Viewpoint:
Cell Stress, Vol. 2, No. 7, pp. 150 - 161; doi: 10.15698/cst2018.07.143
Alzheimer’s disease: amyloid-based pathogenesis and potential therapies

1 Medical College of Soochow University, Soochow University, Suzhou, Jiangsu 215004, China.
2 Jiangsu Key Laboratory of Neuropsychiatric Diseases and Institute of Neuroscience, Soochow University, Suzhou, Jiangsu 215123, China.
Keywords: Alzheimer’s disease (AD), amyloid β (Aβ), synapses, neural circuits, treatment.
Abbreviations:
Aβ – amyloid β,
AD – Alzheimer’s disease,
APP – amyloid precursor protein,
BBB – blood-brain barrier,
FAD – familial AD,
LDLR – low-density lipoprotein receptor,
LTD – long-term depression,
LTP – long-term potentiation,
NMDA – N-methyl-D-aspartic acid
NMDAR – NMDA receptor, PrPc – cellular prion protein.
Received originally: 14/02/2018 Received in revised form: 20/05/2018
Accepted: 24/06/2018
Published: 29/06/2018
Correspondence:
Yaobo Liu, Institute of Neuroscience, Soochow University, 199 Ren-Ai Road, Suzhou, Jiangsu 215123, China; Phone/Fax: 86-512-65882526 liuyaobo@suda.edu.cn
Conflict of interest statement: The authors declare no competing financial interests.
Please cite this article as: Yixiu Zhou, Yuhui Sun, Quan-Hong Ma and Yaobo Liu (2018). Alzheimer’s disease: amyloid-based pathogenesis and potential therapies. Cell Stress 2(7): 150-161. doi: 10.15698/cst2018.07.143
Abstract
Alzheimer’s disease is one of the most severe neurodegenerative diseases among elderly people. Different pathogenic factors for Alzheimer’s disease have been posited and studied in recent decades, but no effective treatment has been found, necessitating further studies. In this Viewpoint article, we assess studies on the mechanisms underlying the accumulation of amyloid b (Aβ) peptide and the formation of Aβ oligomers because their accumulation in amyloid plaques in brain tissue has become a well-studied hallmark of Alzheimer’s disease. We focus on the production of Aβ and its impact on the function of synapses and neural circuits, and also discuss the clinical prospects for amyloid-targeted therapies.
INTRODUCTION
Alzheimer’s disease (AD) is one of the most prevalent forms of dementia; in 2015, for example, AD affected ~46.8 million people worldwide. It is estimated that this number will continue to increase and reach 131.5 million in 2050 [1]. In China in 2014, for instance, ~10% of the population was 60 years of age or older (~212 million people), according to the National Bureau of Statistics of China [2]. Recent increase in life expectancy may greatly expand the future AD burden [3], and indeed, AD will have a larger impact on the economy of China and of the world [3]. Thus, a great deal of effort has been spent on studying the pathological mechanism of AD and on trying to find a treatment to cure AD. In this Viewpoint, we discuss one of the most central hypotheses, namely the amyloid cascade hypothesis, and subsequent research that complements or challenges it. A fundamental aspect of research on AD concerns the involvement of amyloid b (Aβ) in the pathophysiology of the disease and as a possible target for treatment. Despite these efforts, however, no anti-amyloid therapy has yet been established [4]. Hence, this Viewpoint focuses on attempts to research on amyloid-based pathogenesis and develop an amyloid-targeted therapy.
THE AMYLOID CASCADE HYPOTHESIS AND SUBSEQUENT STUDIES
One of the characteristic pathologies of AD is the presence of parenchymal amyloid plaques in the brain tissue of patients [5]. Aβ was first isolated from the meningeal vessels of AD patients in 1984 [6]. One year later, the same peptide was identified as the core of senile plaques observed in the brain tissue of AD patients [7]. These findings called researchers’ attention to the accumulation of the amyloid protein. Moreover, it was discovered that Down syndrome (trisomy 21) patients often develop AD later in life and the amyloid precursor protein (APP) gene is located on chromosome 21 [8]. Thus, the amyloid cascade hypothesis was first posited in 1992, which postulates that the accumulation of Aβ peptides initiates the pathogenesis of AD, leading to neurofibrillary tangles and neurodegeneration that cause memory loss [9]. Hardy et al. proposed that the overproduction of Aβ results from hyperactivation of the β and γ secretases (gain-of-function mechanism), which cleave APP and yield Aβ [8]. In the years since the hypothesis was proposed, the Aβ peptide has been a star molecule in most of the research on the pathophysiology of AD.
–
The amyloid cascade hypothesis has generated a lively discussion whether plaques are neurotoxic or protective. Although it was previously believed that plaques are the initiators of disease pathogenesis, Lee et al. argued that all available data are also consistent with the conclusion that amyloid plaques actually constitute a protective adaptation [10]. Meanwhile, Bishop et al. found that this apparent paradox became evident when Aβ was bound to metal ions, and the resulting complex could be neurotoxic or neuroprotective [11]. Moreover, it has been reported that soluble Aβ oligomers can impair synapse structure and function and that the smallest synaptotoxic species are dimers, whereas neither Aβ monomers nor soluble amyloid plaque cores significantly alter synaptic plasticity [12]. Now it is generally agreed that the soluble Aβ oligomers, rather than amyloid plaques, are synaptotoxic.
–
Aside from the debate concerning amyloid plaques and oligomers, new findings have arisen supporting the amyloid cascade hypothesis. Jonsson et al. made an astonishing discovery that a coding mutation (A673T) in the APP gene could protect against AD and cognitive decline in an elderly population with AD, which indicated that a reduction of b-cleavage of APP might protect against AD [13]. He et al. found that Aβ enhanced tau pathogenesis by creating a unique environment that facilitated tau aggregation at an early stage and helped translocate the tau “seeds” at a later stage [14].
–
However, although the accumulation of Aβ is acknowledged as a key factor in the cognitive deficit observed in AD patients, other studies have pointed out the weakness of the original amyloid cascade hypothesis and pointed out some challenges. Researchers raised concerns about the amyloid cascade hypothesis based on studies of familial AD (FAD), which is attributable to mutations in one of three genes, namely presenilin1 (PSEN1), presenilin2 (PSEN2), or APP [15]. Among them, presenilin1 and presenilin2 are the core components of γ-secretase, which cleaves the C-terminal fragment of APP produced by β-secretase cleavage within the plasma membrane, releasing Aβ [16]. Thus, many studies on FAD have focused on mutations in the gene encoding g-secretase. For instance, Xia et al. suggested that PSEN1 gene mutations could both abolish γ-secretase activity, which decreases the production of Aβ42 and Aβ40, and increase the Aβ42/Aβ40 ratio, which promotes Aβ deposition through a loss-of-function mechanism linked to familial AD onset [17]. Ben-Gedalya et al. reported that inhibition of cyclophilin B leads to presenilin1 misfolding, aggregation, and deposition, which reduces γ-secretase function and thus opposes the gain-of-function mechanism [18]. However, Szaruga et al. expressed doubt about the loss-of-function hypothesis and proposed an alternative view that “pathogenic mutations in PSEN cause disease by qualitative shifts in Aβ profile production (γ-secretase dysfunction)” [19]. In response to these challenges, Hardy et al. later argued that the loss-of-function hypothesis might overlook the elevation of Aβ43 and of other, longer Aβ species [20].
–
Moreover, some researchers have suggested that the simple linear pathway of tracing disease progression from Aβ to AD should be rejected [21]. Some clinical studies have reported that cognitive decline correlates only weakly with changes in Aβ burden and that a window of time exists between Aβ accumulation and AD onset [22]. Moreover, although the antineoplastic drug bexarotene rapidly clears amyloid plaques in mouse brain and reverses the cognitive decline of the mice [23], clinical trials with humans have not proved promising. For instance, the drug AN1792 could eliminate amyloid plaques quite well, but it could not reverse the neurodegeneration [24]. Thus, the opinion that the amyloid cascade hypothesis should be rejected is based on the fact that Aβ accumulation does not correlate with the immediate onset of AD and that elimination of amyloid plaques cannot stop neurodegeneration. However, because Aβ plaques are not necessarily sources of toxicity, as mentioned above, more evidence may be needed before the amyloid cascade hypothesis can be soundly rejected.
–
Also, compared with early-onset FAD, late-onset/sporadic AD, which affects most AD patients, has shown a different mechanism for pathogenesis. Indeed, most sporadic AD cases display normal γ-secretase activity, in contrast to FAD [19]. Moreover, neurofibrillary tangles develop sooner in PSEN-FAD, portending more rapid neuronal demise [25]. However, Thomas et al. suggested that changes in functional connectivity manifest similarly in both types of AD, and therefore early-onset AD might serve as a model for late-onset AD studies [26].
–
Thus, there are different and competing views regarding the amyloid cascade hypothesis, and no definite conclusions can be drawn at this time.
PRODUCTION OF Aβ
The Aβ peptides are proteolytic fragments derived from APP, which is an integral membrane protein found to exhibit both neurotoxic and neurotrophic protective effects [16]. The human APP gene is located on the long arm of the chromosome 21, and alternative splicing can produce various APP mRNAs encoding several isoforms [27]. The most common APP species in the brain is APP695, and it is produced mainly by neurons [28]. APP is synthesized and transported to the plasma membrane via the endoplasmic reticulum-Golgi secretory pathway [29]. Also, it has been proposed that APP can function as a cell-surface receptor, which can be bound by Aβ and regulate the production and downstream signaling of Aβ [27]. APP is transported along axons to presynaptic terminals, where it accumulates and leads to Aβ deposition at synapses [28]. Notably, two pathways are known to process APP, namely the amyloidogenic and non-amyloidogenic pathways [16], and the latter is the principal pathway under physiological conditions [30].
–
In the non-amyloidogenic pathway (Fig. 1A and Fig. 1B), APP is first cleaved by α-secretase, which is a member of the ADAM (A Disintegrin And Metalloproteinase) family and is abundant at the plasma membrane. This cleavage yields the soluble ectodomain sAPPβ and leaves the C-terminal fragment alpha (CTFα) in the plasma membrane. Subsequent cleavage of CTFα by γ-secretase releases a soluble extracellular peptide (p3) and the APP intracellular domain (AICD) [16]. Of note, the APP holoprotein can be bound by various low-density lipoprotein receptors (LDLRs), such as SORL1, which is an APP-specific sorting receptor. Any APP that binds LDLRs can be internalized and enters a recycling pathway. The absence of LDLRs can shunt APP into the β-secretase cleavage pathway (the amyloidogenic pathway) [31].
–
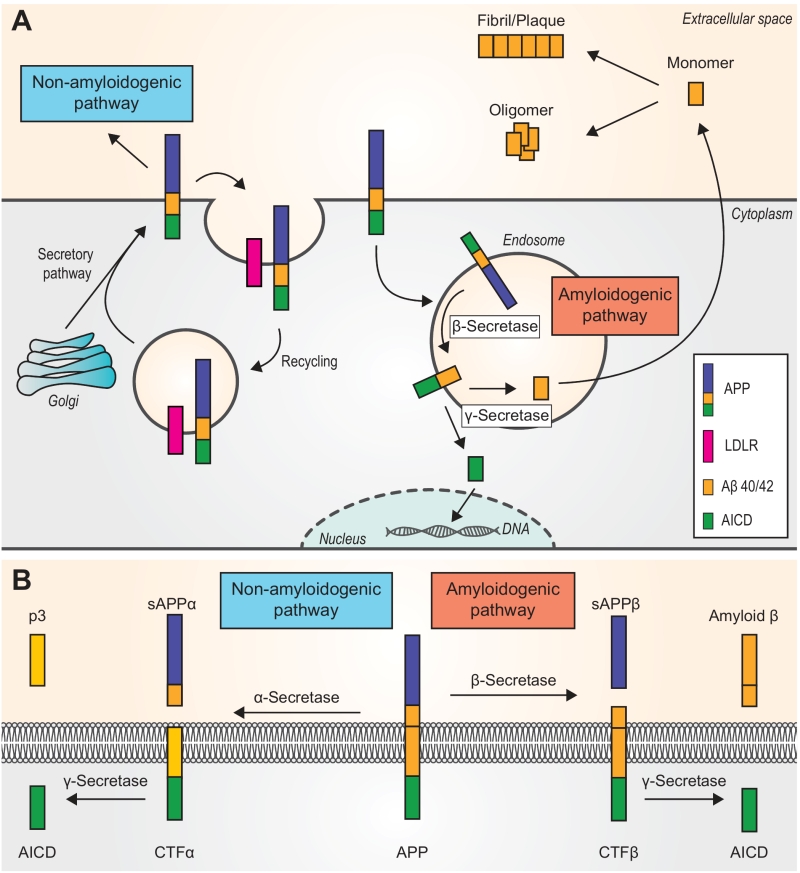 |
FIGURE 1: Two pathways of amyloid β (Aβ) peptides generation. Amyloid precursor protein (APP) is a type I integral membrane protein that is cleaved at different sites to yield various products. (A) An overview of the two pathways. APP is translocated from the cytoplasm into the endoplasmic reticulum where it enters the secretory pathway and then is transported to the neuronal plasma membrane. The majority of APP is processed via the non-amyloidogenic pathway (see panel B). APP can be recycled in endosomes by binding to LDLRs (low-density lipoprotein receptors). The right side of the figure shows the amyloidogenic pathway. Unlike the non-amyloidogenic pathway, which is carried out at the plasma membrane, the amyloidogenic pathway mainly occurs in endosomes. Ultimately, AICD is transferred to the nucleus, where it functions as a transcriptional factor, whereas the Aβ40/42 monomer is removed to the extracellular space. Monomers aggregate either into oligomers or fibrils/plaques. (B) Non-amyloidogenic (left) or amyloidogenic (right) pathway of APP processing. In the non-amyloidogenic pathway, APP is first cleaved by α-secretase, releasing a soluble ectodomain of APP called sAPPα and a membrane-tethered intracellular C-terminal fragment (CTFα). Then, the C-terminal fragment is further cleaved by γ-secretase to produce a 3-kDa peptide (p3) and an APP intracellular domain (AICD). In the amyloidogenic pathway, the products of β-secretase are a soluble ectodomain of APP (sAPPβ) and a C-terminal fragment β (CTFβ). The second step releases Amyloid b and AICD. APP, amyloid precursor protein. CTF alpha/beta, C-terminal fragment alpha/beta. sAPP alpha/beta, soluble ectodomain of APP. p3, 3-kD peptide. AICD, APP intracellular domain. |
–
The amyloidogenic pathway (Fig. 1A and Fig. 1B) involves APP trafficking through the secretory and recycling pathways where APP interacts with b- and γ-secretases [32]. After APP is internalized and delivered to endosomes [30], the first step of the pathway is catalyzed by β-secretase 1 (BACE-1), a single-transmembrane aspartyl protease. BACE-1 cleaves APP and generates the soluble ectodomain sAPPβ and CTFβ (left in the membrane). The subsequent hydrolysis of CTFβ by γ-secretase yields AICDs and Aβ monomers, which have dual physiological effects [16]. The majority of Aβ peptides are secreted to the extracellular space, although a small amount can aggregate inside neurons [30], whereas AICD is transported into the nucleus where it functions as a transcription factor [29]. In addition, γ-secretase cleaves CTFβ at different sites and in multiple sequential steps, which ultimately produces mainly two species of Aβ, namely Aβ40 and Aβ42 [27]. Under basal conditions, Aβ40 comprises ~90% of all Aβ produced [16].
–
The fact that various Aβ species are produced has a pathogenic impact on neurons, and the longer Aβ42 is believed to be more toxic than Aβ40 [33]. In addition, it has been reported that the ratio of Aβ42 to Aβ40 might predict the severity of AD [34], and indeed, this ratio has long been used as a biomarker in AD research. Early studies reported that PSEN mutations increase the Aβ42/Aβ40 ratio [17]. In the amyloidogenic pathway, γ-secretase can trim the epsilon site and gamma site of the transmembrane CTFβ. Thus, both mutations in the domain of the epsilon/gamma sites and the γ-secretase modulators can influence Aβ42 production. Mutations in the transmembrane domain of APP can increase the Aβ42/Aβ40 ratio, leading to aggressive early-onset FAD, whereas γ-secretase modulators can decrease the level of Aβ42 and thus have therapeutic potential [35]. In a recent investigation, Johnson et al. proposed that, under physiological conditions, small Aβ oligomers bound to the plasma membrane and further oligomerized with kinetics depending on the local Aβ42/Aβ40 ratio [36]. Siegel et al. discovered that the ratio was greatest for Aβ’ (the N-terminally truncated Aβ11-x produced from the 89-residue CTF), followed by Aβ and then p3, which provided new insight for the development of γ-secretase modulators [37]. Moreover, others suggested that the ratio could be used as the biomarker for the diagnosis of neurochemical dementia [38][39] as well as in clinical trials targeting cognitively normal individuals with high brain Aβ levels [40].
–
Once the Aβ monomer is produced, it either goes through the degradation process or accumulates to form other species of amyloids, such as oligomers, fibrils, etc. There are two main mechanisms by which amyloids are removed from cells. First, the monomer that forms in endosomes can be transferred to lysosomes in the neuron, where it is degraded. Second, if the monomer is released to the outside of the neuron, microglia can destroy it by releasing insulin-degrading enzyme [29]. Aside from this, there are two models for the formation of Aβ fibrillogenesis. The classic model posits that fibril formation is a nucleation-dependent polymerization process in which monomers give rise to oligomers, from which protofibrils form. Subsequently the protofibrils emanate full-length fibers. The new model, however, implies that protofibrils cannot form fibrils directly. Instead, protofibrils may be the precursors for fibrillogenesis [41]. Also, after studying several species of amyloids, i.e., monomers, oligomers, Aβ*56, etc., Ono noted that “soluble pre-fibrillar aggregates, that is, oligomers of Aβ, are proximate neurotoxins” [41].
–
Compared with Aβ42, which has been studied for many years, Aβ43 has been quite overlooked. A study published in 2011, however, suggested that Aβ43 is potentially toxic and amyloidogenic, perhaps to an extent greater than Aβ42 [42].
–
A new APP processing pathway was recently identified. This pathway generates proteolytic fragments of APP capable of inhibiting neuronal activity within the hippocampus. The cleavage of APP by η-secretase yields CTFη, which is cleaved by ADAM10 and BACE1 into long and short amyloid eta (Aηα, Aηβ). CTFη is abundant in dystrophic neurites, and its generation is usually mediated by membrane-bound matrix metalloproteinase [43]. Also, Aβ42 disrupts the barrier between the blood and cerebrospinal fluid via activation of matrix metalloproteinase [44]. Thus, a connection may exist between these two pathways.
Aβ AFFECTS SYNAPTIC AND NEURAL CIRCUIT FUNCTION
As mentioned above, Aβ peptide plays an important role in AD by influencing synapses and, in turn, neural circuits. Accumulation of Aβ in the brain parenchyma can lead to loss of dendritic spines and synapses as well as alterations in synaptic transmission and neural activity [45].
–
Although overproduction of the Aβ oligomer is pathogenic, a normal level of Aβ helps to maintain physiological homeostasis. A study using mouse/rat hippocampal slices suggested that Aβ might have a normal negative feedback function that regulates APP processing; in that study, Kamenetz et al. proposed that an elevated level of neuronal activity could enhance the activity of β-secretase, which would generate Aβ monomers. Overproduction of these monomers leads to synaptic depression, which in turn suppresses neuronal activity and further reduces Aβ production [46]. This constitutes a protective mechanism that modulates the production of Aβ monomers and can prevent monomer overproduction in nearby neurons. Nevertheless, a study using APP-transfected neurons in rat hippocampal slice cultures revealed that structural plasticity was reversibly impaired in APP-overexpressing cells [47]. Continuous overproduction of Aβ oligomers at either dendrites or axons can lower spine density and plasticity; this “synaptic pruning” in spine number can be reduced by blocking action potentials, nicotinic acetylcholine receptors (nAChRs), or N-methyl-d-aspartic acid (NMDA) receptors (NMDARs) [47]. Aβ-mediated spine loss requires the upregulation of NMDA-type glutamate receptor–dependent activity and the subsequent cascade of signaling that includes cofilin and calcineurin. Shankar et al. experimented on mouse hippocampal neurons and found that activation of NMDARs could induce either long-term depression (LTD) or long-term potentiation (LTP) depending on the Ca2+ concentration. Soluble Aβ oligomers can partially block NMDARs, which either reduces Ca2+ influx or enhances NMDAR-dependent activation of calcineurin. Decreased Ca2+ influx through NMDARs can induce LTD through a calcineurin-dependent pathway [48]. Also, GSK3 activity stimulated by Aβ may lead to NMDAR-dependent LTD and inhibit LTP [49], resulting in loss of synaptic spine and eventually neurodegeneration [50].
–
Because the dendritic spine is part of a synapse, spine loss can also affect synaptic activity. It has been suggested that Aβ oligomers play a central role in controlling neural activity at specific types of synapses which affects the neural circuits [51]. Aside from NMDARs, Aβ oligomers are heterogeneous and have high affinity to specific types of receptors, which activate various signaling pathways leading to inevitable cell death [52]. For instance, Lauren et al. identified the cellular prion protein (PrPC) as an Aβ oligomer receptor by expression cloning and discovered that PrPC mediated the inhibition of LTP in a wild-type mouse hippocampal slice by the binding of Aβ42 oligomers. Presumably, PrPC interacts with NMDAR subunit 2D and inhibits its function by initiating a signaling cascade that modifies synaptic morphology and functions in the brain [53]. Kim et al. observed that murine PirB (paired immunoglobulin-like receptor B) was associated with memory deficits in adult mice as well as loss of synaptic plasticity in the juvenile visual cortex. The selective binding of Aβ42 to PirB could lead to increased interactions between PirB and cofilin or protein phosphatases in APP/presenilin1 mice. They proposed that the human homolog LilrB2 (leukocyte immunoglobulin-like receptor B2) might also enhance cofilin signaling, which is seen in the human AD brain [54]. Yamamoto et al. suggested that Aβ oligomers can lead to nerve growth factor receptor–mediated cell death through the p75 neurotrophin receptor [55]. Zhao et al. reported that Aβ oligomers can impair the function of neuronal insulin receptors in rat hippocampal and cortical neurons, indicating that insulin resistance in the AD brain is a response to the Aβ-derived diffusible ligands (ADDLs) [56]. Moreover, insulin receptor–mediated interference with Aβ production prevented the rapid activation of a specific kinase required for LTP [57]. Also, Magdesian et al. found that Aβ oligomers could bind to the Frizzled cysteine-rich domain at, or in close proximity to, the Wnt-binding site and inhibit the canonical Wnt signaling pathway, which caused tau phosphorylation and neurofibrillary tangles [58]. Moreover, it has even been reported that Aβ peptides can form oligomeric ion channels, assisted by cholesterol, and that these channels can induce an increase of Ca2+ level in neurons [59]. Furthermore, coupled with the increase in membrane permeability induced by Aβ oligomers [60], the channels can disrupt Ca2+ homeostasis and lead to neurodegeneration.
–
Normally, a small increase in Aβ level will increase the probability of releasing synaptic vesicles (Fig. 2A). Such an increase can promote activation of presynaptic acetylcholine receptors, which increases the internal concentration of Ca2+. The high concentration of Ca2+ increases glutamate release and promotes excitatory neural activity [51][61]. Aβ can participate in a positive feedback loop in which it binds to nAChRs that are close to the site of Aβ secretion, resulting in increased intracellular Ca2+. An increase in Ca2+ concentration may increase Aβ production [47]. It is possible that an excessive increase of Aβ will lead to the aforementioned negative feedback loop and eventually to compensatory suppression of the neuronal activity [46]. Palop et al. suggested that Aβ can trigger intermittent and aberrant excitatory neuronal activity in the cortex and hippocampus, which might result in remodeling of the inhibitory circuitry. They proposed that a “high level of Aβ induces aberrant excitatory neuronal activity, which triggers compensatory inhibitory mechanisms to counteract overexcitation”, and both the excitation and inhibition might be involved in AD-related network dysfunction [62]. A high level of Aβ leads to aberrant neuronal activity by enhancing synchrony among the remaining glutamatergic synapses [51]. The acute effects induced by endogenous Aβ in certain studies were exclusively presynaptic. An increase or significant decrease in Aβ level impairs short-term synaptic facilitation [61]. Thus, the level of Aβ must be maintained within an intermediate range, and either a low or high level can negatively impact presynaptic facilitation by decreasing presynaptic efficacy or postsynaptic depression. It has been suggested that excitatory synapses are highly sensitive to changes in Aβ level, whereas inhibitory synapses are relatively immune to the immediate effects of Aβ [61]. The relative decrease in inhibition can lead to hyperactivity as well as abnormal synchronization [63]. In addition, Siskova et al. reported that interneurons can alter their excitability and synchronizing function owing to cell type–specific vulnerability and/or persistently altered input. Thus, dendritic structural dysfunctions may be linked to neuronal hyperexcitability [64].
–
A small increase in Aβ will enhance LTP and memory [61], whereas an acute increase in synaptic Aβ can induce LTD instead [51][65]. A decrease in the density of postsynaptic glutamate receptors, such as AMPARs and NMDARs, and the activation of the calcineurin-dependent pathway, which is involved in Aβ-induced spine loss, is also necessary for LTD [49]. Accumulation of glutamate initially results in postsynaptic depolarization through AMPARs and then the activation of NMDARs [66]. However, long-term activation leads to receptor desensitization and internalization of NMDARs and AMPARs. Moreover, the binding and activation of Aβ on alpha-7 nAChRs induces endocytosis of NMDARs through the action of protein phosphatase 2B [67]. Changes in the number of the NMDARs can affect NMDAR-dependent Ca2+ influx, which is responsible for initiating LTD (Fig. 2B). Activation of perisynaptic NMDARs can lead to LTD [66], and over-activation of extrasynaptic NR2B-containing NMDARs can inhibit hippocampal LTP. Soluble Aβ increases the phosphorylation of p38 MAPK, which contributes to the inhibition of LTP and subsequently impairs the ERK and CREB signaling pathway [68]. Um et al. demonstrated that Aβo/PrPC complexes could activate the Fyn signaling pathway, which increases the density of cell-surface NMDARs and excitotoxicity, resulting in the loss of both dendritic spines and cell-surface receptors [69].
–
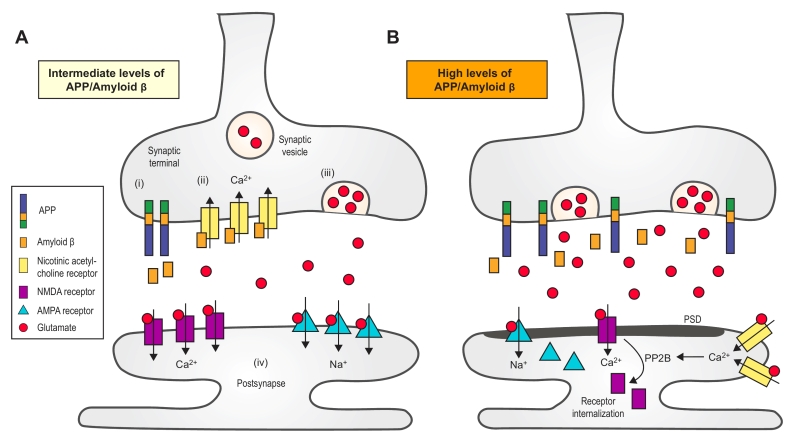 |
FIGURE 2: Synaptic transmission regulated by amyloid b (Aβ). (A) An intermediate increase in Aβ level can only increase the probability of releasing synaptic vesicles. The processing of amy-loid precursor protein (APP) at the synaptic terminal yields Aβ peptides (i), which bind to and activate the presynaptic a7-nAChRs (ii). Moreover, the subsequent influx of Ca2+ is mediated by nAChRs, which, in turn, trigger the release of glutamate from the synaptic vesicles (iii). Glutamate activates both AMPARs and NMDARs, which induce synaptic potentiation (iv). (B) A dramatic increase of Aβ, however, can lead to LTD. First, accumulation of glutamate results in the long-term activation of NMDARs and AMPARs, which facilitates their internalization. Second, Aβ might redistribute NMDARs. Third, the activation of perisynaptic a7-nAChRs activates the protein phosphatase 2B (PP2B), a Ca2+-sensitive enzyme that induces the internalization of NMDARs [67]. a7-nAChR, a7-nicotinic acetylcholine receptor. PSD, post-synaptic density. NMDAR, N-methyl-D-aspartate receptor. AMPAR, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor. |
–
It has been suggested that the formation of neural circuits and memories is impaired by weakened connectivity owing to chronic elevation of Aβ [47]. An increased level of Aβ results in an aberrant excitatory network and compensatory inhibition of learning and memory circuits, which promotes cognitive decline [62]. Long-term accumulation of Aβ, disinhibition of excitatory cells, and synaptic loss lead to neuronal hyperactivity, which may lead to epileptiform activity [45][70]. Coincidently, in some pre-symptomatic individuals who eventually develop AD, neuronal hyperactivity was found in regions – such as hippocampus – that are associated with learning and memory [45]. Additionally, AD impairs slow wave oscillations, which consolidate recently acquired memories in the cortical area, thalamus, and hippocampus [63]. Moreover, excessive oligomeric Aβ binding to cell-surface receptors can induce neuronal apoptosis [52]. Long-term accumulation of Aβ results in oxidative damage to both DNA and proteins, which also leads to cell death [71] and impairs the affected brain regions, although the level of Aβ plateaus before the onset of rapid neurodegeneration and cognitive symptoms [72]. Thus, excessive neuronal activity, hyper-synchrony, impaired oscillations, and cell death, etc., could be key features of AD. It should be noted that aside from Aβ, GABAergic dysfunction also contributes to the formation of the aberrant neural networks that are typical of AD [51]. Thus, diverse mechanisms may contribute to neural network dysfunction in AD.
TREATMENT BASED ON Aβ HAS NOT BEEN ENTIRELY SUCCESSFUL
Owing to its key role in AD, amyloid-targeted therapy has become a major research interest. There are two main ways to reduce excessive level of Aβ in neurons, namely (i) to correct the aberrant generation of Aβ and (ii) repair the faulty clearance mechanism (Fig. 3).
–
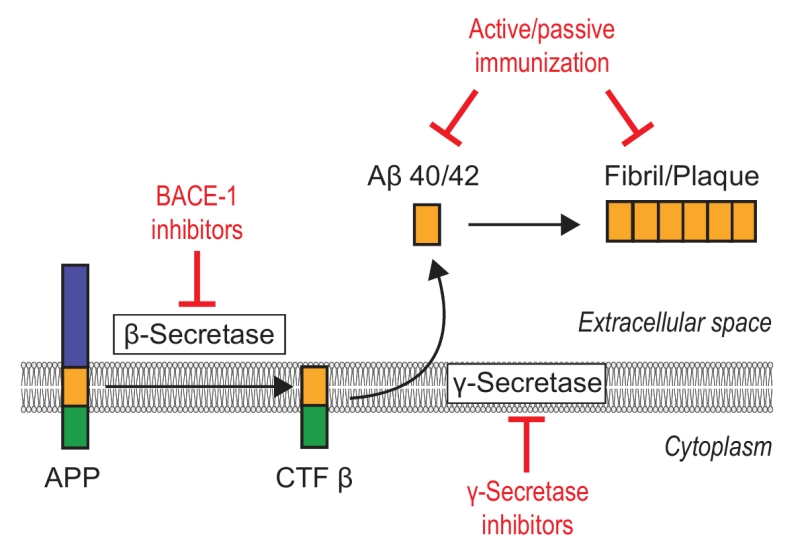 | FIGURE 3: Current treatment strategies based on the amyloid cascade hypothesis. Recent or ongoing clinical trials have mainly focused on inhibition of β-secretase (BACE-1), γ-secretase, or immunization strategies against Aβ monomers or plaques. See text for details. CTFβ, C-terminal fragment beta. |
–
One attractive therapeutic strategy to reduce Aβ production are drugs that modulate the activity of the enzymes β-secretase or γ-secretase, especially inhibitors of β-secretase (BACE-1). Although considerable efforts have been made to develop BACE-1 inhibitors, most trials have failed due to insufficient target specificity, brain permeability, and/or research design without testing cognitive outcomes or measuring Aβ level [71][73]. The first generation of large-molecule drugs failed because of their unfavorable pharmacological properties, such as the inability to cross the blood-brain barrier (BBB), and the second generation of small-molecule compounds still could not effectively penetrate the BBB [71]. The third-generation drugs, such as verubecestat (Merck & Co.), AZD-3293 (AstraZeneca and Eli Lilly), and JNJ-54861911 (Janssen Research & Development), are BACE-1 inhibitors currently in Phase III trials [74]. In February 2017, Merck halted its late-stage trial of verubecestat for mild-to-moderate Alzheimer’s disease (EPOCH) after it was reported as having “virtually no chance of finding a positive clinical effect” according to an independent panel of experts [75]. With respect to patients with prodromal Alzheimer’s disease (APECS), however, the results of Merck’s trial of verubecestat for that purpose are expected in February 2019 [75]. However, AZD-3293 yielded favorable results in Phase I; this compound can cross the BBB and is orally active and well tolerated up to the highest dose given. In September 2014, a large, pivotal phase II/III trial (AMARANTH) started and another Phase III trial (DAYBREAK-ALZ) was initiated in 2016 for AD patients with mild dementia. These trials will end in 2019 and 2021, respectively [76]. As for γ-secretase, the failure to develop an inhibitor is attributable to a limited understanding of the structure of γ-secretase. Inhibitors like semagacestat proved ineffective in clinical trials [73]. Although previous results were not satisfying, peptide-based aggregation inhibitors hold significant promise for future AD therapy owing to their high selectivity and low toxicity, among other attributes [77].
–
Alternatively, both active and passive strategies of immunization with the peptide are potential routes to enhance amyloid plaque clearance in the parenchyma. Active immunization is achieved via immunization with intact Aβ42 peptide or Aβ fragments, whereas passive immunization is achieved with anti-Aβ antibodies [78].
–
Some of the trials that have focused on active immunization with Aβ succeeded in reducing the level of Aβ peptide in patients, but with severe side effects, such as subacute meningoencephalitis (AN1792, Janssen, Pfizer) [79]. Although the trials were halted, subsequent investigations were carried with former participants. Holmes et al. reported that AN1792 could eliminate amyloid plaques fairly well, but the treatment did not stop the process of neurodegeneration [24], which lends credence to the opinion that amyloid plaques are not necessarily neurotoxic. Another study in 2015 showed that AN1792 could accelerate the removal of damaged neurons involving activated microglia [80]. Aside from AN1792, investigators have also developed other active immunization strategies. For instance, Mulder et al. demonstrated that a trivalent vaccine of small Aβ-derived cyclopeptide conjugates could effectively induce a specific antibody response against misfolded Aβ without noticeable side effects [81]. Further, a trial with CAD106 (Novartis Pharmaceuticals Corporation) is now in Phase II/III after five multicenter Phase II trials. Unlike AN1792, CAD106 is generally well tolerated, with no evidence of central nervous system inflammation [82]. Recently, a P-particle–based Aβ epitope vaccine showed promise by reducing amyloid deposition, rescuing memory loss, and restoring Aβ homeostasis in vivo [83]. Thus, active immunization might still be a potential therapy for AD.
–
In recent years, research interests have focused on developing monoclonal antibodies against Aβ; for example, solanezumab against the middle region of Aβ (developed by Ely Lilly), aducanumab against aggregated Aβ (tested by Biogen), and bapineuzumab against the N-terminus of Aβ (directed by OOP & Johnson), among others. The efficacy of Aβ immunization strategies has not been consistent across clinical trials [84]. Although trials of Aβ monoclonal antibodies such as solanezumab and bapineuzumab have proved effective for reducing Aβ level, little improvement in cognition has been achieved for AD patients. In two Phase III clinical trials in 2012, solanezumab, which was once considered the most promising drug for AD treatment, failed to show significant cognition benefits. In a subsequent Phase III trial that ended in November 2016, solanezumab did not show a significant effect on slowing cognitive decline in patients mildly affected with AD [85]. Recently, a more exciting Phase Ib study showed that treatment with aducanumab (BIIB037), a human monoclonal antibody against aggregated Aβ, reduced Aβ deposits in the brain in a dose- and time-dependent manner, as assessed with florbetapir-based PET imaging. This suggested a slowing of clinical progression based on scores from the Clinical Dementia Rating-Sum of Boxes and Mini Mental State Examination [86].
–
All in all, the failure of numerous clinical trials of anti-amyloid agents can be attributed to many factors, such as inadequate preclinical data, poor brain penetration, or poor understanding of amyloid function, among others. In a review, Hardy et al. noted that “AD trials done prior to obligatory amyloid-PET imaging turned out to have up to ~25% of subjects that were amyloid-negative” [20]. Thus, the unsuitable choice of trial candidates may also be one of the factors contributing to the failure of some clinical candidates.
–
Aside from anti-amyloid agents, a study published in 2016 suggested that gamma oscillations may be a prospect for treatment of AD by reducing total amyloid levels via decreased amyloidogenesis and increased amyloid endocytosis by microglia, and the effects were not specific to one animal model [87]. In addition, Sorrentino et al. reported that mitochondrial abnormalities also play a role in the pathogenesis of AD and that boosting mitochondrial function and proteostasis might decrease the formation of Aβ aggregates [88]. Thus, mitochondrial proteostasis may also provide a new insight to amyloid-targeted therapies. Moreover, Ono reported that rosmarinic acid (RA) could inhibit Aβ40/42 oligomerization and decrease oligomer-induced synaptic toxicity [41].
CONCLUSIONS
The discovery of amyloid plaques in the brain tissue of AD patients and subsequent findings concerning APP genes naturally led to the amyloid cascade hypothesis. Unsurprisingly, Aβ peptide plays an important role in the course of AD development. In the more than 20 years since the proposal of the original hypothesis, a substantial number of published reports have helped bolster research on AD and treatment strategies. Serious concerns have been raised about efficacy, however, yet new discoveries have been made. The amyloid cascade hypothesis, although still controversial, continues to help guide AD research. It is agreed that Aβ oligomers, instead of amyloid plaques, constitute the primary cause of toxicity, and Aβ42 seems to be more toxic than Aβ40, as the length of the peptide determines the toxicity [45]. Aβ oligomers disrupt synaptic activity, although Aβ has now been shown not to be responsible for certain pathological effects of AD, as was previously presumed. An intermediate level of Aβ enhances presynaptic excitation whereas elevated or reduced levels depress synaptic function. Moreover, Aβ oligomers have opposite effects on excitatory and inhibitory synapses, and thus their impact on the neural circuitry varies depending on the structures of neuronal networks. A high level of Aβ-induced neuronal hyperexcitability, aberrant neuronal network activity, and dysfunction of slow oscillations can lead to impairment of learning and memory. Many attempts have been made to develop drugs that reduce the level of Aβ. In clinical trials, immunotherapy has been more successful by far than β/γ-secretase inhibitors. However, further studies are needed to improve cognitive outcomes in addition to removing Aβ plaques more efficiently. Nevertheless, AD is multifactorial disease, and a more integrated approach must be applied to increase treatment efficacy. Although immunotherapy holds promise, innovative approaches such as gamma oscillations and mitochondrial proteostasis, among others, have shown promising results. Further research may yield a more efficacious therapy. Notably, the discovery of the connection between amyloid plaques and tau aggregation indicates that future treatment of AD might not be based solely on the amyloid cascade hypothesis.
References
- . Alzheimer’s Disease International, " World Alzheimer Report 2016 summary sheet.", Available: https://www.alz.co.uk/research/worldalzheimerreport2016sheet.pdf. Accessed 03.02.2017, 2016.
- R. Wittenberg, and B. Hu, "The Impact of Alzheimer's Disease in China", EBioMedicine, vol. 4, pp. 22-23, 2016. http://dx.doi.org/10.1016/j.ebiom.2016.02.018
- M.R. Keogh-Brown, H.T. Jensen, H.M. Arrighi, and R.D. Smith, "The Impact of Alzheimer's Disease on the Chinese Economy", EBioMedicine, vol. 4, pp. 184-190, 2016. http://dx.doi.org/10.1016/j.ebiom.2015.12.019
- R. Ricciarelli, and E. Fedele, "The Amyloid Cascade Hypothesis in Alzheimer’s Disease: It’s Time to Change Our Mind", Current Neuropharmacology, vol. 15, 2017. http://dx.doi.org/10.2174/1570159X15666170116143743
- D.M. Holtzman, J.C. Morris, and A.M. Goate, "Alzheimer’s Disease: The Challenge of the Second Century", Science Translational Medicine, vol. 3, 2011. http://dx.doi.org/10.1126/scitranslmed.3002369
- G.G. Glenner, and C.W. Wong, "Alzheimer's disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein", Biochemical and Biophysical Research Communications, vol. 120, pp. 885-890, 1984. http://dx.doi.org/10.1016/s0006-291x(84)80190-4
- C.L. Masters, G. Simms, N.A. Weinman, G. Multhaup, B.L. McDonald, and K. Beyreuther, "Amyloid plaque core protein in Alzheimer disease and Down syndrome.", Proceedings of the National Academy of Sciences, vol. 82, pp. 4245-4249, 1985. http://dx.doi.org/10.1073/pnas.82.12.4245
- J. Hardy, and D.J. Selkoe, "The Amyloid Hypothesis of Alzheimer's Disease: Progress and Problems on the Road to Therapeutics", Science, vol. 297, pp. 353-356, 2002. http://dx.doi.org/10.1126/science.1072994
- J.A. Hardy, and G.A. Higgins, "Alzheimer's Disease: The Amyloid Cascade Hypothesis", Science, vol. 256, pp. 184-185, 1992. http://dx.doi.org/10.1126/science.1566067
- H. LEE, G. CASADESUS, X. ZHU, A. TAKEDA, G. PERRY, and M.A. SMITH, "Challenging the Amyloid Cascade Hypothesis: Senile Plaques and Amyloid‐β as Protective Adaptations to Alzheimer Disease", Annals of the New York Academy of Sciences, vol. 1019, pp. 1-4, 2004. http://dx.doi.org/10.1196/annals.1297.001
- G.M. Bishop, and S.R. Robinson, "The Amyloid Paradox: Amyloid‐β‐Metal Complexes can be Neurotoxic and Neuroprotective", Brain Pathology, vol. 14, pp. 448-452, 2004. http://dx.doi.org/10.1111/j.1750-3639.2004.tb00089.x
- G.M. Shankar, S. Li, T.H. Mehta, A. Garcia-Munoz, N.E. Shepardson, I. Smith, F.M. Brett, M.A. Farrell, M.J. Rowan, C.A. Lemere, C.M. Regan, D.M. Walsh, B.L. Sabatini, and D.J. Selkoe, "Amyloid-β protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory", Nature Medicine, vol. 14, pp. 837-842, 2008. http://dx.doi.org/10.1038/nm1782
- T. Jonsson, J.K. Atwal, S. Steinberg, J. Snaedal, P.V. Jonsson, S. Bjornsson, H. Stefansson, P. Sulem, D. Gudbjartsson, J. Maloney, K. Hoyte, A. Gustafson, Y. Liu, Y. Lu, T. Bhangale, R.R. Graham, J. Huttenlocher, G. Bjornsdottir, O.A. Andreassen, E.G. Jönsson, A. Palotie, T.W. Behrens, O.T. Magnusson, A. Kong, U. Thorsteinsdottir, R.J. Watts, and K. Stefansson, "A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline", Nature, vol. 488, pp. 96-99, 2012. http://dx.doi.org/10.1038/nature11283
- Z. He, J.L. Guo, J.D. McBride, S. Narasimhan, H. Kim, L. Changolkar, B. Zhang, R.J. Gathagan, C. Yue, C. Dengler, A. Stieber, M. Nitla, D.A. Coulter, T. Abel, K.R. Brunden, J.Q. Trojanowski, and V.M. Lee, "Amyloid-β plaques enhance Alzheimer's brain tau-seeded pathologies by facilitating neuritic plaque tau aggregation", Nature Medicine, vol. 24, pp. 29-38, 2017. http://dx.doi.org/10.1038/nm.4443
- J. Williamson, J. Goldman, and K.S. Marder, "Genetic Aspects of Alzheimer Disease", The Neurologist, vol. 15, pp. 80-86, 2009. http://dx.doi.org/10.1097/NRL.0b013e318187e76b
- H.S. Nhan, K. Chiang, and E.H. Koo, "The multifaceted nature of amyloid precursor protein and its proteolytic fragments: friends and foes", Acta Neuropathologica, vol. 129, pp. 1-19, 2014. http://dx.doi.org/10.1007/s00401-014-1347-2
- D. Xia, H. Watanabe, B. Wu, S. Lee, Y. Li, E. Tsvetkov, V. Bolshakov, J. Shen, and R. Kelleher, "Presenilin-1 Knockin Mice Reveal Loss-of-Function Mechanism for Familial Alzheimer’s Disease", Neuron, vol. 85, pp. 967-981, 2015. http://dx.doi.org/10.1016/j.neuron.2015.02.010
- T. Ben‐Gedalya, L. Moll, M. Bejerano‐Sagie, S. Frere, W.A. Cabral, D. Friedmann‐Morvinski, I. Slutsky, T. Burstyn‐Cohen, J.C. Marini, and E. Cohen, "Alzheimer's disease‐causing proline substitutions lead to presenilin 1 aggregation and malfunction", The EMBO Journal, vol. 34, pp. 2820-2839, 2015. http://dx.doi.org/10.15252/embj.201592042
- M. Szaruga, S. Veugelen, M. Benurwar, S. Lismont, D. Sepulveda-Falla, A. Lleo, N.S. Ryan, T. Lashley, N.C. Fox, S. Murayama, H. Gijsen, B. De Strooper, and L. Chávez-Gutiérrez, "Qualitative changes in human γ-secretase underlie familial Alzheimer’s disease", Journal of Experimental Medicine, vol. 212, pp. 2003-2013, 2015. http://dx.doi.org/10.1084/jem.20150892
- D.J. Selkoe, and J. Hardy, "The amyloid hypothesis of Alzheimer's disease at 25 years", EMBO Molecular Medicine, vol. 8, pp. 595-608, 2016. http://dx.doi.org/10.15252/emmm.201606210
- K. Herrup, "The case for rejecting the amyloid cascade hypothesis", Nature Neuroscience, vol. 18, pp. 794-799, 2015. http://dx.doi.org/10.1038/nn.4017
- V.L. Villemagne, K.E. Pike, G. Chételat, K.A. Ellis, R.S. Mulligan, P. Bourgeat, U. Ackermann, G. Jones, C. Szoeke, O. Salvado, R. Martins, G. O'Keefe, C.A. Mathis, W.E. Klunk, D. Ames, C.L. Masters, and C.C. Rowe, "Longitudinal assessment of Aβ and cognition in aging and Alzheimer disease", Annals of Neurology, vol. 69, pp. 181-192, 2011. http://dx.doi.org/10.1002/ana.22248
- P.E. Cramer, J.R. Cirrito, D.W. Wesson, C.Y.D. Lee, J.C. Karlo, A.E. Zinn, B.T. Casali, J.L. Restivo, W.D. Goebel, M.J. James, K.R. Brunden, D.A. Wilson, and G.E. Landreth, "ApoE-Directed Therapeutics Rapidly Clear β-Amyloid and Reverse Deficits in AD Mouse Models", Science, vol. 335, pp. 1503-1506, 2012. http://dx.doi.org/10.1126/science.1217697
- C. Holmes, D. Boche, D. Wilkinson, G. Yadegarfar, V. Hopkins, A. Bayer, R.W. Jones, R. Bullock, S. Love, J.W. Neal, E. Zotova, and J.A. Nicoll, "Long-term effects of Aβ42 immunisation in Alzheimer's disease: follow-up of a randomised, placebo-controlled phase I trial", The Lancet, vol. 372, pp. 216-223, 2008. http://dx.doi.org/10.1016/S0140-6736(08)61075-2
- A.E. Roher, C.L. Maarouf, and T.A. Kokjohn, "Familial Presenilin Mutations and Sporadic Alzheimer’s Disease Pathology: Is the Assumption of Biochemical Equivalence Justified?", Journal of Alzheimer's Disease, vol. 50, pp. 645-658, 2016. http://dx.doi.org/10.3233/JAD-150757
- J.B. Thomas, M.R. Brier, R.J. Bateman, A.Z. Snyder, T.L. Benzinger, C. Xiong, M. Raichle, D.M. Holtzman, R.A. Sperling, R. Mayeux, B. Ghetti, J.M. Ringman, S. Salloway, E. McDade, M.N. Rossor, S. Ourselin, P.R. Schofield, C.L. Masters, R.N. Martins, M.W. Weiner, P.M. Thompson, N.C. Fox, R.A. Koeppe, C.R. Jack, C.A. Mathis, A. Oliver, T.M. Blazey, K. Moulder, V. Buckles, R. Hornbeck, J. Chhatwal, A.P. Schultz, A.M. Goate, A.M. Fagan, N.J. Cairns, D.S. Marcus, J.C. Morris, and B.M. Ances, "Functional Connectivity in Autosomal Dominant and Late-Onset Alzheimer Disease", JAMA Neurology, vol. 71, pp. 1111, 2014. http://dx.doi.org/10.1001/jamaneurol.2014.1654
- H. Zheng, and E.H. Koo, "Biology and pathophysiology of the amyloid precursor protein", Molecular Neurodegeneration, vol. 6, pp. 27, 2011. http://dx.doi.org/10.1186/1750-1326-6-27
- M.P. Mattson, "Pathways towards and away from Alzheimer's disease", Nature, vol. 430, pp. 631-639, 2004. http://dx.doi.org/10.1038/nature02621
- M.J. Berridge, "Calcium hypothesis of Alzheimer’s disease", Pflügers Archiv - European Journal of Physiology, vol. 459, pp. 441-449, 2009. http://dx.doi.org/10.1007/s00424-009-0736-1
- G. Bu, "Apolipoprotein E and its receptors in Alzheimer's disease: pathways, pathogenesis and therapy", Nature Reviews Neuroscience, vol. 10, pp. 333-344, 2009. http://dx.doi.org/10.1038/nrn2620
- E. Rogaeva, Y. Meng, J.H. Lee, Y. Gu, T. Kawarai, F. Zou, T. Katayama, C.T. Baldwin, R. Cheng, H. Hasegawa, F. Chen, N. Shibata, K.L. Lunetta, R. Pardossi-Piquard, C. Bohm, Y. Wakutani, L.A. Cupples, K.T. Cuenco, R.C. Green, L. Pinessi, I. Rainero, S. Sorbi, A. Bruni, R. Duara, R.P. Friedland, R. Inzelberg, W. Hampe, H. Bujo, Y. Song, O.M. Andersen, T.E. Willnow, N. Graff-Radford, R.C. Petersen, D. Dickson, S.D. Der, P.E. Fraser, G. Schmitt-Ulms, S. Younkin, R. Mayeux, L.A. Farrer, and P. St George-Hyslop, "The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease", Nature Genetics, vol. 39, pp. 168-177, 2007. http://dx.doi.org/10.1038/ng1943
- X. Zhang, and W. Song, "The role of APP and BACE1 trafficking in APP processing and amyloid-β generation", Alzheimer's Research & Therapy, vol. 5, pp. 46, 2013. http://dx.doi.org/10.1186/alzrt211
- D.M. Walsh, and D.J. Selkoe, "Aβ Oligomers – a decade of discovery", Journal of Neurochemistry, vol. 101, pp. 1172-1184, 2007. http://dx.doi.org/10.1111/j.1471-4159.2006.04426.x
- C. De Jonghe, "Pathogenic APP mutations near the gamma-secretase cleavage site differentially affect Abeta secretion and APP C-terminal fragment stability", Human Molecular Genetics, vol. 10, pp. 1665-1671, 2001. http://dx.doi.org/10.1093/hmg/10.16.1665
- M. Dimitrov, J. Alattia, T. Lemmin, R. Lehal, A. Fligier, J. Houacine, I. Hussain, F. Radtke, M. Dal Peraro, D. Beher, and P.C. Fraering, "Alzheimer’s disease mutations in APP but not γ-secretase modulators affect epsilon-cleavage-dependent AICD production", Nature Communications, vol. 4, 2013. http://dx.doi.org/10.1038/ncomms3246
- R. Johnson, J. Schauerte, C. Chang, K. Wisser, J. Althaus, C. Carruthers, M. Sutton, D. Steel, and A. Gafni, "Single-Molecule Imaging Reveals Aβ42:Aβ40 Ratio-Dependent Oligomer Growth on Neuronal Processes", Biophysical Journal, vol. 104, pp. 894-903, 2013. http://dx.doi.org/10.1016/j.bpj.2012.12.051
- G. Siegel, H. Gerber, P. Koch, O. Bruestle, P.C. Fraering, and L. Rajendran, "The Alzheimer’s Disease γ-Secretase Generates Higher 42:40 Ratios for β-Amyloid Than for p3 Peptides", Cell Reports, vol. 19, pp. 1967-1976, 2017. http://dx.doi.org/10.1016/j.celrep.2017.05.034
- J. Wiltfang, H. Esselmann, M. Bibl, M. Hüll, H. Hampel, H. Kessler, L. Frölich, J. Schröder, O. Peters, F. Jessen, C. Luckhaus, R. Perneczky, H. Jahn, M. Fiszer, J.M. Maler, R. Zimmermann, R. Bruckmoser, J. Kornhuber, and P. Lewczuk, "Amyloid β peptide ratio 42/40 but not Aβ42 correlates with phospho‐Tau in patients with low‐ and high‐CSF Aβ40 load", Journal of Neurochemistry, vol. 101, pp. 1053-1059, 2006. http://dx.doi.org/10.1111/j.1471-4159.2006.04404.x
- P.E. Spies, D. Slats, J.M.C. Sjögren, B.P.H. Kremer, F.R.J. Verhey, M.G.M.O. Rikkert, and M.M. Verbeek, "The cerebrospinal fluid amyloid beta42/40 ratio in the differentiation of Alzheimer's disease from non-Alzheimer's dementia.", Current Alzheimer research, 2010. http://www.ncbi.nlm.nih.gov/pubmed/20043812
- N. Fandos, V. Pérez‐Grijalba, P. Pesini, S. Olmos, M. Bossa, V.L. Villemagne, J. Doecke, C. Fowler, C.L. Masters, M. Sarasa, and . , "Plasma amyloid β 42/40 ratios as biomarkers for amyloid β cerebral deposition in cognitively normal individuals", Alzheimer's & Dementia: Diagnosis, Assessment & Disease Monitoring, vol. 8, pp. 179-187, 2017. http://dx.doi.org/10.1016/j.dadm.2017.07.004
- K. Ono, "Alzheimer's disease as oligomeropathy", Neurochemistry International, vol. 119, pp. 57-70, 2018. http://dx.doi.org/10.1016/j.neuint.2017.08.010
- T. Saito, T. Suemoto, N. Brouwers, K. Sleegers, S. Funamoto, N. Mihira, Y. Matsuba, K. Yamada, P. Nilsson, J. Takano, M. Nishimura, N. Iwata, C. Van Broeckhoven, Y. Ihara, and T.C. Saido, "Potent amyloidogenicity and pathogenicity of Aβ43", Nature Neuroscience, vol. 14, pp. 1023-1032, 2011. http://dx.doi.org/10.1038/nn.2858
- M. Willem, S. Tahirovic, M.A. Busche, S.V. Ovsepian, M. Chafai, S. Kootar, D. Hornburg, L.D.B. Evans, S. Moore, A. Daria, H. Hampel, V. Müller, C. Giudici, B. Nuscher, A. Wenninger-Weinzierl, E. Kremmer, M.T. Heneka, D.R. Thal, V. Giedraitis, L. Lannfelt, U. Müller, F.J. Livesey, F. Meissner, J. Herms, A. Konnerth, H. Marie, and C. Haass, "η-Secretase processing of APP inhibits neuronal activity in the hippocampus", Nature, vol. 526, pp. 443-447, 2015. http://dx.doi.org/10.1038/nature14864
- M. Brkic, S. Balusu, E. Van Wonterghem, N. Gorlé, I. Benilova, A. Kremer, I. Van Hove, L. Moons, B. De Strooper, S. Kanazir, C. Libert, and R.E. Vandenbroucke, "Amyloid β Oligomers Disrupt Blood–CSF Barrier Integrity by Activating Matrix Metalloproteinases", The Journal of Neuroscience, vol. 35, pp. 12766-12778, 2015. http://dx.doi.org/10.1523/JNEUROSCI.0006-15.2015
- R.G. Canter, J. Penney, and L. Tsai, "The road to restoring neural circuits for the treatment of Alzheimer's disease", Nature, vol. 539, pp. 187-196, 2016. http://dx.doi.org/10.1038/nature20412
- F. Kamenetz, T. Tomita, H. Hsieh, G. Seabrook, D. Borchelt, T. Iwatsubo, S. Sisodia, and R. Malinow, "APP Processing and Synaptic Function", Neuron, vol. 37, pp. 925-937, 2003. http://dx.doi.org/10.1016/s0896-6273(03)00124-7
- W. Wei, L.N. Nguyen, H.W. Kessels, H. Hagiwara, S. Sisodia, and R. Malinow, "Amyloid beta from axons and dendrites reduces local spine number and plasticity", Nature Neuroscience, vol. 13, pp. 190-196, 2009. http://dx.doi.org/10.1038/nn.2476
- G.M. Shankar, B.L. Bloodgood, M. Townsend, D.M. Walsh, D.J. Selkoe, and B.L. Sabatini, "Natural Oligomers of the Alzheimer Amyloid-β Protein Induce Reversible Synapse Loss by Modulating an NMDA-Type Glutamate Receptor-Dependent Signaling Pathway", The Journal of Neuroscience, vol. 27, pp. 2866-2875, 2007. http://dx.doi.org/10.1523/JNEUROSCI.4970-06.2007
- M. Sheng, B.L. Sabatini, and T.C. Sudhof, "Synapses and Alzheimer's Disease", Cold Spring Harbor Perspectives in Biology, vol. 4, pp. a005777-a005777, 2012. http://dx.doi.org/10.1101/cshperspect.a005777
- G.L. Collingridge, S. Peineau, J.G. Howland, and Y.T. Wang, "Long-term depression in the CNS", Nature Reviews Neuroscience, vol. 11, pp. 459-473, 2010. http://dx.doi.org/10.1038/nrn2867
- J.J. Palop, and L. Mucke, "Amyloid-β–induced neuronal dysfunction in Alzheimer's disease: from synapses toward neural networks", Nature Neuroscience, vol. 13, pp. 812-818, 2010. http://dx.doi.org/10.1038/nn.2583
- R. Kayed, and C.A. Lasagna-Reeves, "Molecular Mechanisms of Amyloid Oligomers Toxicity", Journal of Alzheimer's Disease, vol. 33, pp. S67-S78, 2012. http://dx.doi.org/10.3233/JAD-2012-129001
- J. Laurén, D.A. Gimbel, H.B. Nygaard, J.W. Gilbert, and S.M. Strittmatter, "Cellular prion protein mediates impairment of synaptic plasticity by amyloid-β oligomers", Nature, vol. 457, pp. 1128-1132, 2009. http://dx.doi.org/10.1038/nature07761
- T. Kim, G.S. Vidal, M. Djurisic, C.M. William, M.E. Birnbaum, K.C. Garcia, B.T. Hyman, and C.J. Shatz, "Human LilrB2 Is a β-Amyloid Receptor and Its Murine Homolog PirB Regulates Synaptic Plasticity in an Alzheimer’s Model", Science, vol. 341, pp. 1399-1404, 2013. http://dx.doi.org/10.1126/science.1242077
- N. Yamamoto, E. Matsubara, S. Maeda, H. Minagawa, A. Takashima, W. Maruyama, M. Michikawa, and K. Yanagisawa, "A Ganglioside-induced Toxic Soluble Aβ Assembly", Journal of Biological Chemistry, vol. 282, pp. 2646-2655, 2007. http://dx.doi.org/10.1074/jbc.M606202200
- W. Zhao, F.G. De Felice, S. Fernandez, H. Chen, M.P. Lambert, M.J. Quon, G.A. Krafft, and W.L. Klein, "Amyloid beta oligomers induce impairment of neuronal insulin receptors", The FASEB Journal, vol. 22, pp. 246-260, 2007. http://dx.doi.org/10.1096/fj.06-7703com
- M. Townsend, T. Mehta, and D.J. Selkoe, "Soluble Aβ Inhibits Specific Signal Transduction Cascades Common to the Insulin Receptor Pathway", Journal of Biological Chemistry, vol. 282, pp. 33305-33312, 2007. http://dx.doi.org/10.1074/jbc.M610390200
- M.H. Magdesian, M.M.V. Carvalho, F.A. Mendes, L.M. Saraiva, M.A. Juliano, L. Juliano, J. Garcia-Abreu, and S.T. Ferreira, "Amyloid-β Binds to the Extracellular Cysteine-rich Domain of Frizzled and Inhibits Wnt/β-Catenin Signaling", Journal of Biological Chemistry, vol. 283, pp. 9359-9368, 2008. http://dx.doi.org/10.1074/jbc.M707108200
- C. Di Scala, J. Troadec, C. Lelièvre, N. Garmy, J. Fantini, and H. Chahinian, "Mechanism of cholesterol‐assisted oligomeric channel formation by a short Alzheimer β‐amyloid peptide", Journal of Neurochemistry, vol. 128, pp. 186-195, 2013. http://dx.doi.org/10.1111/jnc.12390
- R. Kayed, Y. Sokolov, B. Edmonds, T.M. McIntire, S.C. Milton, J.E. Hall, and C.G. Glabe, "Permeabilization of Lipid Bilayers Is a Common Conformation-dependent Activity of Soluble Amyloid Oligomers in Protein Misfolding Diseases", Journal of Biological Chemistry, vol. 279, pp. 46363-46366, 2004. http://dx.doi.org/10.1074/jbc.C400260200
- E. Abramov, I. Dolev, H. Fogel, G.D. Ciccotosto, E. Ruff, and I. Slutsky, "Amyloid-β as a positive endogenous regulator of release probability at hippocampal synapses", Nature Neuroscience, vol. 12, pp. 1567-1576, 2009. http://dx.doi.org/10.1038/nn.2433
- J.J. Palop, J. Chin, E.D. Roberson, J. Wang, M.T. Thwin, N. Bien-Ly, J. Yoo, K.O. Ho, G. Yu, A. Kreitzer, S. Finkbeiner, J.L. Noebels, and L. Mucke, "Aberrant Excitatory Neuronal Activity and Compensatory Remodeling of Inhibitory Hippocampal Circuits in Mouse Models of Alzheimer's Disease", Neuron, vol. 55, pp. 697-711, 2007. http://dx.doi.org/10.1016/j.neuron.2007.07.025
- M.A. Busche, and A. Konnerth, "Impairments of neural circuit function in Alzheimer's disease", Philosophical Transactions of the Royal Society B: Biological Sciences, vol. 371, pp. 20150429, 2016. http://dx.doi.org/10.1098/rstb.2015.0429
- Z. Šišková, D. Justus, H. Kaneko, D. Friedrichs, N. Henneberg, T. Beutel, J. Pitsch, S. Schoch, A. Becker, H. von der Kammer, and S. Remy, "Dendritic Structural Degeneration Is Functionally Linked to Cellular Hyperexcitability in a Mouse Model of Alzheimer’s Disease", Neuron, vol. 84, pp. 1023-1033, 2014. http://dx.doi.org/10.1016/j.neuron.2014.10.024
- D.M. Walsh, I. Klyubin, J.V. Fadeeva, W.K. Cullen, R. Anwyl, M.S. Wolfe, M.J. Rowan, and D.J. Selkoe, "Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo", Nature, vol. 416, pp. 535-539, 2002. http://dx.doi.org/10.1038/416535a
- D.M. Kullmann, and K.P. Lamsa, "Long-term synaptic plasticity in hippocampal interneurons", Nature Reviews Neuroscience, vol. 8, pp. 687-699, 2007. http://dx.doi.org/10.1038/nrn2207
- E.M. Snyder, Y. Nong, C.G. Almeida, S. Paul, T. Moran, E.Y. Choi, A.C. Nairn, M.W. Salter, P.J. Lombroso, G.K. Gouras, and P. Greengard, "Regulation of NMDA receptor trafficking by amyloid-β", Nature Neuroscience, vol. 8, pp. 1051-1058, 2005. http://dx.doi.org/10.1038/nn1503
- S. Li, M. Jin, T. Koeglsperger, N.E. Shepardson, G.M. Shankar, and D.J. Selkoe, "Soluble Aβ Oligomers Inhibit Long-Term Potentiation through a Mechanism Involving Excessive Activation of Extrasynaptic NR2B-Containing NMDA Receptors", The Journal of Neuroscience, vol. 31, pp. 6627-6638, 2011. http://dx.doi.org/10.1523/JNEUROSCI.0203-11.2011
- J.W. Um, H.B. Nygaard, J.K. Heiss, M.A. Kostylev, M. Stagi, A. Vortmeyer, T. Wisniewski, E.C. Gunther, and S.M. Strittmatter, "Alzheimer amyloid-β oligomer bound to postsynaptic prion protein activates Fyn to impair neurons", Nature Neuroscience, vol. 15, pp. 1227-1235, 2012. http://dx.doi.org/10.1038/nn.3178
- K.A. Vossel, A.J. Beagle, G.D. Rabinovici, H. Shu, S.E. Lee, G. Naasan, M. Hegde, S.B. Cornes, M.L. Henry, A.B. Nelson, W.W. Seeley, M.D. Geschwind, M.L. Gorno-Tempini, T. Shih, H.E. Kirsch, P.A. Garcia, B.L. Miller, and L. Mucke, "Seizures and Epileptiform Activity in the Early Stages of Alzheimer Disease", JAMA Neurology, vol. 70, pp. 1158, 2013. http://dx.doi.org/10.1001/jamaneurol.2013.136
- R. Vassar, "BACE1 inhibitor drugs in clinical trials for Alzheimer’s disease", Alzheimer's Research & Therapy, vol. 6, 2014. http://dx.doi.org/10.1186/s13195-014-0089-7
- C.R. Jack, V.J. Lowe, S.D. Weigand, H.J. Wiste, M.L. Senjem, D.S. Knopman, M.M. Shiung, J.L. Gunter, B.F. Boeve, B.J. Kemp, M. Weiner, and R.C. Petersen, "Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer's disease: implications for sequence of pathological events in Alzheimer's disease", Brain, vol. 132, pp. 1355-1365, 2009. http://dx.doi.org/10.1093/brain/awp062
- B. De Strooper, "Lessons from a Failed γ-Secretase Alzheimer Trial", Cell, vol. 159, pp. 721-726, 2014. http://dx.doi.org/10.1016/j.cell.2014.10.016
- S.O. Bachurin, E.V. Bovina, and A.A. Ustyugov, "Drugs in Clinical Trials for Alzheimer's Disease: The Major Trends", Medicinal Research Reviews, vol. 37, pp. 1186-1225, 2017. http://dx.doi.org/10.1002/med.21434
- A. Mullard, "BACE inhibitor bust in Alzheimer trial", Nature Reviews Drug Discovery, vol. 16, pp. 155-155, 2017. http://dx.doi.org/10.1038/nrd.2017.43
- G. Cebers, R.C. Alexander, S.B. Haeberlein, D. Han, R. Goldwater, L. Ereshefsky, T. Olsson, N. Ye, L. Rosen, M. Russell, J. Maltby, S. Eketjäll, and A.R. Kugler, "AZD3293: Pharmacokinetic and Pharmacodynamic Effects in Healthy Subjects and Patients with Alzheimer’s Disease", Journal of Alzheimer's Disease, vol. 55, pp. 1039-1053, 2016. http://dx.doi.org/10.3233/jad-160701
- D. Goyal, S. Shuaib, S. Mann, and B. Goyal, "Rationally Designed Peptides and Peptidomimetics as Inhibitors of Amyloid-β (Aβ) Aggregation: Potential Therapeutics of Alzheimer’s Disease", ACS Combinatorial Science, vol. 19, pp. 55-80, 2017. http://dx.doi.org/10.1021/acscombsci.6b00116
- D. Schenk, "Amyloid-β immunotherapy for Alzheimer's disease: the end of the beginning", Nature Reviews Neuroscience, vol. 3, pp. 824-828, 2002. http://dx.doi.org/10.1038/nrn938
- J. Orgogozo, S. Gilman, J. Dartigues, B. Laurent, M. Puel, L.C. Kirby, P. Jouanny, B. Dubois, L. Eisner, S. Flitman, B.F. Michel, M. Boada, A. Frank, and C. Hock, "Subacute meningoencephalitis in a subset of patients with AD after Aβ42 immunization", Neurology, vol. 61, pp. 46-54, 2003. http://dx.doi.org/10.1212/01.wnl.0000073623.84147.a8
- C. Paquet, J. Amin, F. Mouton‐Liger, M. Nasser, S. Love, F. Gray, R.M. Pickering, J.A. Nicoll, C. Holmes, J. Hugon, and D. Boche, "Effect of active Aβ immunotherapy on neurons in human Alzheimer's disease", The Journal of Pathology, vol. 235, pp. 721-730, 2015. http://dx.doi.org/10.1002/path.4491
- C.K. Mulder, Y. Dong, H.F. Brugghe, H.A. Timmermans, W. Tilstra, J. Westdijk, E. van Riet, H. van Steeg, P. Hoogerhout, and U.L. Eisel, "Immunization with Small Amyloid-β-derived Cyclopeptide Conjugates Diminishes Amyloid-β-Induced Neurodegeneration in Mice", Journal of Alzheimer's Disease, vol. 52, pp. 1111-1123, 2016. http://dx.doi.org/10.3233/JAD-151136
- R. Vandenberghe, M. Riviere, A. Caputo, J. Sovago, R.P. Maguire, M. Farlow, G. Marotta, R. Sanchez‐Valle, P. Scheltens, J.M. Ryan, and A. Graf, "Active Aβ immunotherapy CAD106 in Alzheimer's disease: A phase 2b study", Alzheimer's & Dementia: Translational Research & Clinical Interventions, vol. 3, pp. 10-22, 2016. http://dx.doi.org/10.1016/j.trci.2016.12.003
- L. Fu, Y. Li, Y. Hu, Y. Zheng, B. Yu, H. Zhang, J. Wu, H. Wu, X. Yu, and W. Kong, "Norovirus P particle-based active Aβ immunotherapy elicits sufficient immunogenicity and improves cognitive capacity in a mouse model of Alzheimer’s disease", Scientific Reports, vol. 7, 2017. http://dx.doi.org/10.1038/srep41041
- M. Pohanka, "Vaccination to Alzheimer Disease. Is it a Promising Tool or a Blind Way?", Current Medicinal Chemistry, vol. 23, pp. 1432-1441, 2016. http://dx.doi.org/10.2174/0929867323666160418114733
- C.A. Sacks, J. Avorn, and A.S. Kesselheim, "The Failure of Solanezumab — How the FDA Saved Taxpayers Billions", New England Journal of Medicine, vol. 376, pp. 1706-1708, 2017. http://dx.doi.org/10.1056/NEJMp1701047
- J. Sevigny, P. Chiao, T. Bussière, P.H. Weinreb, L. Williams, M. Maier, R. Dunstan, S. Salloway, T. Chen, Y. Ling, J. O’Gorman, F. Qian, M. Arastu, M. Li, S. Chollate, M.S. Brennan, O. Quintero-Monzon, R.H. Scannevin, H.M. Arnold, T. Engber, K. Rhodes, J. Ferrero, Y. Hang, A. Mikulskis, J. Grimm, C. Hock, R.M. Nitsch, and A. Sandrock, "The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease", Nature, vol. 537, pp. 50-56, 2016. http://dx.doi.org/10.1038/nature19323
- H.F. Iaccarino, A.C. Singer, A.J. Martorell, A. Rudenko, F. Gao, T.Z. Gillingham, H. Mathys, J. Seo, O. Kritskiy, F. Abdurrob, C. Adaikkan, R.G. Canter, R. Rueda, E.N. Brown, E.S. Boyden, and L. Tsai, "Gamma frequency entrainment attenuates amyloid load and modifies microglia", Nature, vol. 540, pp. 230-235, 2016. http://dx.doi.org/10.1038/nature20587
- V. Sorrentino, M. Romani, L. Mouchiroud, J.S. Beck, H. Zhang, D. D’Amico, N. Moullan, F. Potenza, A.W. Schmid, S. Rietsch, S.E. Counts, and J. Auwerx, "Enhancing mitochondrial proteostasis reduces amyloid-β proteotoxicity", Nature, vol. 552, pp. 187-193, 2017. http://dx.doi.org/10.1038/nature25143
ACKNOWLEDGMENTS
This study was financially supported by the National Natural Sciences Foundation of China (numbers 81330026, 81771330, 31271259), the National Key Basic Research Development Program of the Ministry of Science and Technology of China (973 Program, 2013CB945600), and a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.
COPYRIGHT
© 2018

Alzheimer’s disease: amyloid-based pathogenesis and potential therapies by Zhou et al. is licensed under a Creative Commons Attribution 4.0 International License.


