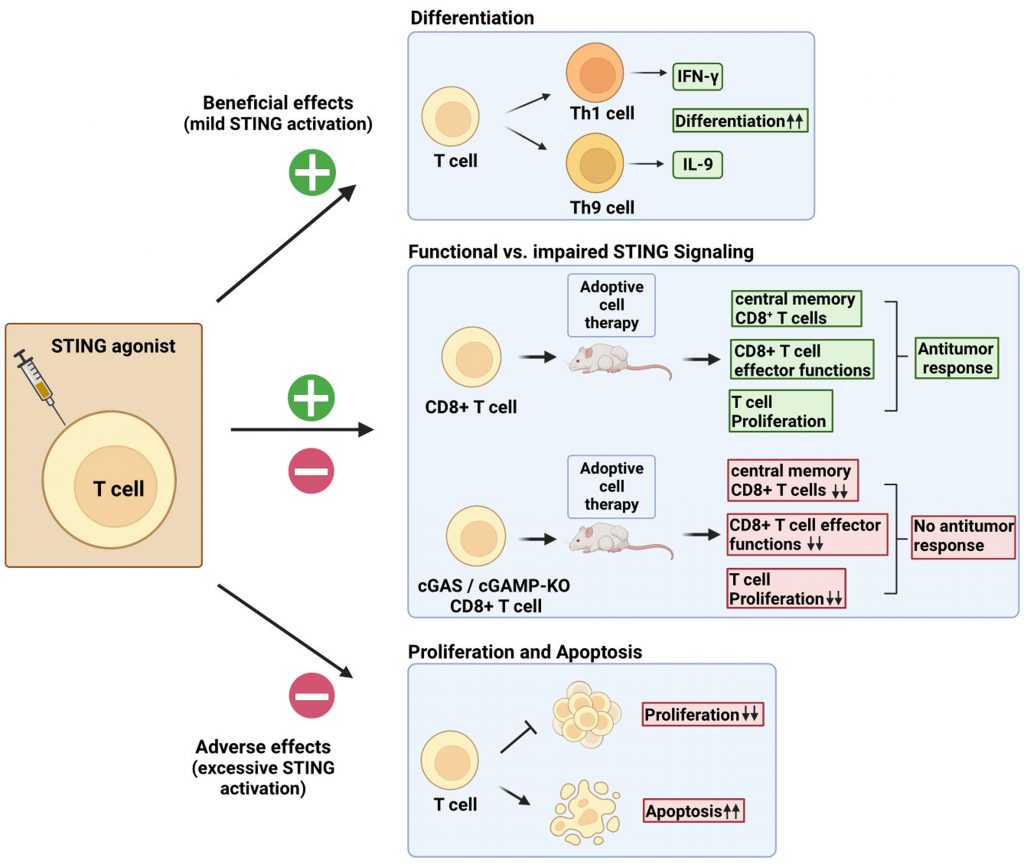 | FIGURE 2: Cell-intrinsic STING activation effects on T cells. Direct stimulation of T cells by STING ligands can produce contrasting effects. Mild activation levels of STING signaling can be exploited to boost TH1 and TH9 differentiation [55]. Impaired STING signaling in T cells results in reduced frequency of central memory CD8+ T cells, reduced proliferation, and reduced effector functions upon adoptive transfer [56]. Potent STING signaling results in reduced proliferation and induction of cell death by apoptosis [53][54]. |
–
Alexander Poltroak’s team studies on mouse T cells showed that the STING signaling pathway, already shown to be active in macrophages, was also functional in T cells. In T cells, STING induction with DMXAA leads to IFN-I production, prevention of cell proliferation, and induction of proapoptotic genes. However, these cell-adverse effects were not observable with low doses of DMXAA [53]. Andrea Ablasser’s group confirmed the cell death-inducing effect of the STING agonists CMA and DMXAA, but additionally found that this was due to an intensified STING signaling response in T cells as compared to macrophages and DCs [53][54]. It should be noted that those findings were mostly established using synthetic STING ligands such as CMA and DMXAA. By contrast, other studies reported limited apoptosis induction upon treatment of T cells with 2’3′-cGAMP [42][55]. Liufu Deng’s laboratory instead investigated whether STING signaling in CD8+ T cells was necessary for their antitumor effects. They found that the cGAS-STING pathway was required in CD8+ T cells to induce an antitumor response [56]. The use of murine tumor-specific T cells lacking either cGAS or STING showed reduced antitumor activity as compared to controls. Likewise, conditional knockout of STING in CD4Cre+-STINGflox/flox mice resulted in accelerated tumor progression in the mouse T cell lymphoma model EG7 and mouse glioblastoma model GL261. Examination of proliferative capacity and effector functions revealed that cGAS- and STING-deficient CD8+ T cells displayed impaired proliferation rate after adoptive transfer as compared to controls. In addition, the number of TNF-α+ and IFN-γ+ CD8+ T cells in tumor draining-lymph nodes was significantly reduced, suggesting dysfunctional effector functions. Finally, the authors showed that cGAS or STING deficiency in CD8+ T cells led to an accumulation of effector CD8+ T cells at the expense of self-renewing and persistent central memory CD8+ T cells. cGAS- or STING-deficient CD8+ T cells also exhibited a terminally exhausted phenotype.
–
Collectively, the authors’ findings support the fundamental role of the cGAS-STING axis in CD8+ T cells for ACT [56]. Importantly, in line with the observations of other investigators [42][55], STING treatment did not affect the cell viability of the human CD8+ T cells used [56]. In summary, an intact cGAS-STING axis in CD8+ T cells is required for their anticancer effector functions. Because excessive STING activation in T cells may trigger cell death, it remains important to carefully consider the dose of the STING ligands used to harness T cell effector functions without compromising their proliferation or viability. We have accordingly shown that STING ligands can enhance T cell effector functions and the differentiation of TH1 and TH9 cells [55].
–
TH9 cells are defined as cells lacking Foxp3 but secreting high levels of IL-9 [57]. They also secrete IL-10 and IL-21 [58][59]. Polarization of naive T cells into TH9 cells is initiated by IL-4 and TGF-β and requires a complex interaction of a network of transcription factors, including IRF4, GATA3, BATF3, and PU.1 [60]. TH9 cells are particularly promising in the context of ACT, as Puwar and colleagues demonstrated superior antitumor properties of TH9 cells upon adoptive transfer as compared to other CD4+ subsets in a mouse model of melanoma [61]. Their superiority was subsequently independently demonstrated by multiple laboratories [58][62][63]. Adoptively transferred tumor Ag-specific murine TH9 cells were shown to provide superior antitumor immunity by eliminating variants with antigen loss [64]. All these findings provide impetus to investigating the relevance of TH9 cells in the ACT of cancer.
–
Investigations conducted by our group using different subsets of CD4+ T cells, including TH1, TH9, and TH17 cells, which were directly activated with different STING ligands, revealed that the differentiation and effector functions of TH1 and TH9 cells could be enhanced by STING activation [55].
–
We further demonstrated that TH1 and TH9 cells respond differently to STING ligands, as illustrated by our observation that TH1 cells were more sensitive to STING ligand-induced apoptosis than TH9 cells. STING activation enhanced human TH1 and TH9 polarization, and resulted in increased expression and secretion of IFN-γ and IL-9, the respective TH1 and TH9 signature cytokines [55]. These results showing that ligands of STING enhance TH9 cell differentiation are in line with published investigations indicating that pro-inflammatory components such as glucocorticoid-induced TNFR-related protein (GITR), IL-1β, TNFα, OX40L, and TL1A support TH9 effector functions [58][65][66][67][68]. It is noteworthy that distinct mechanisms were contributing to the cell-intrinsic STING-driven enhancement of TH1 and TH9 differentiation. IRF3 activation was essential for the STING-mediated induction of TH1 cell differentiation, while mTOR signaling accounts for the increased TH9 cell differentiation following STING activation [55]. We tested the in vivo functions of TH9 cells treated with 2’3′-cGAMP in the B16-OVA melanoma model. For both subcutaneously or intravenously injected B16-OVA cells, we demonstrated that adoptively transferred 2’3′-cGAMP-stimulated tumor Ag-specific TH9 cells secreted more IL-9 and triggered better antitumor immunity compared to controls without STING agonists [55]. These results show that STING activation can enhance the anticancer efficacy of adoptively transferred T cells (Figure 2).
CONCLUSIONS
While STING function was initially characterized in fibroblasts [69], it is now clear that STING shapes the biology of multiple immune cell types, including T cells. STING agonists have the potential to address some of the critical challenges of adoptive cell therapy. STING agonists can indeed enhance T cell infiltration and reduce tumor-induced immunosuppression [42][43]. Despite these promising advances, the disappointing results obtained when combining STING agonists with ICIs underscore the challenge to translate the use of STING agonists into the clinic. Documented issues such as toxicity, low bioavailability, and related difficulties of administration likely prevent the clinical implementation of STING ligands against cancer (discussed in [70]). In that regard, the recent work of Jneid et al. that relies on the use of virus-like particles to deliver cGAMP in the TME highlights an elegant venue to circumvent some of these issues [71]. Recent results suggest that direct T cell activation by STING agonists can be exploited in the context of ACT. Both Liufu Deng’s laboratory and ours have shown that T cells can be directly activated with STING agonists without triggering marked cell death [55][56]. A thoughtful selection and careful use of STING ligands will allow harnessing of T cell anticancer functions without compromising their fitness. Further research is warranted to translate the therapeutic use of STING ligands in the setting of ACT.
REFERENCES
- Kroemer G, Zitvogel L (2021). Immune checkpoint inhibitors. J Exp Med 218(3): e20201979. 10.1084/jem.20201979
- Pardoll DM (2012). The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 12(4): 252-264. 10.1038/nrc3239
- Sharma P, Goswami S, Raychaudhuri D, Siddiqui BA, Singh P, Nagarajan A, Liu J, Subudhi SK, Poon C, Gant KL, Herbrich SM, Anandhan S, Islam S, Amit M, Anandappa G, Allison JP (2023). Immune checkpoint therapy-current perspectives and future directions. Cell 186(8): 1652-1669. 10.1016/j.cell.2023.03.006
- Bagchi S, Yuan R, Engleman EG (2021). Immune Checkpoint Inhibitors for the Treatment of Cancer: Clinical Impact and Mechanisms of Response and Resistance. Annu Rev Pathol 16(223-249. 10.1146/annurev-pathol-042020-042741
- Zhou C, Cheng X, Tu S (2021). Current status and future perspective of immune checkpoint inhibitors in colorectal cancer. Cancer Lett 521: 119-129. 10.1016/j.canlet.2021.07.023
- Rosenberg SA, Restifo NP (2015). Adoptive cell transfer as personalized immunotherapy for human cancer. Science 348(6230): 62-68. 10.1126/science.aaa4967
- Kverneland AH, Pedersen M, Westergaard MCW, Nielsen M, Borch TH, Olsen LR, Aasbjerg G, Santegoets SJ, van der Burg SH, Milne K, Nelson BH, Met Ö, Donia M, Svane IM (2020). Adoptive cell therapy in combination with checkpoint inhibitors in ovarian cancer. Oncotarget 11(22): 2092-2105. 10.18632/oncotarget.27604
- Mullinax JE, Hall M, Prabhakaran S, Weber J, Khushalani N, Eroglu Z, Brohl AS, Markowitz J, Royster E, Richards A, Stark V, Zager JS, Kelley L, Cox C, Sondak VK, Mulé JJ, Pilon-Thomas S, Sarnaik AA (2018). Combination of Ipilimumab and Adoptive Cell Therapy with Tumor-Infiltrating Lymphocytes for Patients with Metastatic Melanoma. Front Oncol 8: 44. 10.3389/fonc.2018.00044
- Kodumudi KN, Siegel J, Weber AM, Scott E, Sarnaik AA, Pilon-Thomas S (2016). Immune Checkpoint Blockade to Improve Tumor Infiltrating Lymphocytes for Adoptive Cell Therapy. PLoS One 11(4): e0153053. 10.1371/journal.pone.0153053
- Lövgren T, Wolodarski M, Wickström S, Edbäck U, Wallin M, Martell E, Markland K, Blomberg P, Nyström M, Lundqvist A, Jacobsson H, Ullenhag G, Ljungman P, Hansson J, Masucci G, Tell R, Poschke I, Adamson L, Mattsson J, Kiessling R (2020). Complete and long-lasting clinical responses in immune checkpoint inhibitor-resistant, metastasized melanoma treated with adoptive T cell transfer combined with DC vaccination. Oncoimmunology 9(1): 1792058. 10.1080/2162402x.2020.1792058
- Andersen R, Borch TH, Draghi A, Gokuldass A, Rana MAH, Pedersen M, Nielsen M, Kongsted P, Kjeldsen JW, Westergaard MCW, Radic HD, Chamberlain CA, Hölmich LR, Hendel HW, Larsen MS, Met Ö, Svane IM, Donia M (2018). T cells isolated from patients with checkpoint inhibitor-resistant melanoma are functional and can mediate tumor regression. Ann Oncol 29(7): 1575-1581. 10.1093/annonc/mdy139
- Besser MJ, Shapira-Frommer R, Itzhaki O, Treves AJ, Zippel DB, Levy D, Kubi A, Shoshani N, Zikich D, Ohayon Y, Ohayon D, Shalmon B, Markel G, Yerushalmi R, Apter S, Ben-Nun A, Ben-Ami E, Shimoni A, Nagler A, Schachter J (2013). Adoptive transfer of tumor-infiltrating lymphocytes in patients with metastatic melanoma: intent-to-treat analysis and efficacy after failure to prior immunotherapies. Clin Cancer Res 19(17): 4792-4800. 10.1158/1078-0432.Ccr-13-0380
- Forget MA, Haymaker C, Hess KR, Meng YJ, Creasy C, Karpinets T, Fulbright OJ, Roszik J, Woodman SE, Kim YU, Sakellariou-Thompson D, Bhatta A, Wahl A, Flores E, Thorsen ST, Tavera RJ, Ramachandran R, Gonzalez AM, Toth CL, Wardell S, Mansaray R, Patel V, Carpio DJ, Vaughn C, Farinas CM, Velasquez PG, Hwu WJ, Patel SP, Davies MA, Diab A, et al. (2018). Prospective Analysis of Adoptive TIL Therapy in Patients with Metastatic Melanoma: Response, Impact of Anti-CTLA4, and Biomarkers to Predict Clinical Outcome. Clin Cancer Res 24(18): 4416-4428. 10.1158/1078-0432.Ccr-17-3649
- Kirtane K, Elmariah H, Chung CH, Abate-Daga D (2021). Adoptive cellular therapy in solid tumor malignancies: review of the literature and challenges ahead. J Immunother Cancer 9(7): e002723. 10.1136/jitc-2021-002723
- Farhood B, Najafi M, Mortezaee K (2019). CD8(+) cytotoxic T lymphocytes in cancer immunotherapy: A review. J Cell Physiol 234(6): 8509-8521. 10.1002/jcp.27782
- Apetoh L, Smyth MJ, Drake CG, Abastado JP, Apte RN, Ayyoub M, Blay JY, Bonneville M, Butterfield LH, Caignard A, Castelli C, Cavallo F, Celis E, Chen L, Colombo MP, Comin-Anduix B, Coukos G, Dhodapkar MV, Dranoff G, Frazer IH, Fridman WH, Gabrilovich DI, Gilboa E, Gnjatic S, Jäger D, Kalinski P, Kaufman HL, Kiessling R, Kirkwood J, Knuth A, et al. (2015). Consensus nomenclature for CD8(+) T cell phenotypes in cancer. Oncoimmunology 4(4): e998538. 10.1080/2162402x.2014.998538
- Borst J, Ahrends T, Bąbała N, Melief CJM, Kastenmüller W (2018). CD4(+) T cell help in cancer immunology and immunotherapy. Nat Rev Immunol 18(10): 635-647. 10.1038/s41577-018-0044-0
- Melssen M, Slingluff CL, Jr. (2017). Vaccines targeting helper T cells for cancer immunotherapy. Curr Opin Immunol 47: 85-92. 10.1016/j.coi.2017.07.004
- Melenhorst JJ, Chen GM, Wang M, Porter DL, Chen C, Collins MA, Gao P, Bandyopadhyay S, Sun H, Zhao Z, Lundh S, Pruteanu-Malinici I, Nobles CL, Maji S, Frey NV, Gill SI, Loren AW, Tian L, Kulikovskaya I, Gupta M, Ambrose DE, Davis MM, Fraietta JA, Brogdon JL, Young RM, Chew A, Levine BL, Siegel DL, Alanio C, Wherry EJ, et al. (2022). Decade-long leukaemia remissions with persistence of CD4(+) CAR T cells. Nature 602(7897): 503-509. 10.1038/s41586-021-04390-6
- Schreiber RD, Old LJ, Smyth MJ (2011). Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science 331(6024): 1565-1570. 10.1126/science.1203486
- Man SM, Jenkins BJ (2022). Context-dependent functions of pattern recognition receptors in cancer. Nat Rev Cancer 22(7): 397-413. 10.1038/s41568-022-00462-5
- Chen Q, Sun L, Chen ZJ (2016). Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol 17(10): 1142-1149. 10.1038/ni.3558
- Decout A, Katz JD, Venkatraman S, Ablasser A (2021). The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat Rev Immunol 21(9): 548-569. 10.1038/s41577-021-00524-z
- Zhu Y, An X, Zhang X, Qiao Y, Zheng T, Li X (2019). STING: a master regulator in the cancer-immunity cycle. Mol Cancer 18(1): 152. 10.1186/s12943-019-1087-y
- Flood BA, Higgs EF, Li S, Luke JJ, Gajewski TF (2019). STING pathway agonism as a cancer therapeutic. Immunol Rev 290(1): 24-38. 10.1111/imr.12765
- Jiang X, Wang J, Zheng X, Liu Z, Zhang X, Li Y, Wilhelm J, Cao J, Huang G, Zhang J, Sumer B, Lea J, Lu Z, Gao J, Luo M (2022). Intratumoral administration of STING-activating nanovaccine enhances T cell immunotherapy. J Immunother Cancer 10(5): e003960. 10.1136/jitc-2021-003960
- Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, Duggan R, Wang Y, Barber GN, Fitzgerald KA, Alegre ML, Gajewski TF (2014). STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 41(5): 830-842. 10.1016/j.immuni.2014.10.017
- Tian Z, Zeng Y, Peng Y, Liu J, Wu F (2022). Cancer immunotherapy strategies that target the cGAS-STING pathway. Front Immunol 13: 996663. 10.3389/fimmu.2022.996663
- Dorta-Estremera S, Hegde VL, Slay RB, Sun R, Yanamandra AV, Nicholas C, Nookala S, Sierra G, Curran MA, Sastry KJ (2019). Targeting interferon signaling and CTLA-4 enhance the therapeutic efficacy of anti-PD-1 immunotherapy in preclinical model of HPV(+) oral cancer. J Immunother Cancer 7(1): 252. 10.1186/s40425-019-0728-4
- Lv M, Chen M, Zhang R, Zhang W, Wang C, Zhang Y, Wei X, Guan Y, Liu J, Feng K, Jing M, Wang X, Liu YC, Mei Q, Han W, Jiang Z (2020). Manganese is critical for antitumor immune responses via cGAS-STING and improves the efficacy of clinical immunotherapy. Cell Res 30(11): 966-979. 10.1038/s41422-020-00395-4
- Demaria O, De Gassart A, Coso S, Gestermann N, Di Domizio J, Flatz L, Gaide O, Michielin O, Hwu P, Petrova TV, Martinon F, Modlin RL, Speiser DE, Gilliet M (2015). STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc Natl Acad Sci USA 112(50): 15408-15413. 10.1073/pnas.1512832112
- Ager CR, Reilley MJ, Nicholas C, Bartkowiak T, Jaiswal AR, Curran MA (2017). Intratumoral STING Activation with T-cell Checkpoint Modulation Generates Systemic Antitumor Immunity. Cancer Immunol Res 5(8): 676-684. 10.1158/2326-6066.CIR-17-0049
- Motedayen Aval L, Pease JE, Sharma R, Pinato DJ (2020). Challenges and Opportunities in the Clinical Development of STING Agonists for Cancer Immunotherapy. J Clin Med 9(10): 3323. 10.3390/jcm9103323
- Ghosh M, Saha S, Bettke J, Nagar R, Parrales A, Iwakuma T, van der Velden AWM, Martinez LA (2021). Mutant p53 suppresses innate immune signaling to promote tumorigenesis. Cancer Cell 39(4): 494-508 e495. 10.1016/j.ccell.2021.01.003
- Carozza JA, Brown JA, Bohnert V, Fernandez D, AlSaif Y, Mardjuki RE, Smith M, Li L (2020). Structure-Aided Development of Small-Molecule Inhibitors of ENPP1, the Extracellular Phosphodiesterase of the Immunotransmitter cGAMP. Cell Chem Biol 27(11): 1347-1358 e1345. 10.1016/j.chembiol.2020.07.007
- Zhao F, Liu A, Gong X, Chen H, Wei J, Chen B, Chen S, Yang R, Fan Y, Mao R (2022). Hypoxia-induced RNASEH2A limits activation of cGAS-STING signaling in HCC and predicts poor prognosis. Tumori 108(1): 63-76. 10.1177/03008916211026019
- de Mingo Pulido Á, Hänggi K, Celias DP, Gardner A, Li J, Batista-Bittencourt B, Mohamed E, Trillo-Tinoco J, Osunmakinde O, Peña R, Onimus A, Kaisho T, Kaufmann J, McEachern K, Soliman H, Luca VC, Rodriguez PC, Yu X, Ruffell B (2021). The inhibitory receptor TIM-3 limits activation of the cGAS-STING pathway in intra-tumoral dendritic cells by suppressing extracellular DNA uptake. Immunity 54(6): 1154-1167.e1157. 10.1016/j.immuni.2021.04.019
- Chen DS, Mellman I (2013). Oncology meets immunology: the cancer-immunity cycle. Immunity 39(1): 1-10. 10.1016/j.immuni.2013.07.012
- Cheng N, Watkins-Schulz R, Junkins RD, David CN, Johnson BM, Montgomery SA, Peine KJ, Darr DB, Yuan H, McKinnon KP, Liu Q, Miao L, Huang L, Bachelder EM, Ainslie KM, Ting JP (2018). A nanoparticle-incorporated STING activator enhances antitumor immunity in PD-L1-insensitive models of triple-negative breast cancer. JCI Insight 3(22): e120638. 10.1172/jci.insight.120638
- Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, Woo SR, Lemmens E, Banda T, Leong JJ, Metchette K, Dubensky TW, Jr., Gajewski TF (2015). Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep 11(7): 1018-1030. 10.1016/j.celrep.2015.04.031
- Ohkuri T, Kosaka A, Ishibashi K, Kumai T, Hirata Y, Ohara K, Nagato T, Oikawa K, Aoki N, Harabuchi Y, Celis E, Kobayashi H (2017). Intratumoral administration of cGAMP transiently accumulates potent macrophages for anti-tumor immunity at a mouse tumor site. Cancer Immunol Immunother 66(6): 705-716. 10.1007/s00262-017-1975-1
- Conde E, Vercher E, Soria-Castellano M, Suarez-Olmos J, Mancheño U, Elizalde E, Rodriguez ML, Glez-Vaz J, Casares N, Rodríguez-García E, Hommel M, González-Aseguinolaza G, Uranga-Murillo I, Pardo J, Alkorta G, Melero I, Lasarte J, Hervas-Stubbs S (2021). Epitope spreading driven by the joint action of CART cells and pharmacological STING stimulation counteracts tumor escape via antigen-loss variants. J Immunother Cancer 9(11): e003351. 10.1136/jitc-2021-003351
- Xu N, Palmer DC, Robeson AC, Shou P, Bommiasamy H, Laurie SJ, Willis C, Dotti G, Vincent BG, Restifo NP, Serody JS (2021). STING agonist promotes CAR T cell trafficking and persistence in breast cancer. J Exp Med 218(2): e20200844. 10.1084/jem.20200844
- Tay RE, Richardson EK, Toh HC (2021). Revisiting the role of CD4(+) T cells in cancer immunotherapy-new insights into old paradigms. Cancer Gene Ther 28(1-2): 5-17. 10.1038/s41417-020-0183-x
- Kim HJ, Cantor H (2014). CD4 T-cell subsets and tumor immunity: the helpful and the not-so-helpful. Cancer Immunol Res 2(2): 91-98. 10.1158/2326-6066.Cir-13-0216
- Bailey SR, Nelson MH, Himes RA, Li Z, Mehrotra S, Paulos CM (2014). Th17 cells in cancer: the ultimate identity crisis. Front Immunol 5: 276. 10.3389/fimmu.2014.00276
- Martin-Orozco N, Muranski P, Chung Y, Yang XO, Yamazaki T, Lu S, Hwu P, Restifo NP, Overwijk WW, Dong C (2009). T helper 17 cells promote cytotoxic T cell activation in tumor immunity. Immunity 31(5): 787-798. 10.1016/j.immuni.2009.09.014
- Knochelmann HM, Dwyer CJ, Smith AS, Bowers JS, Wyatt MM, Nelson MH, Rangel Rivera GO, Horton JD, Krieg C, Armeson K, Lesinski GB, Rubinstein MP, Li Z, Paulos CM (2020). IL6 Fuels Durable Memory for Th17 Cell-Mediated Responses to Tumors. Cancer Res 80(18): 3920-3932. 10.1158/0008-5472.Can-19-3685
- Muranski P, Boni A, Antony PA, Cassard L, Irvine KR, Kaiser A, Paulos CM, Palmer DC, Touloukian CE, Ptak K, Gattinoni L, Wrzesinski C, Hinrichs CS, Kerstann KW, Feigenbaum L, Chan CC, Restifo NP (2008). Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood 112(2): 362-373. 10.1182/blood-2007-11-120998
- Guéry L, Hugues S (2015). Th17 Cell Plasticity and Functions in Cancer Immunity. Biomed Res Int 2015: 314620. 10.1155/2015/314620
- Muranski P, Restifo NP (2013). Essentials of Th17 cell commitment and plasticity. Blood 121(13): 2402-2414. 10.1182/blood-2012-09-378653
- Muranski P, Borman ZA, Kerkar SP, Klebanoff CA, Ji Y, Sanchez-Perez L, Sukumar M, Reger RN, Yu Z, Kern SJ, Roychoudhuri R, Ferreyra GA, Shen W, Durum SK, Feigenbaum L, Palmer DC, Antony PA, Chan CC, Laurence A, Danner RL, Gattinoni L, Restifo NP (2011). Th17 cells are long lived and retain a stem cell-like molecular signature. Immunity 35(6): 972-985. 10.1016/j.immuni.2011.09.019
- Larkin B, Ilyukha V, Sorokin M, Buzdin A, Vannier E, Poltorak A (2017). Cutting Edge: Activation of STING in T Cells Induces Type I IFN Responses and Cell Death. J Immunol 199(2): 397-402. 10.4049/jimmunol.1601999
- Gulen MF, Koch U, Haag SM, Schuler F, Apetoh L, Villunger A, Radtke F, Ablasser A (2017). Signalling strength determines proapoptotic functions of STING. Nat Commun 8(1): 427. 10.1038/s41467-017-00573-w
- Benoit-Lizon I, Jacquin E, Rivera Vargas T, Richard C, Roussey A, Dal Zuffo L, Martin T, Melis A, Vinokurova D, Shahoei SH, Baeza Garcia A, Pignol C, Giorgiutti S, Carapito R, Boidot R, Végran F, Flavell RA, Ryffel B, Nelson ER, Soulas-Sprauel P, Lawrence T, Apetoh L (2022). CD4 T cell-intrinsic STING signaling controls the differentiation and effector functions of T(H)1 and T(H)9 cells. J Immunother Cancer 10(1): e003459. 10.1136/jitc-2021-003459
- Li W, Lu L, Lu J, Wang X, Yang C, Jin J, Wu L, Hong X, Li F, Cao D, Yang Y, Wu M, Su B, Cheng J, Yang X, Di W, Deng L (2020). cGAS-STING-mediated DNA sensing maintains CD8(+) T cell stemness and promotes antitumor T cell therapy. Sci Transl Med 12(549): eaay9013. 10.1126/scitranslmed.aay9013
- Dardalhon V, Awasthi A, Kwon H, Galileos G, Gao W, Sobel RA, Mitsdoerffer M, Strom TB, Elyaman W, Ho IC, Khoury S, Oukka M, Kuchroo VK (2008). IL-4 inhibits TGF-beta-induced Foxp3+ T cells and, together with TGF-beta, generates IL-9+ IL-10+ Foxp3(-) effector T cells. Nat Immunol 9(12): 1347-1355. 10.1038/ni.1677
- Végran F, Berger H, Boidot R, Mignot G, Bruchard M, Dosset M, Chalmin F, Rébé C, Dérangère V, Ryffel B, Kato M, Prévost-Blondel A, Ghiringhelli F, Apetoh L (2014). The transcription factor IRF1 dictates the IL-21-dependent anticancer functions of TH9 cells. Nat Immunol 15(8): 758-766. 10.1038/ni.2925
- Benoit-Lizon I, Apetoh L (2021). Harnessing T(H)9 cells in cancer immunotherapy. Semin Immunol 52: 101477. 10.1016/j.smim.2021.101477
- Kaplan MH, Hufford MM, Olson MR (2015). The development and in vivo function of T helper 9 cells. Nat Rev Immunol 15(5): 295-307. 10.1038/nri3824
- Purwar R, Schlapbach C, Xiao S, Kang HS, Elyaman W, Jiang X, Jetten AM, Khoury SJ, Fuhlbrigge RC, Kuchroo VK, Clark RA, Kupper TS (2012). Robust tumor immunity to melanoma mediated by interleukin-9-producing T cells. Nat Med 18(8): 1248-1253. 10.1038/nm.2856
- Lu Y, Wang Q, Xue G, Bi E, Ma X, Wang A, Qian J, Dong C, Yi Q (2018). Th9 Cells Represent a Unique Subset of CD4(+) T Cells Endowed with the Ability to Eradicate Advanced Tumors. Cancer Cell 33(6): 1048-1060.e1047. 10.1016/j.ccell.2018.05.004
- Chen T, Xue Y, Wang S, Lu J, Zhou H, Zhang W, Zhou Z, Li B, Li Y, Wang Z, Li C, Eloy Y, Sun H, Shen Y, Diarra MD, Ge C, Chai X, Mou H, Lin P, Yu X, Ye Z (2023). Enhancement of T cell infiltration via tumor-targeted Th9 cell delivery improves the efficacy of antitumor immunotherapy of solid tumors. Bioact Mater 23: 508-523. 10.1016/j.bioactmat.2022.11.022
- Xue G, Zheng N, Fang J, Jin G, Li X, Dotti G, Yi Q, Lu Y (2021). Adoptive cell therapy with tumor-specific Th9 cells induces viral mimicry to eliminate antigen-loss-variant tumor cells. Cancer Cell 39(12): 1610-1622.e1619. 10.1016/j.ccell.2021.09.011
- Jiang Y, Chen J, Bi E, Zhao Y, Qin T, Wang Y, Wang A, Gao S, Yi Q, Wang S (2019). TNF-α enhances Th9 cell differentiation and antitumor immunity via TNFR2-dependent pathways. J Immunother Cancer 7(1): 28. 10.1186/s40425-018-0494-8
- Kim IK, Kim BS, Koh CH, Seok JW, Park JS, Shin KS, Bae EA, Lee GE, Jeon H, Cho J, Jung Y, Han D, Kwon BS, Lee HY, Chung Y, Kang CY (2015). Glucocorticoid-induced tumor necrosis factor receptor-related protein co-stimulation facilitates tumor regression by inducing IL-9-producing helper T cells. Nat Med 21(9): 1010-1017. 10.1038/nm.3922
- Xiao X, Balasubramanian S, Liu W, Chu X, Wang H, Taparowsky EJ, Fu YX, Choi Y, Walsh MC, Li XC (2012). OX40 signaling favors the induction of T(H)9 cells and airway inflammation. Nat Immunol 13(10): 981-990. 10.1038/ni.2390
- Richard AC, Tan C, Hawley ET, Gomez-Rodriguez J, Goswami R, Yang XP, Cruz AC, Penumetcha P, Hayes ET, Pelletier M, Gabay O, Walsh M, Ferdinand JR, Keane-Myers A, Choi Y, O’Shea JJ, Al-Shamkhani A, Kaplan MH, Gery I, Siegel RM, Meylan F (2015). The TNF-family ligand TL1A and its receptor DR3 promote T cell-mediated allergic immunopathology by enhancing differentiation and pathogenicity of IL-9-producing T cells. J Immunol 194(8): 3567-3582. 10.4049/jimmunol.1401220
- Ishikawa H, Barber GN (2008). STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455(7213): 674-678. 10.1038/nature07317
- Garland KM, Sheehy TL, Wilson JT (2022). Chemical and Biomolecular Strategies for STING Pathway Activation in Cancer Immunotherapy. Chem Rev 122(6): 5977-6039. 10.1021/acs.chemrev.1c00750
- Jneid B, Bochnakian A, Hoffmann C, Delisle F, Djacoto E, Sirven P, Denizeau J, Sedlik C, Gerber-Ferder Y, Fiore F, Akyol R, Brousse C, Kramer R, Walters I, Carlioz S, Salmon H, Malissen B, Dalod M, Piaggio E, Manel N (2023). Selective STING stimulation in dendritic cells primes antitumor T cell responses. Sci Immunol 8(79): eabn6612. 10.1126/sciimmunol.abn6612
–
ACKNOWLEDGMENTS
This work received funding by the Marie Sklodowska-Curie Training Network for Optimizing Adoptive T Cell Therapy of Cancer (funded by the Horizon 2020 programme of the European Union; grant 955575). This work was supported in part by the Brown Center for Immunotherapy at Indiana University Melvin and Bren Simon Comprehensive Cancer Center. All figures were created with Biorender.com.
COPYRIGHT
© 2023

STING-driven activation of T cells: relevance for the adoptive cell therapy of cancer by Richter et al. is licensed under a Creative Commons Attribution 4.0 International License.


