Reviews:
Cell Stress, Vol. 2, No. 11, pp. 282 - 291; doi: 10.15698/cst2018.11.161
Mechanisms and therapeutic significance of autophagy modulation by antipsychotic drugs

1 Institute for Biological Research, University of Belgrade, Belgrade, Serbia.
2 Clinic of Psychiatry, Clinical Centre of Serbia and School of Medicine, University of Belgrade, Belgrade, Serbia.
3 Institute of Microbiology and Immunology, School of Medicine, University of Belgrade, Dr. Subotica 1, 11000 Belgrade, Serbia.
Keywords: autophagy, antipsychotics, schizophrenia, neurodegeneration, cancer.
Abbreviations:
AMPK – AMP-activated protein kinase,
ATG – autophagy-related,
LC3 – light chain 3,
mTOR – mammalian target of rapamycin,
ROS – reactive oxygen species,
TFEB – transcription factor EB,
ULK – Unc-51-like autophagy activating kinase.
Received originally: 13/07/2018 Received in revised form: 05/10/2018
Accepted: 08/10/2018
Published: 25/10/2018
Correspondence:
Vladimir Trajkovic, Institute of Microbiology and Immunology, School of Medicine, University of Belgrade, Dr. Subotica 1, 11000 Belgrade, Serbia vladimir.trajkovic@med.bg.ac.rs
Conflict of interest statement: The authors declare no conflict of interest.
Please cite this article as: Ljubica Vucicevic, Maja Misirkic-Marjanovic, Ljubica Harhaji-Trajkovic, Nadja Maric and Vladimir Trajkovic (2018). Mechanisms and therapeutic significance of autophagy modulation by antipsychotic drugs. Cell Stress 2(11): 282-291. doi: 10.15698/cst2018.11.161
Abstract
In this review we analyze the ability of antipsychotic medications to modulate macroautophagy, a process of controlled lysosomal digestion of cellular macromolecules and organelles. We focus on its molecular mechanisms, consequences for the function/survival of neuronal and other cells, and the contribution to the beneficial and side-effects of antipsychotics in the treatment of schizophrenia, neurodegeneration, and cancer. A wide range of antipsychotics was able to induce neuronal autophagy as a part of the adaptive stress response apparently independent of mammalian target of rapamycin and dopamine receptor blockade. Autophagy induction by antipsychotics could contribute to reducing neuronal dysfunction in schizophrenia, but also to the adverse effects associated with their long-term use, such as brain volume loss and weight gain. In neurodegenerative diseases, antipsychotic-stimulated autophagy might help to increase the clearance and reduce neurotoxicity of aggregated proteotoxins. However, the possibility that some antipsychotics might block autophagic flux and potentially contribute to proteotoxin-mediated neurodegeneration must be considered. Finally, the anticancer effects of autophagy induction by antipsychotics make plausible their repurposing as adjuncts to standard cancer therapy.
INTRODUCTION
The term autophagy (‘self-eating’ in Greek) refers to a group of evolutionarily conserved homeostatic mechanisms employed by eukaryotic cells to eliminate aged, unused, and damaged cytoplasmic components through lysosomal degradation [1]. Three main types of autophagy that differ in the mechanisms of delivery of autophagic substrates to lysosomes have been termed microautophagy, chaperone-mediated autophagy, and macroautophagy [2]. Macroautophagy (hereafter autophagy), the best characterized form of autophagy in mammalian cells, relies on sequestration of autophagic cargo by double-membraned vesicles known as autophagosomes, which fuse with lysosomes to form autolysosomes [3]. In addition to its physiological role in maintaining intracellular homeostasis, autophagy is an important part of the adaptive protective response against metabolic, hypoxic, oxidative, infectious, inflammatory, proteotoxic, and drug-induced stress [4]. On the other hand, when extensive or activated inappropriately, autophagy can contribute to apoptotic/necrotic cell demise or function as an alternative programmed cell-death pathway [5].
–
Neurons, being post-mitotic, heavily rely on autophagic clearance of proteotoxins and damaged mitochondria, and are thus particularly vulnerable to autophagy disturbances [6][7]. In addition to controlling neuronal survival and death during stress, autophagy has been shown to regulate presynaptic neurotransmission [8]. Moreover, dysregulation of neuronal autophagy has recently been implicated in the pathogenesis of schizophrenia [9], a chronic mental disorder in which the abnormalities in social behavior and perception of reality are associated with altered neurotransmission [10]. Antipsychotic drugs used to manage psychosis in schizophrenia mainly act by reducing dopaminergic neurotransmission [11]. They could be broadly classified as typical and atypical, the latter being developed more recently, and possessing additional anti-serotonergic activity. However, the complexity and beneficial/adverse consequences of dopamine-dependent and -independent actions of antipsychotics on neurons and other cell types are far from being completely elucidated. More than three decades ago it has been reported that administration of now discontinued antipsychotic reserpine caused autophagy in rat liver [12]. Many years later, a screening of 480 bioactive small molecules for autophagy-inducing capacity revealed that three among eight most efficient compounds were FDA-approved antipsychotic drugs trifluoperazine, fluspirilene, and pimozide [13]. A number of studies subsequently confirmed the ability of various typical and atypical antipsychotic drugs to modulate autophagy in different cell types, including neurons.
–
In this concise review, we analyze autophagy regulation by antipsychotic medications, focusing on its molecular mechanisms and consequences for the neuronal function/survival and treatment of schizophrenia and neurodegeneration. The autophagy modulation by antipsychotics in other cell types and cancer is also briefly discussed.
ANTIPSYCHOTIC-MEDIATED MODULATION OF NEURONAL/BRAIN AUTOPHAGY
Autophagy induction is mainly assessed by measuring the conversion of autophagic protein microtubule-associated light chain 3 (LC3)-I to its lipidated, autophagosome-localized form LC3-II [14]. However, as LC3-II is subsequently degraded in lysosomes, its increase can also result from autophagy inhibition [14]. To avoid confusion due to inadequate interpretation of LC3 data, we have focused on the studies that assessed autophagic turnover (flux) and/or used pharmacological/genetic modulation of autophagy to confirm its biological effects [14]. However, due to known technical difficulties with assessing autophagic flux in vivo [15], the available in vivo data are presented with the limitations explicitly noted.
–
Activation of neuronal autophagy by antipsychotics
In most of the in vitro studies, antipsychotics stimulated autophagic flux in neuronal cells (Table 1), as confirmed by the increase in LC3 conversion in the presence of lysosomal inhibitors, appearance of autophagosomes and their fusion with lysosomes, and/or degradation of selective autophagic targets, such as autophagic cargo receptor sequestosome 1/p62. Pimozide, sertindole, olanzapine, and trifluoperazine induced autophagy in human neuronal cell line SH-SY5Y [16][17][18][19], while trifluoperazine, fluphenazine, and methotrimeprazine efficiently increased autophagic flux in human or rat primary neurons [20][21]. A structure-activity study in rat striatal neurons revealed that an amino-containing substituent at the N10 position was important for LC3-II increase by tricyclic antipsychotics trifluoperazine, promazine, chlorpromazine, triflupromazine, thioridazine, and mesoridazine, although no flux data were provided [22]. Pimozide and fluspirilene, having a biphenyl core and a 3–4 carbon linker to a tertiary amine, display structural similarities to tricyclics, which further emphasizes the importance of this structural scaffold in neuronal autophagy induction by antipsychotics [22]. Risperidone and haloperidol increased the numbers of acidic, presumably autophagic vesicles, in the cytoplasm of SH-SY5Y cells [18], but their ability to stimulate autophagic flux was not directly confirmed. Importantly, antipsychotics were also able to increase autophagic markers in the brains of experimental animals in vivo. Pimozide, olanzapine, and clozapine increased LC3-II conversion, autophagosome numbers, autophagic degradation of p62, and/or expression of autophagy genes in mouse or rat frontal cortex and/or hippocampus [16][18][23], while trifluoperazine increased LC3 conversion in a zebrafish model of Parkinson’s disease caused by the deficiency of phosphatase and tensin homolog-induced kinase 1 [19].
–
Table 1. Modulation of neuronal/brain autophagy by antipsychotics. |
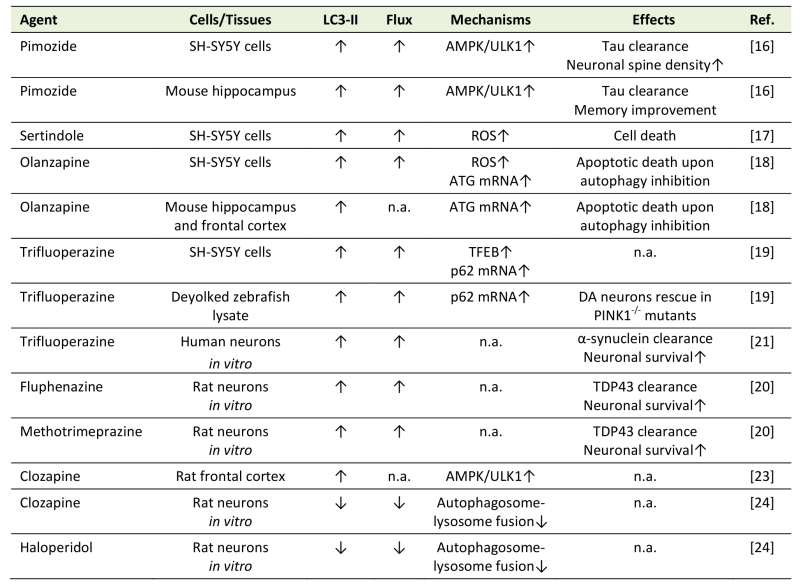 |
“Flux” refers to autophagic flux; ↑ denotes increase/activation; ↓ denotes de-crease/inhibition; n.a. – not assessed; AMPK – AMP-activated protein kinase; ATG – autophagy related; DA – dopaminergic; PINK1 – Phosphatase and tensin homolog -induced kinase 1; ROS – reactive oxygen species; TDP43 – TAR DNA-binding protein 43; TFEB – transcription factor EB; ULK1 – Unc-51 like autophagy activating kinase. [16][17][18][19][20][21][23][24] |
–
Inhibition of neuronal autophagy by antipsychotics
Similarly to the above findings, the in vitro treatment with haloperidol and clozapine increased LC3-II levels in rat primary neurons [24]. However, this was attributed to a decrease in LC3-II degradation due to a block of autophagosome-lysosome fusion and subsequent inhibition of autophagic proteolysis [24]. While these findings indicate a unique ability of haloperidol and clozapine to prevent autophagy completion, they also imply that the in vivo increase in steady-state LC3-II levels by clozapine [23] might reflect inhibition, rather than induction of autophagic flux. It is also possible that haloperidol and/or clozapine actually triggered an autophagic response, but later blocked it at the lysosomal stage. An assessment of the earlier stages of autophagy and autophagy-inducing signaling pathways, as well as recently developed methods for measuring neuronal autophagic flux in vivo [15][25] could be employed to resolve these issues.
MECHANISMS OF ANTIPSYCHOTIC-MEDIATED MODULATION OF NEURONAL/BRAIN AUTOPHAGY
The mechanisms underlying the modulation of neuronal autophagy by antipsychotics are delineated below and schematically depicted in Fig. 1. Autophagy is executed through hierarchical activation of autophagy-related (ATG) proteins organized in functional complexes that control different stages of autophagy process [26]. Autophagosome biogenesis is initiated by the functional complex containing the mammalian ATG1 homologue Unc-51-like kinase (ULK)1, ATG13, and FIP200, which is recruited to the phagophore assembly site. Vesicle nucleation continues through the generation of phosphatidylinositol 3-phosphate, catalyzed by ULK1-activated complex containing the lipid kinase VPS34, beclin-1, and ATG14. The next step, vesicle elongation, is mediated by the two ubiquitin-like conjugation systems: ATG7 and ATG10 conjugate ATG5 to ATG12, and then ATG4, ATG7, and ATG3 conjugate phosphatidylethanolamine to LC3. LC3 (mammalian ATG8 homologue) is the signature autophagosome protein promoting the expansion of the autophagosomal membrane and its closure and fusion with the lysosome (maturation step), where the ubiquitinated cytoplasmic material bound to autophagic cargo receptor p62 is eventually degraded.
–
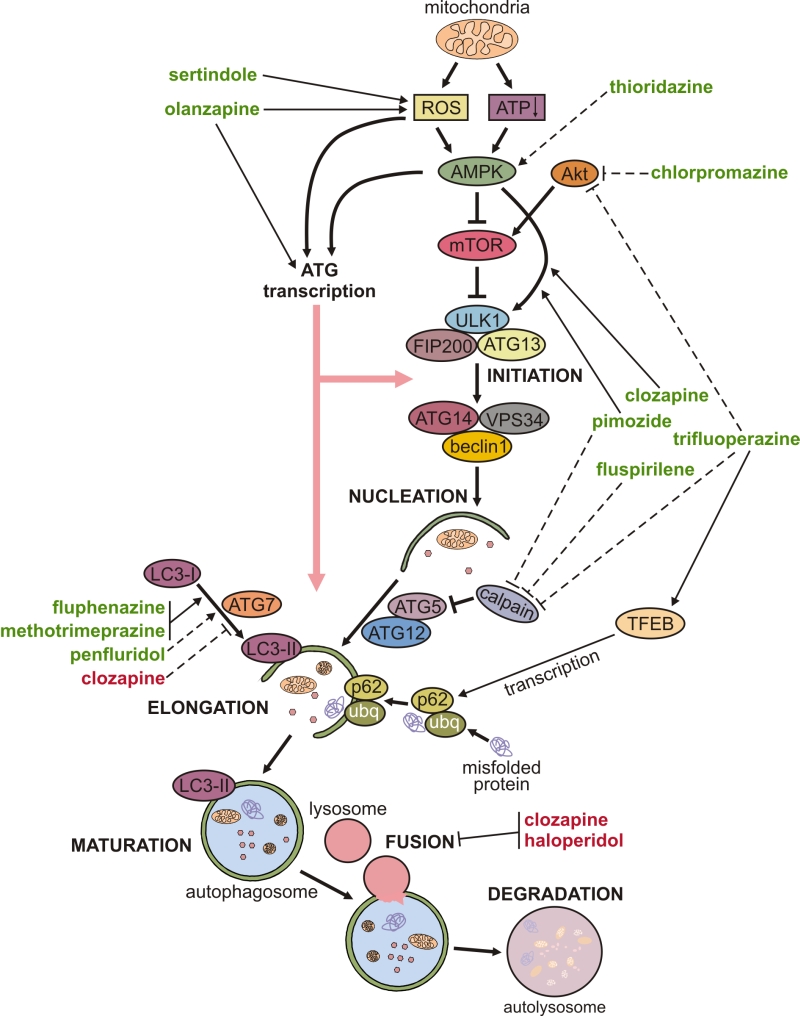 | Figure 1: Mechanisms of autophagy regulation by antipsychotics. A simplified map of autophagy regulation is presented, including the sites of action of antipsychotic drugs shown as arrow-headed (activation/increase) or bar-headed lines (inhibition/decrease). The full and dashed lines refer to modulation in neurons/brain and non-neuronal cells, respectively. The drug names are marked green for autophagy stimulation, or red for autophagy inhibition. Some drugs are shown to directly modulate LC3 conversion only because no specific underlying mechanism was described. (ubq – ubiquitin). |
–
Autophagy is mainly regulated post-transcriptionally, through phosphorylation, ubiquitination, and/or acetylation of ATG proteins and their regulators, which alters their functional activity or changes their structure to modulate the affinity for binding partners [26]. The initiation of autophagy is controlled by mammalian target of rapamycin (mTOR)-containing mTOR complex 1 (mTORC1), which inhibits autophagy by phosphorylating Unc-51-like autophagy activating kinase (ULK1, mammalian ATG1) [26]. mTORC1 is activated by Akt and inhibited by AMP-activated protein kinase (AMPK), which also activates autophagy by directly activating ULK1 [27]. The induction of autophagy by olanzapine or pimozide was apparently independent of mTORC1 modulation, as neither drug was able to affect the phosphorylation of mTOR or mTORC1 substrate ribosomal p70S6 kinase [16][18]. On the other hand, pimozide- and clozapine-triggered autophagy was associated with activation of AMPK and AMPK-dependent phosphorylation of ULK1 in SH-SY5Y neurons and in mouse hippocampus or rat frontal cortex in vivo [16][23]. A genetic AMPK knockdown in vitro, and pharmacological AMPK suppression in vivo, reduced pimozide- or clozapine-triggered ULK1 phosphorylation and subsequent induction of autophagy [16][23], thus further supporting a role for AMPK-ULK1 interaction in the mTORC1-independent autophagy triggered by antipsychotics. AMPK-dependent autophagy is mainly activated by oxidative stress and the energy deficit reflected in the increase of AMP/ATP ratio, both resulting from mitochondrial dysfunction [27][28]. Moreover, reactive oxygen species (ROS) can directly stimulate autophagy through redox modification of ATG proteins [29]. Accordingly, sertindole-induced autophagy in SH-SY5Y neuronal cells was caused by ROS [17][18], while olanzapine-mediated mitochondrial depolarization led to oxidative stress-dependent autophagic clearance of damaged mitochondria in SH-SY5Y cells [18].
–
Autophagy is also regulated transcriptionally [26], and both AMPK and oxidative stress can stimulate transcription of ATG genes by activating various transcription factors such as transcription factor EB (TFEB), forkhead box O1/3, activating transcription factor 4, nuclear factor-κB, nuclear factor (erythroid-derived 2)-like 2, and p53 [4][30]. Transcriptional regulation of autophagy by antipsychotics is supported by the ability of olanzapine to increase the mRNA expression of ATG4, 5, 7, 12, and beclin-1 in SH-SY5Y neurons in vitro and in mouse brain in vivo [18]. Similarly, trifluoperazine increased TFEB-dependent transcription of autophagic cargo receptor p62 in mitochondrially-stressed SH-SY5Y cells [19]. Post-transcriptional regulation of autophagy by microRNA-mediated ATG mRNA cleavage and/or translational arrest has also been described [26]. While there is presently no available direct evidence of post-transcriptional regulation of neuronal autophagy by antipsychotic drugs, it has been reported that penfluridol can downregulate the expression of microRNA-17 and -20a [31], which are shown to inhibit autophagic response by blocking the expression of ULK1 and ATG7, respectively [32][33].
–
The above delineated connections between antipsychotic-triggered neuronal autophagy and ROS, energy balance sensor AMPK, and stress-induced transcriptional activator TFEB indicate that autophagy is a part of the integrated stress response [34] of neurons to antipsychotic treatment. However, it remains on future studies to explore the exact connections between autophagy and other parts of the adaptive response to antipsychotics, as well as their role in neuronal homeostasis and function. Finally, it should be noted that some antipsychotics, such as haloperidol and clozapine, have been reported to suppress autophagy at the stage of autophagosome-lysosome fusion [24]. It was proposed that haloperidol-mediated phosphorylation of the microtubule-associated protein tau and subsequent neurocytoskeletal disorganization [35] could impair autophagosome fusion with lysosomes, but this possibility was not experimentally tested.
–
The role of dopaminergic/serotonergic blockade in autophagy modulation by antipsychotics
It has recently been shown that dopamine receptor subtypes differently regulate autophagy, with D2 and D3 (D2-like family) being positive, and D1 and D5 (D1-like family) negative regulators [36]. The stimulation of D2 and D3 receptors was also able to induce beclin-1-dependent autophagy in neurons [37]. Since most antipsychotic drugs are primarily antagonists of the D2-like family receptors, it seems unlikely that dopaminergic blockade was involved in their ability to induce autophagy. Accordingly, a phenoxazine-derived compound structurally related to phenotiazine antipsychotics, but lacking their antidopaminergic activity, readily induced neuronal autophagy [22], while D2 antagonist sulpiride failed to exert a similar effect [16]. Also, the ability of both typical (fluphenazine, methotrimeprazine, pimozide, trifluoperazine) and atypical antipsychotics (olanzapine, sertindole) to stimulate neuronal autophagy argues against the involvement of serotonergic blockade in autophagy induction by the latter. Finally, as the inhibition of autophagic flux by haloperidol and clozapine was not mimicked by other antipsychotics, it is unlikely that it was mediated by dopaminergic/serotonergic blockade. It remains to be examined if chronic interference with dopaminergic/serotonergic signaling by antipsychotics might affect neuronal autophagy in vivo.
CLINICAL SIGNIFICANCE OF NEURONAL AUTOPHAGY MODULATION BY ANTIPSYCHOTICS
The above findings indicate modulation of neuronal autophagy as a common denominator of intracellular action of antipsychotic drugs, raising the possibility that their therapeutic and/or side-effects might partly rely on this property. Indeed, a possible link between autophagy and schizophrenia has recently emerged, as the expression of autophagy-regulating genes, including autophagy-essential beclin-1, was found to be reduced in postmortem cortical and hippocampal tissues of schizophrenic individuals [38][39][40]. Autophagy impairment might contribute to schizophrenia by interfering with the role of autophagy in synaptic plasticity [41] and removal of neurotoxic protein aggregates, such as those formed by the products of Disrupted-in-schizophrenia-1 gene [42]. Accordingly, autophagy dysregulation has also been demonstrated in different animal models of schizophrenia [43][44][45] and pharmacological restoration of autophagic activity by microtubule assembly-promoting peptide davunetide was associated with behavioral improvements [44]. It is therefore plausible that autophagy stimulation by antipsychotics such as trifluoperazine, fluphenazine, methotrimeprazine, olanzapine, pimozide, or sertindole might contribute to their beneficial effects by improving neuronal function.
–
Some studies suggest that long-term antipsychotic treatment may cause neurotoxicity and contribute to brain tissue volume loss associated with schizophrenia [46][47], but the underlying mechanisms are unknown. Autophagy activation by antipsychotic drugs has been shown to influence survival and death of neuronal cells in various experimental settings. The induction of autophagy by sertindole caused neuronal death in vitro [17], indicating a possible involvement of autophagy in the neurotoxicity of antipsychotics. On the other hand, olanzapine-mediated autophagy was associated with oxidative stress and mitochondrial dysfunction in the absence of overt neurotoxicity [18]. Moreover, pharmacological and/or genetic inhibition of olanzapine-triggered autophagy led to impaired clearance of damaged mitochondria and apoptotic neuronal death both in vitro and in vivo, thus unmasking the neurotoxic action of the drug [18]. These data suggest that the neurotoxicity of antipsychotics might be increased in patients with impaired autophagy due to aging, neuroinflamation, and/or neurodegeneration [48].
–
Understanding the possible involvement of antipsychotics in neurodegeneration is even more important having in mind their increasing use in the treatment of psychotic or related behavioral disturbances in patients with neurodegenerative diseases [49]. Interestingly, a line of recent evidence indicates that autophagy induction by antipsychotics might actually have beneficial effects on mitochondrial dysfunction and proteotoxic aggregation in these disorders. Trifluoperazine-induced autophagy rescued human dopaminergic neurons from the toxic effects of α-synuclein accumulation in vitro [21], and reduced dopaminergic neuron loss in a zebrafish model of Parkinson’s disease [19]. Autophagy stimulation by fluphenazine and methotrimeprazine enhanced survival of primary rat neurons and human stem cell–derived neurons by clearing mutant TAR DNA-binding protein 43 in the in vitro model of amiotrophic lateral sclerosis [20]. Pimozide-triggered autophagy decreased abnormally phosphorylated tau aggregates in neuronal cells in vitro and in vivo, leading to an improvement of memory deficit in a mouse model of Alzheimer’s disease [16]. Similarly, haloperidol protected striatal neurons from dysfunction induced by accumulation of mutated huntingtin in vivo, although the role of autophagy in this effect was not investigated [50].
–
The above findings provide a proof of concept for using autophagy-activating drugs to improve clearance of proteotoxic aggregates in neurodegenerative diseases. Moreover, as mTORC1 inhibition in neurons does not always trigger autophagic response [51], mTORC1-independent autophagy induction by antipsychotics might be a more reliable therapeutic option than the use of classic mTORC1-inhibiting drugs like rapamycin and its analogues. Such an approach would ideally require elimination of the antidopaminergic activity and the related side-effects that could compromise the use of antipsychotics in neurodegenerative disorders. On the other hand, treatment with antipsychotics was associated with a significant increase in neurofibrillary tangles and amyloid plaques in the frontal lobe cortex of patients suffering from dementia [52]. Therefore, the possibility that some antipsychotics, as shown for haloperidol and clozapine, might block autophagic proteolysis [24], thus potentially contributing to accumulation of proteotoxins and neurodegeneration, must be carefully considered.
ANTIPSYCHOTIC-MEDIATED MODULATION OF AUTOPHAGY IN NON-NEURONAL CELLS
In addition to modulating autophagy in neurons, antipsychotics have been shown to affect autophagy induction in other cell types (Table 2) through a variety of mechanisms (Fig. 1). Similar to findings obtained in neurons, antipsychotics mostly stimulated autophagy in non-neuonal cells, but inhibitory effects were also observed.
–
Table 2. Modulation of autophagy by antipsychotics in non-neuronal cells. |
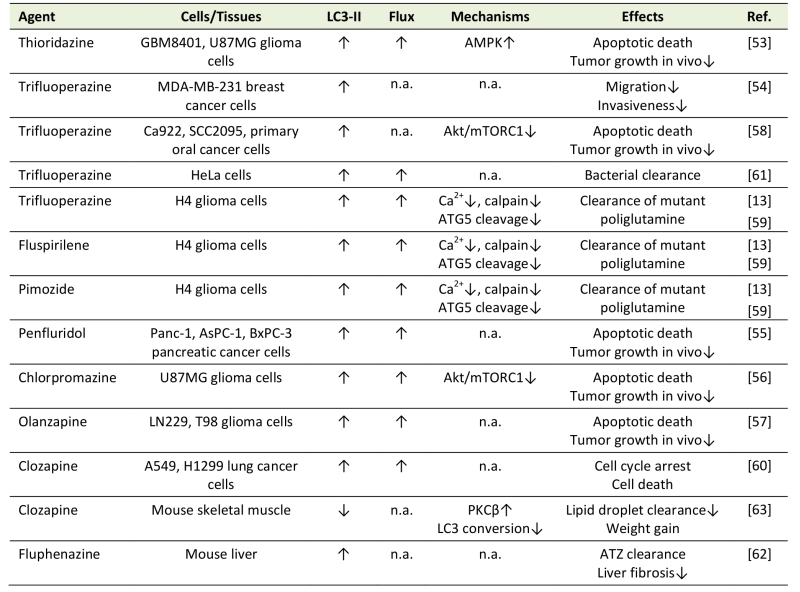 |
“Flux” refers to autophagic flux; ↑ denotes increase/activation; ↓ denotes de-crease/inhibition; n.a. – not assessed; ATZ - α1-antitrypsin Z; ER – endoplasmic reticulum; PKC – protein kinase C. [13][53][54][55][56][57][58][59][60][61][62][63] |
–
Autophagy induction by antipsychotics in non-neuronal cells
Phenothiazine antipsychotics trifluoperazine, thioridazine, and chlorpromazine, as well as fluspirilene, pimozide, penfluridol, olanzapine, and clozapine all induced autophagy in various cancer cell types, including glioma, breast, lung, pancreatic, and oral cancer cells [13][53][54][55][56][57][58][59][60]. Autophagy induction by antipsychotics reduced migration and invasiveness of cancer cells [54], or cause their apoptotic/autophagic death in vitro and in vivo [53][55][56][57][58][59][60]. Moreover, trifluoperazine was used as a lead compound to develop autophagy-inducing anticancer agents with improved efficacy and reduced toxicity, thus providing a proof-of-concept for designing phenothiazine-derived anticancer drugs [58]. Trifluoperazine, fluspirilene, and pimozide increased autophagic clearance of mutant poliglutamine in H4 glial cells [13], trifluoperazine-induced autophagy promoted bacterial clearance in HeLa cells [61], and fluphenazine-triggered autophagy reduced the accumulation of mutant α1-antitrypsin in hepatocytes and subsequent liver fibrosis [62]. Chlorpromazine- and trifluoperazine-mediated autophagy in cancer cells was dependent on Akt inhibition-mediated mTORC1 suppression [56][58], while mTORC1-unrelated AMPK activation was apparently responsible for thioridazine-induced autophagy [53]. Fluspirilene, pimozide, and trifluoperazine stimulated autophagy by inhibiting Ca2+-dependent cleavage of ATG5 by calpain, which in turn increased the levels of ATG5-ATG12 conjugate required for autophagosome formation [59]. Moreover, a structure-activity relationship study of 83 fluspirilene derivatives demonstrated that only those inhibiting Ca2+ channels and subsequent calpain-mediated ATG5 cleavage were also able to induce autophagy [59], suggesting a critical role of this mechanism in autophagy induction by antipsychotics.
–
Autophagy inhibition by antipsychotics in non-neuronal cells
In contrast to the above data, clozapine-mediated protein kinase Cβ activation inhibited LC3 conversion and autophagic clearance of lipid droplets in the mouse skeletal muscle in vivo [63], thus providing a novel insight into the mechanisms underlying the clozapine-induced weight gain. Also, N-n-butyl haloperidol iodide protected cardiomyocytes from hypoxia/reoxygenation injury by reducing beclin-1 and ATG5 levels and inhibiting autophagy [64], but it was not assessed whether these effects were actually due to the chemical modification of the parental compound.
–
While it would be interesting to compare the mechanisms of antipsychotic-mediated autophagy modulation in neurons and non-neuronal cells, the number of studies providing a mechanistic insight required for such a comparison is presently rather limited. Nevertheless, it appears that autophagy induction by antipsychotics in non-neuronal cells might more often rely on mTORC1 inhibition, which is consistent with the relative insensitivity of neuronal autophagy to mTORC1 suppression [51]. Also, having in mind the critical role of calcium signaling in neuronal function [65], it would be important to investigate if the inhibition of Ca2+ channels by antipsychotics, causing autophagy in cancer cells [59], might affect autophagy in neurons as well. Further studies are required to directly compare signaling events involved in autophagy modulation by antipsychotics in neurons and other cell types, thus facilitating the development of selective modulators of neuronal autophagy.
CONCLUSIONS
The present review demonstrates that many typical (trifluoperazine, fluphenazine, fluspirilene, chlorpromazine, methotrimeprazine, thioridazine, pimozide, penfluridol) and atypical antipsychotics (olanzapine, sertindole) induce autophagy in neuronal or other cell types, mainly through AMPK-mediated mTORC1-independent or mTORC1-dependent mechanisms, respectively. On the other hand, antipsychotics such as clozapine and haloperidol in certain conditions caused inhibition of autophagic flux, the mechanisms and significance of which remain to be explored. The induction of autophagic response by antipsychotic drugs might contribute to reducing neuronal dysfunction in schizophrenia, but also to the adverse effects associated with their long-term use, including brain volume loss and weight gain. If antipsychotics, in accordance with their ability to induce autophagy in glioma cells, could influence normal glia in a similar way, this might have further implications for schizophrenia therapy. Moreover, having in mind the increasing use of antipsychotics in disorders other than schizophrenia, understanding the effects that autophagy activation by these drugs might have on the brain has important clinical implications beyond schizophrenia treatment. This seems particularly important in neurodegenerative diseases, where mTORC1-independent autophagy induction by antipsychotic derivatives devoid of anti-dopaminergic activity might help to increase the clearance and reduce neurotoxicity of aggregated proteotoxins. Moreover, the ability of different antipsychotics to preferentially cross the blood-brain barrier in certain brain areas [66] could be exploited to enable brain region-selective autophagy activation. Finally, the anticancer effects of autophagy induction by antipsychotics make plausible their repurposing as adjuncts to standard cancer therapy. A further in-depth investigation of the mechanisms and effects of autophagy triggered in neuronal and other cells/tissues by antipsychotics is required for understanding the biological actions, increasing the beneficial/side-effect ratio, and broadening the therapeutic scope of this important class of drugs.
References
- N. Mizushima, "A brief history of autophagy from cell biology to physiology and disease", Nature Cell Biology, vol. 20, pp. 521-527, 2018. http://dx.doi.org/10.1038/s41556-018-0092-5
- L. Galluzzi, E.H. Baehrecke, A. Ballabio, P. Boya, J.M. Bravo‐San Pedro, F. Cecconi, A.M. Choi, C.T. Chu, P. Codogno, M.I. Colombo, A.M. Cuervo, J. Debnath, V. Deretic, I. Dikic, E. Eskelinen, G.M. Fimia, S. Fulda, D.A. Gewirtz, D.R. Green, M. Hansen, J.W. Harper, M. Jäättelä, T. Johansen, G. Juhasz, A.C. Kimmelman, C. Kraft, N.T. Ktistakis, S. Kumar, B. Levine, C. Lopez‐Otin, F. Madeo, S. Martens, J. Martinez, A. Melendez, N. Mizushima, C. Münz, L.O. Murphy, J.M. Penninger, M. Piacentini, F. Reggiori, D.C. Rubinsztein, K.M. Ryan, L. Santambrogio, L. Scorrano, A.K. Simon, H. Simon, A. Simonsen, N. Tavernarakis, S.A. Tooze, T. Yoshimori, J. Yuan, Z. Yue, Q. Zhong, and G. Kroemer, "Molecular definitions of autophagy and related processes", The EMBO Journal, vol. 36, pp. 1811-1836, 2017. http://dx.doi.org/10.15252/embj.201796697
- Z. Yin, C. Pascual, and D. Klionsky, "Autophagy: machinery and regulation", Microbial Cell, vol. 3, pp. 588-596, 2016. http://dx.doi.org/10.15698/mic2016.12.546
- F. Pietrocola, V. Izzo, M. Niso-Santano, E. Vacchelli, L. Galluzzi, M.C. Maiuri, and G. Kroemer, "Regulation of autophagy by stress-responsive transcription factors", Seminars in Cancer Biology, vol. 23, pp. 310-322, 2013. http://dx.doi.org/10.1016/j.semcancer.2013.05.008
- G. Mariño, M. Niso-Santano, E.H. Baehrecke, and G. Kroemer, "Self-consumption: the interplay of autophagy and apoptosis", Nature Reviews Molecular Cell Biology, vol. 15, pp. 81-94, 2014. http://dx.doi.org/10.1038/nrm3735
- A.R. Ariosa, and D.J. Klionsky, "Autophagy core machinery: overcoming spatial barriers in neurons", Journal of Molecular Medicine, vol. 94, pp. 1217-1227, 2016. http://dx.doi.org/10.1007/s00109-016-1461-9
- L. Galluzzi, J.M. Bravo-San Pedro, K. Blomgren, and G. Kroemer, "Autophagy in acute brain injury", Nature Reviews Neuroscience, vol. 17, pp. 467-484, 2016. http://dx.doi.org/10.1038/nrn.2016.51
- D. Hernandez, C. Torres, W. Setlik, C. Cebrián, E. Mosharov, G. Tang, H. Cheng, N. Kholodilov, O. Yarygina, R. Burke, M. Gershon, and D. Sulzer, "Regulation of Presynaptic Neurotransmission by Macroautophagy", Neuron, vol. 74, pp. 277-284, 2012. http://dx.doi.org/10.1016/j.neuron.2012.02.020
- J.L. Schneider, A.M. Miller, and M.E. Woesner, "Autophagy and Schizophrenia: A Closer Look at How Dysregulation of Neuronal Cell Homeostasis Influences the Pathogenesis of Schizophrenia", Einstein Journal of Biology and Medicine, vol. 31, pp. 34, 2017. http://dx.doi.org/10.23861/EJBM201631752
- J. Kesby, D. Eyles, J. McGrath, and J. Scott, "Dopamine, psychosis and schizophrenia: the widening gap between basic and clinical neuroscience", Translational Psychiatry, vol. 8, 2018. http://dx.doi.org/10.1038/s41398-017-0071-9
- J. Lally, and J.H. MacCabe, "Antipsychotic medication in schizophrenia: a review", British Medical Bulletin, vol. 114, pp. 169-179, 2015. http://dx.doi.org/10.1093/bmb/ldv017
- M. Salas, B. Tuchweber, and P. Kourounakis, "Liver Ultrastructure During Acute Stress", Pathology - Research and Practice, vol. 167, pp. 217-233, 1980. http://dx.doi.org/10.1016/S0344-0338(80)80052-5
- L. Zhang, J. Yu, H. Pan, P. Hu, Y. Hao, W. Cai, H. Zhu, A.D. Yu, X. Xie, D. Ma, and J. Yuan, "Small molecule regulators of autophagy identified by an image-based high-throughput screen", Proceedings of the National Academy of Sciences, vol. 104, pp. 19023-19028, 2007. http://dx.doi.org/10.1073/pnas.0709695104
- D.C. Rubinsztein, A.M. Cuervo, B. Ravikumar, S. Sarkar, V.I. Korolchuk, S. Kaushik, and D.J. Klionsky, "In search of an “autophagomometer”", Autophagy, vol. 5, pp. 585-589, 2009. http://dx.doi.org/10.4161/auto.5.5.8823
- L. Esteban-Martínez, and P. Boya, "Autophagic flux determination in vivo and ex vivo", Methods, vol. 75, pp. 79-86, 2015. http://dx.doi.org/10.1016/j.ymeth.2015.01.008
- Y.D. Kim, E.I. Jeong, J. Nah, S. Yoo, W.J. Lee, Y. Kim, S. Moon, S. Hong, and Y. Jung, "Pimozide reduces toxic forms of tau in TauC3 mice via 5′ adenosine monophosphate‐activated protein kinase‐mediated autophagy", Journal of Neurochemistry, vol. 142, pp. 734-746, 2017. http://dx.doi.org/10.1111/jnc.14109
- J.H. Shin, S.J. Park, E.S. Kim, Y.K. Jo, J. Hong, and D. Cho, "Sertindole, a Potent Antagonist at Dopamine D<sub>2</sub> Receptors, Induces Autophagy by Increasing Reactive Oxygen Species in SH-SY5Y Neuroblastoma Cells", Biological and Pharmaceutical Bulletin, vol. 35, pp. 1069-1075, 2012. http://dx.doi.org/10.1248/bpb.b12-00009
- L. Vucicevic, M. Misirkic-Marjanovic, V. Paunovic, T. Kravic-Stevovic, T. Martinovic, D. Ciric, N. Maric, S. Petricevic, L. Harhaji-Trajkovic, V. Bumbasirevic, and V. Trajkovic, "Autophagy inhibition uncovers the neurotoxic action of the antipsychotic drug olanzapine", Autophagy, vol. 10, pp. 2362-2378, 2014. http://dx.doi.org/10.4161/15548627.2014.984270
- Y. Zhang, D.T. Nguyen, E.M. Olzomer, G.P. Poon, N.J. Cole, A. Puvanendran, B.R. Phillips, and D. Hesselson, "Rescue of Pink1 Deficiency by Stress-Dependent Activation of Autophagy", Cell Chemical Biology, vol. 24, pp. 471-480.e4, 2017. http://dx.doi.org/10.1016/j.chembiol.2017.03.005
- S.J. Barmada, A. Serio, A. Arjun, B. Bilican, A. Daub, D.M. Ando, A. Tsvetkov, M. Pleiss, X. Li, D. Peisach, C. Shaw, S. Chandran, and S. Finkbeiner, "Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models", Nature Chemical Biology, vol. 10, pp. 677-685, 2014. http://dx.doi.org/10.1038/nchembio.1563
- M. Höllerhage, J.N. Goebel, A. de Andrade, T. Hildebrandt, A. Dolga, C. Culmsee, W.H. Oertel, B. Hengerer, and G.U. Höglinger, "Trifluoperazine rescues human dopaminergic cells from wild-type α-synuclein-induced toxicity", Neurobiology of Aging, vol. 35, pp. 1700-1711, 2014. http://dx.doi.org/10.1016/j.neurobiolaging.2014.01.027
- A.S. Tsvetkov, J. Miller, M. Arrasate, J.S. Wong, M.A. Pleiss, and S. Finkbeiner, "A small-molecule scaffold induces autophagy in primary neurons and protects against toxicity in a Huntington disease model", Proceedings of the National Academy of Sciences, vol. 107, pp. 16982-16987, 2010. http://dx.doi.org/10.1073/pnas.1004498107
- S.H. Kim, S. Park, H.S. Yu, K.H. Ko, H.G. Park, and Y.S. Kim, "The antipsychotic agent clozapine induces autophagy via the AMPK-ULK1-Beclin1 signaling pathway in the rat frontal cortex", Progress in Neuro-Psychopharmacology and Biological Psychiatry, vol. 81, pp. 96-104, 2018. http://dx.doi.org/10.1016/j.pnpbp.2017.10.012
- J. Park, S. Chung, H. An, J. Kim, J. Seo, D. Kim, and S. Yoon, "Haloperidol and clozapine block formation of autophagolysosomes in rat primary neurons", Neuroscience, vol. 209, pp. 64-73, 2012. http://dx.doi.org/10.1016/j.neuroscience.2012.02.035
- K. Castillo, V. Valenzuela, M. Oñate, and C. Hetz, "A Molecular Reporter for Monitoring Autophagic Flux in Nervous System In Vivo", Methods in Enzymology, pp. 109-131, 2017. http://dx.doi.org/10.1016/bs.mie.2016.09.077
- Y. Feng, Z. Yao, and D.J. Klionsky, "How to control self-digestion: transcriptional, post-transcriptional, and post-translational regulation of autophagy", Trends in Cell Biology, vol. 25, pp. 354-363, 2015. http://dx.doi.org/10.1016/j.tcb.2015.02.002
- D.G. Hardie, "AMPK and autophagy get connected", The EMBO Journal, vol. 30, pp. 634-635, 2011. http://dx.doi.org/10.1038/emboj.2011.12
- G. Filomeni, D. De Zio, and F. Cecconi, "Oxidative stress and autophagy: the clash between damage and metabolic needs", Cell Death & Differentiation, vol. 22, pp. 377-388, 2014. http://dx.doi.org/10.1038/cdd.2014.150
- J. Lee, S. Giordano, and J. Zhang, "Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling", Biochemical Journal, vol. 441, pp. 523-540, 2011. http://dx.doi.org/10.1042/BJ20111451
- H.R. Shin, H. Kim, S. Oh, J. Lee, M. Kee, H. Ko, M. Kweon, K. Won, and S.H. Baek, "AMPK–SKP2–CARM1 signalling cascade in transcriptional regulation of autophagy", Nature, vol. 534, pp. 553-557, 2016. http://dx.doi.org/10.1038/nature18014
- E. Hedrick, X. Li, and S. Safe, "Penfluridol Represses Integrin Expression in Breast Cancer through Induction of Reactive Oxygen Species and Downregulation of Sp Transcription Factors", Molecular Cancer Therapeutics, vol. 16, pp. 205-216, 2017. http://dx.doi.org/10.1158/1535-7163.MCT-16-0451
- S. Comincini, G. Allavena, S. Palumbo, M. Morini, F. Durando, F. Angeletti, L. Pirtoli, and C. Miracco, "microRNA-17 regulates the expression of ATG7 and modulates the autophagy process, improving the sensitivity to temozolomide and low-dose ionizing radiation treatments in human glioblastoma cells", Cancer Biology & Therapy, vol. 14, pp. 574-586, 2013. http://dx.doi.org/10.4161/cbt.24597
- H. Wu, F. Wang, S. Hu, C. Yin, X. Li, S. Zhao, J. Wang, and X. Yan, "MiR-20a and miR-106b negatively regulate autophagy induced by leucine deprivation via suppression of ULK1 expression in C2C12 myoblasts", Cellular Signalling, vol. 24, pp. 2179-2186, 2012. http://dx.doi.org/10.1016/j.cellsig.2012.07.001
- G. Kroemer, G. Mariño, and B. Levine, "Autophagy and the Integrated Stress Response", Molecular Cell, vol. 40, pp. 280-293, 2010. http://dx.doi.org/10.1016/j.molcel.2010.09.023
- G. Benítez-King, L. Ortíz-López, G. Jiménez-Rubio, and G. Ramírez-Rodríguez, "Haloperidol causes cytoskeletal collapse in N1E-115 cells through tau hyperphosphorylation induced by oxidative stress: Implications for neurodevelopment", European Journal of Pharmacology, vol. 644, pp. 24-31, 2010. http://dx.doi.org/10.1016/j.ejphar.2010.06.057
- D. Wang, X. Ji, J. Liu, Z. Li, and X. Zhang, "Dopamine Receptor Subtypes Differentially Regulate Autophagy", International Journal of Molecular Sciences, vol. 19, pp. 1540, 2018. http://dx.doi.org/10.3390/ijms19051540
- J. Wang, Y. Cao, Q. Li, Y. Yang, M. Jin, D. Chen, F. Wang, G. Wang, Z. Qin, L. Hu, and C. Liu, "A pivotal role of FOS-mediated BECN1/Beclin 1 upregulation in dopamine D2 and D3 receptor agonist-induced autophagy activation", Autophagy, vol. 11, pp. 2057-2073, 2015. http://dx.doi.org/10.1080/15548627.2015.1100930
- M.R. Barnes, J. Huxley‐Jones, P.R. Maycox, M. Lennon, A. Thornber, F. Kelly, S. Bates, A. Taylor, J. Reid, N. Jones, J. Schroeder, C.A. Scorer, C. Davies, J.J. Hagan, J.N. Kew, C. Angelinetta, T. Akbar, S. Hirsch, A.M. Mortimer, T.R. Barnes, and J. de Belleroche, "Transcription and pathway analysis of the superior temporal cortex and anterior prefrontal cortex in schizophrenia", Journal of Neuroscience Research, vol. 89, pp. 1218-1227, 2011. http://dx.doi.org/10.1002/jnr.22647
- Y. Horesh, P. Katsel, V. Haroutunian, and E. Domany, "Gene expression signature is shared by patients with Alzheimer’s disease and schizophrenia at the superior temporal gyrus", European Journal of Neurology, vol. 18, pp. 410-424, 2011. http://dx.doi.org/10.1111/j.1468-1331.2010.03166.x
- A. Merenlender-Wagner, A. Malishkevich, Z. Shemer, M. Udawela, A. Gibbons, E. Scarr, B. Dean, J. Levine, G. Agam, and I. Gozes, "Autophagy has a key role in the pathophysiology of schizophrenia", Molecular Psychiatry, vol. 20, pp. 126-132, 2013. http://dx.doi.org/10.1038/mp.2013.174
- M. Shehata, and K. Inokuchi, "Does autophagy work in synaptic plasticity and memory?", Reviews in the Neurosciences, vol. 25, 2014. http://dx.doi.org/10.1515/revneuro-2014-0002
- T.A. Atkin, N.J. Brandon, and J.T. Kittler, "Disrupted in Schizophrenia 1 forms pathological aggresomes that disrupt its function in intracellular transport", Human Molecular Genetics, vol. 21, pp. 2017-2028, 2012. http://dx.doi.org/10.1093/hmg/dds018
- G. Jevtić, T. Nikolić, A. Mirčić, T. Stojković, M. Velimirović, V. Trajković, I. Marković, A.M. Trbovich, N.V. Radonjić, and N.D. Petronijević, "Mitochondrial impairment, apoptosis and autophagy in a rat brain as immediate and long-term effects of perinatal phencyclidine treatment — influence of restraint stress", Progress in Neuro-Psychopharmacology and Biological Psychiatry, vol. 66, pp. 87-96, 2016. http://dx.doi.org/10.1016/j.pnpbp.2015.11.014
- A. Merenlender-Wagner, Z. Shemer, O. Touloumi, R. Lagoudaki, E. Giladi, A. Andrieux, N.C. Grigoriadis, and I. Gozes, "New horizons in schizophrenia treatment: autophagy protection is coupled with behavioral improvements in a mouse model of schizophrenia", Autophagy, vol. 10, pp. 2324-2332, 2014. http://dx.doi.org/10.4161/15548627.2014.984274
- W. Song, H. Zukor, S. Lin, J. Hascalovici, A. Liberman, A. Tavitian, J. Mui, H. Vali, X. Tong, S.K. Bhardwaj, L.K. Srivastava, E. Hamel, and H.M. Schipper, "Schizophrenia-Like Features in Transgenic Mice Overexpressing Human HO-1 in the Astrocytic Compartment", The Journal of Neuroscience, vol. 32, pp. 10841-10853, 2012. http://dx.doi.org/10.1523/JNEUROSCI.6469-11.2012
- R.M. Bonelli, P. Hofmann, A. Aschoff, G. Niederwieser, C. Heuberger, G. Jirikowski, and H. Kapfhammer, "The influence of psychotropic drugs on cerebral cell death: female neurovulnerability to antipsychotics", International Clinical Psychopharmacology, vol. 20, pp. 145-149, 2005. http://dx.doi.org/10.1097/00004850-200505000-00004
- B. Ho, N.C. Andreasen, S. Ziebell, R. Pierson, and V. Magnotta, "Long-term Antipsychotic Treatment and Brain Volumes", Archives of General Psychiatry, vol. 68, pp. 128, 2011. http://dx.doi.org/10.1001/archgenpsychiatry.2010.199
- B. Caballero, and A. Coto-Montes, "An insight into the role of autophagy in cell responses in the aging and neurodegenerative brain.", Histology and histopathology, 2012. http://www.ncbi.nlm.nih.gov/pubmed/22237704
- G.P. Reynolds, "Antipsychotic drug use in neurodegenerative disease in the elderly: problems and potential from a pharmacological perspective", Expert Opinion on Pharmacotherapy, vol. 2, pp. 543-548, 2001. http://dx.doi.org/10.1517/14656566.2.4.543
- D. Charvin, E. Roze, V. Perrin, C. Deyts, S. Betuing, C. Pagès, E. Régulier, R. Luthi-Carter, E. Brouillet, N. Déglon, and J. Caboche, "Haloperidol protects striatal neurons from dysfunction induced by mutated huntingtin in vivo", Neurobiology of Disease, vol. 29, pp. 22-29, 2008. http://dx.doi.org/10.1016/j.nbd.2007.07.028
- Z. Yue, L. Friedman, M. Komatsu, and K. Tanaka, "The cellular pathways of neuronal autophagy and their implication in neurodegenerative diseases", Biochimica et Biophysica Acta (BBA) - Molecular Cell Research, vol. 1793, pp. 1496-1507, 2009. http://dx.doi.org/10.1016/j.bbamcr.2009.01.016
- C.G. Ballard, R.H. Perry, I.G. McKeith, and E.K. Perry, "Neuroleptics are associated with more severe tangle pathology in dementia with Lewy bodies", International Journal of Geriatric Psychiatry, vol. 20, pp. 872-875, 2005. http://dx.doi.org/10.1002/gps.1378
- H. Cheng, Y. Liang, Y. Kuo, C. Chuu, C. Lin, M. Lee, A.T.H. Wu, C. Yeh, E.I. Chen, J. Whang-Peng, C. Su, and C. Huang, "Identification of thioridazine, an antipsychotic drug, as an antiglioblastoma and anticancer stem cell agent using public gene expression data", Cell Death & Disease, vol. 6, pp. e1753-e1753, 2015. http://dx.doi.org/10.1038/cddis.2015.77
- M. Indelicato, B. Pucci, L. Schito, V. Reali, M. Aventaggiato, M.C. Mazzarino, F. Stivala, M. Fini, M.A. Russo, and M. Tafani, "Role of hypoxia and autophagy in MDA‐MB‐231 invasiveness", Journal of Cellular Physiology, vol. 223, pp. 359-368, 2010. http://dx.doi.org/10.1002/jcp.22041
- A. Ranjan, and S.K. Srivastava, "Penfluridol suppresses pancreatic tumor growth by autophagy-mediated apoptosis", Scientific Reports, vol. 6, 2016. http://dx.doi.org/10.1038/srep26165
- S.Y. Shin, K.S. Lee, Y. Choi, H.J. Lim, H.G. Lee, Y. Lim, and Y.H. Lee, "The antipsychotic agent chlorpromazine induces autophagic cell death by inhibiting the Akt/mTOR pathway in human U-87MG glioma cells", Carcinogenesis, vol. 34, pp. 2080-2089, 2013. http://dx.doi.org/10.1093/carcin/bgt169
- Y. Wang, S. Xu, X. Chen, R. Liu, and Z. Liang, "Autophagy Involvement in Olanzapine-Mediated Cytotoxic Effects in Human Glioma Cells", Asian Pacific Journal of Cancer Prevention, vol. 15, pp. 8107-8113, 2014. http://dx.doi.org/10.7314/APJCP.2014.15.19.8107
- C. Wu, L. Bai, M. Tsai, P. Chu, C. Chiu, M.Y. Chen, S. Chiu, J. Chiang, and J. Weng, "Pharmacological exploitation of the phenothiazine antipsychotics to develop novel antitumor agents–A drug repurposing strategy", Scientific Reports, vol. 6, 2016. http://dx.doi.org/10.1038/srep27540
- H. Xia, L. Zhang, G. Chen, T. Zhang, J. Liu, M. Jin, X. Ma, D. Ma, and J. Yuan, "Control of basal autophagy by calpain1 mediated cleavage of ATG5", Autophagy, vol. 6, pp. 61-66, 2010. http://dx.doi.org/10.4161/auto.6.1.10326
- Y. Yin, C. Lin, T. Chen, J. Chen, H. Tsai, C. Wang, and S. Chen, "Clozapine Induces Autophagic Cell Death in Non-Small Cell Lung Cancer Cells", Cellular Physiology and Biochemistry, vol. 35, pp. 945-956, 2015. http://dx.doi.org/10.1159/000369751
- K.L. Conway, P. Kuballa, J. Song, K.K. Patel, A.B. Castoreno, O.H. Yilmaz, H.B. Jijon, M. Zhang, L.N. Aldrich, E.J. Villablanca, J.M. Peloquin, G. Goel, I. Lee, E. Mizoguchi, H.N. Shi, A.K. Bhan, S.Y. Shaw, S.L. Schreiber, H.W. Virgin, A.F. Shamji, T.S. Stappenbeck, H. Reinecker, and R.J. Xavier, "Atg16l1 is Required for Autophagy in Intestinal Epithelial Cells and Protection of Mice From Salmonella Infection", Gastroenterology, vol. 145, pp. 1347-1357, 2013. http://dx.doi.org/10.1053/j.gastro.2013.08.035
- J. Li, S.C. Pak, L.P. O’Reilly, J.A. Benson, Y. Wang, T. Hidvegi, P. Hale, C. Dippold, M. Ewing, G.A. Silverman, and D.H. Perlmutter, "Fluphenazine Reduces Proteotoxicity in C. elegans and Mammalian Models of Alpha-1-Antitrypsin Deficiency", PLoS ONE, vol. 9, pp. e87260, 2014. http://dx.doi.org/10.1371/journal.pone.0087260
- A. Rimessi, C. Pavan, E. Ioannidi, F. Nigro, C. Morganti, A. Brugnoli, F. Longo, C. Gardin, L. Ferroni, M. Morari, V. Vindigni, B. Zavan, and P. Pinton, "Protein Kinase C β: a New Target Therapy to Prevent the Long-Term Atypical Antipsychotic-Induced Weight Gain", Neuropsychopharmacology, vol. 42, pp. 1491-1501, 2017. http://dx.doi.org/10.1038/npp.2017.20
- B. Wang, S. Zhong, F. Zheng, Y. Zhang, F. Gao, Y. Chen, B. Lu, H. Xu, and G. Shi, "N-n-butyl haloperidol iodide protects cardiomyocytes against hypoxia/reoxygenation injury by inhibiting autophagy", Oncotarget, vol. 6, pp. 24709-24721, 2015. http://dx.doi.org/10.18632/oncotarget.5077
- M. Brini, T. Calì, D. Ottolini, and E. Carafoli, "Neuronal calcium signaling: function and dysfunction", Cellular and Molecular Life Sciences, vol. 71, pp. 2787-2814, 2014. http://dx.doi.org/10.1007/s00018-013-1550-7
- I. Loryan, E. Melander, M. Svensson, M. Payan, F. König, B. Jansson, and M. Hammarlund-Udenaes, "In-depth neuropharmacokinetic analysis of antipsychotics based on a novel approach to estimate unbound target-site concentration in CNS regions: link to spatial receptor occupancy", Molecular Psychiatry, vol. 21, pp. 1527-1536, 2016. http://dx.doi.org/10.1038/mp.2015.229
ACKNOWLEDGMENTS
The study was supported by the Ministry of Education, Science, and Technological Development of the Republic of Serbia (Grant No. 41025 and 173053).
COPYRIGHT
© 2018

Mechanisms and therapeutic significance of autophagy modulation by antipsychotic drugs by Vucicevic et al. is licensed under a Creative Commons Attribution 4.0 International License.


