Reviews:
Cell Stress, Vol. 2, No. 8, pp. 200 - 212; doi: 10.15698/cst2018.07.148
T lymphocytes against solid malignancies: winning ways to defeat tumours

1 Dept. of Pediatric Onco-Hematology and cell and gene therapy, IRCCS Bambino Gesù Children’s Hospital, Rome, Italy.
2 Dept. of Biology, University of Rome Tor Vergata, Rome, Italy.
3 IRCCS, Santa Lucia Foundation, Rome, Italy.
§,# These authors contributed equally to this work.
Keywords: cancer immuno-therapy, immune cell infiltration, tumour microenvironment, extracellular matrix, immune-suppression.
Abbreviations:
CAR – chimeric antigen receptor,
DC – dendritic cell,
ECM – extracellular matrix,
ILC – innate lymphoid cell,
MDSC – myeloid-derived suppressor cell,
MHC – major-histocompatibility complex
NK – natural killer,
OXPHOS – oxidative phosphorylation,
SLO – secondary lymphoid organ,
TAMa – tumour-associated macrophage,
TCR – T-cell receptor,
TIL – tumour infiltrating T lymphocytes,
TME – tumour microenvironment,
Treg – regulatory T-cell.
Received originally: 29/04/2018 Received in revised form: 10/07/2018
Accepted: 11/07/2018
Published: 26/07/2018
Correspondence:
Silvia Campello, Department of Biology, University of Rome Tor Vergata, Via della Ricerca Scientifica, 00133 Rome, Italy; Phone: +390672594227 silvia.campello@uniroma2.it
Conflict of interest statement: The authors declare no conflict of interest.
Please cite this article as: Ignazio Caruana, Luca Simula, Franco Locatelli and Silvia Campello (2018). T lymphocytes against solid malignancies: winning ways to defeat tumours. Cell Stress 2(8): 200-212. doi: 10.15698/cst2018.07.148
Abstract
In the last decades, a novel field has emerged in the cure of cancer, by boosting the ability of the patient’s immune system to recognize and kill tumour cells. Although excellent and encouraging results, exploiting the effect of genetically modified T cells, have been obtained, it is now evident that tumour malignancies can evolve several mechanisms to escape such immune responses, thus continuing their growth in the body. These mechanisms are in part due to tumour cell metabolic or genetic alterations, which can render the target invisible to the immune system or can favour the generation of an extracellular milieu preventing immune cell infiltration or cytotoxicity. Such mechanisms may also involve the accumulation inside the tumour microenvironment of different immune-suppressive cell types, which further down-regulate the activity of cytotoxic immune cells either directly by interacting with them or indirectly by releasing suppressive molecules. In this review, we will first focus on describing several mechanisms by which tumour cells may dampen or abrogate the immune response inside the tumour microenvironment and, second, on current strategies that are adopted to cope with and possibly overcome such alterations, thus ameliorating the efficacy of the current-in-use anti-cancer immuno-therapies.
INTRODUCTION
When a tumour mass starts growing in the body, T lymphocytes are activated inside secondary lymphoid organs (SLOs) and mount an immune response against immuno-stimulating tumour antigens presented by professional antigen presenting cells. This stimulation allows T lymphocytes to leave SLOs, thus reaching the tumour mass and eliminating malignant cells. Although the capability of the immune system to prevent tumour growth may last for years, tumour cells may eventually escape immune-surveillance [1] and create a chronic tumour microenvironment (TME), which predisposes to the generation of highly-inefficient, “dysfunctional” T cells, with impaired metabolic activity and cytotoxic functionality [2].
In this Review, we will focus on the mechanisms by which alterations in the architecture of the TME predispose to a dysfunctionality of T cells, once inside the tumour, thus limiting their ability to cope with a growing number of cancer cells.
THE SEVERAL IMMUNE SUBTYPES INSIDE THE TME
Most subtypes of the innate and adaptive immune cells infiltrate into a solid tumour mass and can be generally divided for their pro- or anti-tumor role (Table 1). Tumour-infiltrating T lymphocytes are commonly known as TILs. Cytotoxic CD8+ TILs are major player in the immune response against tumour growth and their recognition of tumour-specific antigens allows killing of malignant cells through Fas ligand or perforin/granzyme pathways. Usually, helper CD4+ TILs mainly produce several inflammatory cytokines, which can support the activation and cytotoxicity of CD8+ T cells and of other pro-inflammatory immune cells [3]. However, a subgroup of CD4+ T cells, the regulatory T cells (Treg), play an anti-inflammatory and immune-suppressive role inside the TME, thus favouring tumour growth, by inhibiting dendritic cell (DC) presentation of tumour antigens to T cells and by producing membrane-bound or soluble factors, which impair T-cell activation and cytotoxicity [4]. The role of tumour infiltrating B lymphocytes (TIBLs) in cancer progression is more debated. Although some reports indicate that TIBLs can have an anti-tumor effect, for example producing antibodies which favour tumour cell recognition by phagocytic macrophages, they can also differentiate towards a regulatory phenotype (Bregs), which produce pro-tumor factors, such as lymphotoxin (which promotes neo-angiogenesis) and immune suppressive cytokines [5].
–
TABLE 1. Immune subtypes infiltrating solid tumors. List of innate and adaptive immune subtypes infiltrating solid tumour microenvironment. For each one, the pro- or anti-tumoral role (activity), the prevalent metabolism, and the main released factors -as soluble molecules or exposed on the cell surface, are reported. |
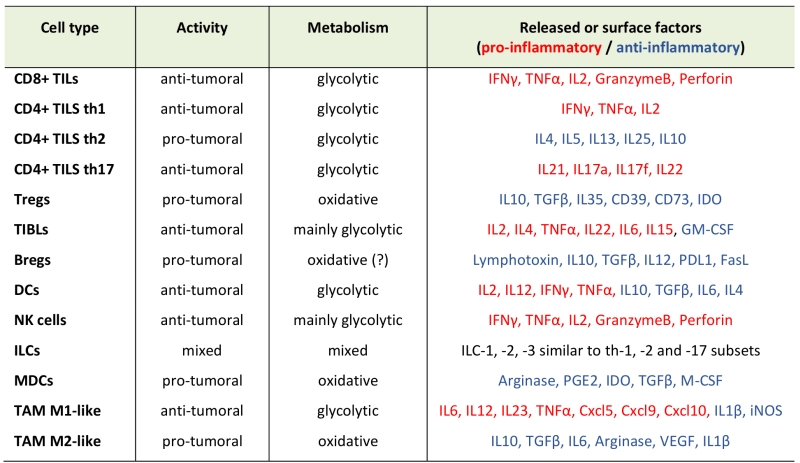 |
Abbreviations: TILs: tumor-infiltrating-T-lymphocytes; TIBLs: tumor-infiltrating-B-lymphocytes; DCs: dendritic cells; NK: natural Killer; ILCs: innate lymphoid cells (other than NK cells); MDSCs: myeloid-derived-suppressor-cells; TAM: tumor-associated-macrophages; IDO: Indoleamine-2,3-dioxygenase; iNOS: inducible Nitric Oxide Synthase; VEGF: vascular endothelial growth factor. |
–
DCs are required to uptake and present tumour-derived antigens to adaptive T cells into secondary lymphoid organs (SLOs). Therefore, their infiltration into tumour mass is required to mount an efficient adaptive immune response against tumours, and to sustain the generation of tumour-specific T lymphocytes for an efficient elimination of malignant cells [6]. Innate lymphoid cells (ILCs) are a large family of innate cells whose subdivision is very similar to that of T lymphocytes, although they lack antigen-specific receptors and are mainly activated in response to common innate signals. Indeed, among them, we can find cytotoxic natural killer (NK) cells (equivalent of CD8+ T lymphocytes) and helper-like ILCs, which further subdivide into ILC-1, -2, and -3 (which are functionally similar to Th1, Th2 and Th17 CD4+ T cells, respectively) [7]. NK cells are cytotoxic cells which can recognize and kill tumour cells that lack the expression of major-histocompatibility-complex (MHC) molecules, and therefore cannot present antigens on cell surface, this rendering them invisible to cytotoxic CD8+ T cells [8]. Between regular α/β T lymphocytes and NK cells, a subgroup of T cells named γ/δ T lymphocytes combine both innate and adaptive characteristics. Their potent cytotoxic activity against bacteria, virus and tumours makes them particularly attractive for adoptive immunotherapy approaches. Differently from αβ T cells, these cells recognize their ligands in an MHC-independent manner showing a significant reduced allo-reactivity as compared to αβ T cells, this making them appealing for clinical translation [9][10]. Contrary to NK cells and γ/δ T lymphocytes, the role of other ILCs in cancer has only recently been discovered and few related data are available. However, it seems that ILC2 and ILC3 subtypes may favour tumour growth by secretion of specific immune-suppressive cytokines [7]. Similarly, myeloid-derived-suppressor-cells (MDSCs) are pro-tumoral cells, producing factors that inhibit T-cell activity and also promote tumour growth by remodelling extracellular matrix and blood capillaries [11]. Last, macrophages can differentiate into different subtypes inside the TME, where they are known as tumor-associated macrophages (TAM). Briefly, an anti-tumor M1-type TAM can produce inflammatory cytokines and phagocyte tumour cells [12]. However, most TAMs have a pro-tumor M2-like phenotype, favouring tissue remodelling and angiogenesis and producing immune suppressive cytokines, which dampen T cell response [12].
–
It is worth noting that the amount and subtypes of immune cells present in TME may vary widely between different tumours. This heterogeneity has led to the development of a classification into “immune-desert”, “immune-inflamed” and “immune-excluded” tumours [13], based on the quality and quantity of immune infiltrates. Immune-desert tumours show poor infiltration of T cells and are characterized by high number of myeloid suppressor cells, which also produce immune-suppressive cytokines. Immune-excluded tumours are characterized by a high amount of infiltrating immune cells, both immune-suppressive (pro-tumor) or cytotoxic (anti-tumor). However, these cells do not frequently penetrate inside the tumour parenchyma but remain in the surrounding stroma and therefore cannot efficiently kill malignant cells. Last, immune-inflamed tumors are characterized by high amounts of infiltrating T cells, which efficiently reach tumour cells for their killing, and by abundant production of pro-inflammatory cytokines [13].
THE FIRST SIDE OF THE COIN: TME IN SOLID MALIGNANCIES
Tumours have been recognized as complex disorganized and chaotic organs, where cancer cells co-exist and co-evolve with their stroma. The interface between malignant and non-transformed cells defines the TME [1][14]. The importance of TME for tumorigenesis is now widely recognized in both solid and haematological malignancies [15]. However, in solid tumours, the TME has a more relevant impact on tumour growth being able to offer protection with respect of the action of the immune system. During solid tumour development, the TME initiates to organize itself supporting tumour growth directly, but also erects chemical and physical barriers capable to defend the tumour from the activity of an intact immune system, thereby preventing cancer immune surveillance. In this context, TME appears to be a complex ecosystem containing a tight interstitial extracellular matrix (ECM), where various stromal, endothelial and inflammatory cells are recruited from the surrounding tissues. The interaction between these different components modulates phenotype and behaviour of the tumour and may affect cancer progression, as well as the formation of metastases [16][17][18]. Specifically, it has been reported that, in this environment, cancer cells show self-sufficiency in growth signals, resistance to programmed cell death, limitless replicative potential, and ability to induce angiogenesis, invasion, and metastasis formation [19][20]. Recently, the role of ECM in the regulation of many of these cellular responses has been recognized. ECM has a fundamental role in cell behaviour and fate, not only sustaining and interconnecting cells, but also influencing many cellular mechanics and functions, such as differentiation and migration, in both physiological and pathological conditions.
–
Cancer cells are also able to develop mechanisms to blunt detection and eradication by immune cells. These strategies include: i) a reduction of tumour immunogenicity, due to loss of expression of tumour-associated antigens or MHC class I molecules, ii) acquired DNA copy number alterations and oncogenic signaling -equipping them with an uncontrolled proliferative capacity and insensitivity to negative feedback from microenvironment, iii) an up-regulation of cellular immune check-points – such as the programmed death ligand 1 (PDL1, which inhibits T-cell activation by stimulating PD-1 receptor on T-cell surface), indoleamine 2,3-dioxygenase, and finally iv) an altered metabolism producing a low pH and secretion of various metabolites, which inhibit the effector cell recruitment, persistence and activity [21][22][23][24]. In particular, tumour cells can evolve mechanisms which actively induce T-cell apoptosis, by up-regulating the expression of pro-apoptotic molecules on their surface, such as Galectin-1 [25], TRAIL (TNF-Related Apoptosis-Inducing Ligand) [26] and Fas Ligand (FasL) [27], which promote T-cell death by interacting with the corresponding receptors on T-cell surface. Moreover, in the TME, a competition for nutrient availability is frequently observed between tumour and immune cells. Although cancer stem cells may rely on an oxidative metabolism for their survival [28], in most cases, tumour cells switch their metabolism from oxidative phosphorylation (OXPHOS) to glycolysis even in presence of high-oxygen tension to sustain a high proliferation rate (a process known as Warburg effect [29]). Since this switch is also observed for T-cell upon activation [30], this generates a competition for glucose availability between these cell types. However, tumour cells frequently win the battle probably because of a faster glycolytic rate in tumour cells [31] and a further down-regulation of glycolytic flux in immune cells by tumour cells-released extracellular lactate [32][33]. As mentioned above, tumour cells may up-regulate PDL1 on their surface [34][35], which engages the PD-1 receptor on activated T lymphocytes, thereby activating a signalling cascade that inhibits PI3K/Akt/mTOR axis [36], essential to induce glycolysis in effector T cells. Interestingly, while these considerations apply to effector cytotoxic T cells, immune Tregs actively maintain an OXPHOS-based catabolism instead of glycolysis [37][38]. This confers to Tregs a metabolic advantage compared to effector/cytotoxic T cells [39]. To further increase the suppressive TME, several other molecules are secreted by tumour cells and by other immune-cells recruited by the tumour. For example, in a preclinical study it has been reported how the secretion of prostaglandin-E2 (PGE2) and adenosine by endothelial tumour-associated cells selectively kills effector T cells and how their inhibition resulted in down-regulation of FasL and CD8 T-cell influx [40].
–
Besides Tregs, other cells of the immune system are recruited to the TME as MDSC, TAMs and neutrophils. All these cells boost the tumour survival-promoting environment [41][42][43][44] i) by reducing, for example, L-arginine concentration, which is required for long-lasting survival of infiltrating memory T cells [45], ii) by producing reactive nitrogen species that hamper T-cell proliferation and function, and iii) by expressing molecules on their surface capable to activate check-point inhibitor receptors expressed by T lymphocytes (Galactin9) [44][45][46][47]. The described TME with all these elements is able to regenerate and stabilize itself.
–
Making matters worse, tumour cells and their TME are able to initiate and promote angiogenesis. This phenomenon induces the formation of new vessels capable to support blood supply to the tumour; the resulting vessel network is leaky, chaotically organized, immature, thin-walled and ill-perfused. Such an aberrant angiogenesis contributes to the maintenance of the pro-tumorigenic and immunosuppressive TME and profoundly influences how cancer cells escape the anti-cancer immune surveillance, metastasize, and respond to immunotherapy [48][49][50][51]. This occurs by preventing, for instance, a correct inflow of the immune system to the tumour site. Furthermore, this reduced influx of nutrients and gaseous exchange strongly decreases the quality and number of effector cells in the TME, thus increasing tumour growth as well as the possibility of invasiveness of tumour cells [52][53]. For these reasons, the TME represents an inhospitable and inaccessible environment for effector immune cells, due to the generation of a hypoxic atmosphere, low nutrient supply and a high concentration of metabolic acids. These conditions facilitate the selection of cancer cells with genetic and epigenetic alterations, which enhance their aggressiveness. In the meanwhile, they increase activation-induced autophagy processes and stress in immune cells, which makes cytotoxic lymphocytes unable to proliferate and produce cytokines [54][55]. Hypoxia in the TME can dampen T-cell functionality through different mechanisms, such as: i) by exacerbating glucose deprivation, ii) by reducing cytosolic levels of Ca2+, which is essential for cytokine production [56], or iii) by promoting excessive formation of reactive oxygen species [57]. In addition, it has been proven that hypoxia can up-regulate PDL1 on tumour cells [58], which in turn dampens T-cell functionality by interacting with the inhibitory receptor PD1 on T-cell surface. Importantly, hypoxia and acidosis, besides reducing the cytotoxic activity of tumour-infiltrating effector T cells, also facilitate the attraction and/or development of immuno-suppressive immune cells, and hamper delivery of chemotherapeutics and immunotherapeutic entities, as well as cancer cell killing in response to radio/chemotherapy and immunotherapy.
–
The recruitment of particular cell types into TME and their contact with tumour cells has been described to produce an immuno-suppressive microenvironment, for example, due to the secretion of PGE2 and adenosine.
–
To further protect itself, the tumour establishes strong interactions with the corrupted stromal cells, by also initiating the production of a physical barrier remodelling the ECM. This is achieved, for example, by the modification of soluble factors (cytokines, growth factors, hormones), type of cells, and structural proteins (collagens, laminins, fibronectins, proteoglycans and hyaluronans), with the latter altering the normal stiffness and adhesion strength of the ECM [59][60][61]. In summary, these biomechanical changes involve not only cancer cells but also their ECM and the entire TME components. The increase of ECM stiffness, for instance, promotes cancer invasion and progression [62]. Moreover, the recruitment of other cells including fibroblasts, myofibroblasts, granulocytes, macrophages, mesenchymal stem cells and lymphocytes in the surrounding stroma, could also be responsible for the hard consistency of tumours at a macroscopic scale. Also, cancer-associated fibroblasts (CAFs) reorganize the stroma by secreting new ECM elements and enzymes that covalently cross-link collagen fibres and pull the collagen network closer together.
THE OPPOSITE FACE OF THE COIN: THE STRESS OF T LYPMPHOCYTES IN THE ATTEMPT TO SURVIVE, REACH AND KILL TUMOUR CELLS
An efficient T-cell response depends on several aspects related to both tumour cells, as described, as well as on factors associated with T-cell activation and functionality. In order to be effective, T lymphocytes require three signals: i) the interaction of the antigenic peptide–MHC complex with the T-cell receptor (TCR), ii) the binding with the co-stimulatory or co-inhibitory ligand, provided by antigen-presenting cells, and iii) the stimulation/proliferation mediated by extracellular cytokines such as interleukin (IL)-2 and IL-15 [63]. Among these signals, the second one determines the promotion or inhibition of T-cell cytokine production and effector function; appropriate co-inhibitory signals dampen inflammation to avoid tissue damage due to an excessive immune reaction, whereas durative and excessive co-inhibitory signals lead to T-cell hypo-responsiveness [64]. Then, in many cases, tumour antigens are weakly immunogenic self-molecules and most tumour-specific T cells have low precursor frequencies and low TCR affinity. This phenomenon occurs because T cells with high avidity against self-molecules, including also tumour self-antigens, are normally deleted during thymic T cell education [65]. In addition, it has been proven that the antigen presentation process is strongly impaired in TME, this leading to insufficient priming and boosting of T lymphocytes [66]. As mentioned above, down-regulation of MHC proteins makes tumour cells “invisible” to infiltrating effector T cells, this resulting into a dysfunction of their anti-tumour activity. This effect is further enhanced by a lack of costimulatory molecules in several solid and haematopoietic tumours [67].
–
The presence of anti-inflammatory soluble factors released by tumour-associated corrupted cells, such as IL-10, transforming-growth-factor-β (TGFβ), cyclooxygenase-2 (COX2), inducible nitric oxide synthase (iNOS) and PGE2, induces the expression of several negative ligands (FasL, PDL1, PDL2, Galactin 9, ect.) on the cells present inside the TME, including T lymphocytes, whose response is therefore inhibited (Table 2). T lymphocytes infiltrating the TME may thus undergo functional exhaustion. This “exhausted” signature is progressively acquired with time, mainly due to a continuous stimulation of the TCR, followed by a progressive increase of expression of co-inhibitory receptors (PD-1, LAG-3, TIM3, CTLA-4, BTLA and TIGIT) [68][69][70][71], and by a decrease of cytokine production and proliferation potential. All these modifications make exhausted T cells unable to differentiate back into functional memory cells, even if antigen stimulation is removed [72]. Interestingly, this “exhausted” state is hard-wired to epigenetic modifications into the T cell genome. Given the frequent chronic nature of a cancer, it is not surprising that exhausted T cells have been found in several tumours [73][74][75]. In this scenario, also TAMs, MDSC [76] and Treg [77] facilitate the generation of exhausted cells in the TME [78]. Also, the fine-tuning control of ionic balance in the TME is an additional check-point for an efficient T-cell functionality. Indeed, potassium ions, released by necrotic cells in the extracellular milieu, can be internalized by infiltrating T cells, thus inhibiting their effector functions by downregulation of the Akt/mTOR signalling, downstream of TCR stimulation [79]. Similarly, tumour-infiltrating human T cells expressing high levels of the calcium channel Kv1.3 can sustain their calcium influx upon TCR stimulation also in environment with low-calcium levels, thus improving their cytotoxicity against tumour cells [80]. The presence of an immune-suppressive environment and the absence of adequate chemotactic factors significantly reduce recruitment of new immune cells from the periphery or from lymphatic organs. To migrate towards a particular tissue, immune cells need the presence of a particular environment produced by cytokines or chemokines, these creating the in situ correct attractive chemical gradient. Every TME produces a specific offset of cytokines and chemokines capable to attract or repel different cell types. Chemokine gene expression profiles and immune cell infiltration have been investigated in different tumour types [81][82][83]. To hinder T cell migration to the tumour site, tumour-derived chemokines may misdirect activated T cells to the tumour surrounding stromal cells [84], and cancer cells can further post-transcriptionally modify their chemokine expression profile. For example, CCL2 nitrosylation can reduce its chemoattractive effect on effector T cells, but not on MDSCs [85].
–
TABLE 2. Factors regulating CD8+ TIL functionality inside the TME. List of molecules that can inhibit, or sustain, tumour-infiltrating-T-lymphocyte (TIL) functionality inside the tumour microenvironment. The corresponding receptors on TIL surface of the indicated check-point molecules are reported in brackets. |
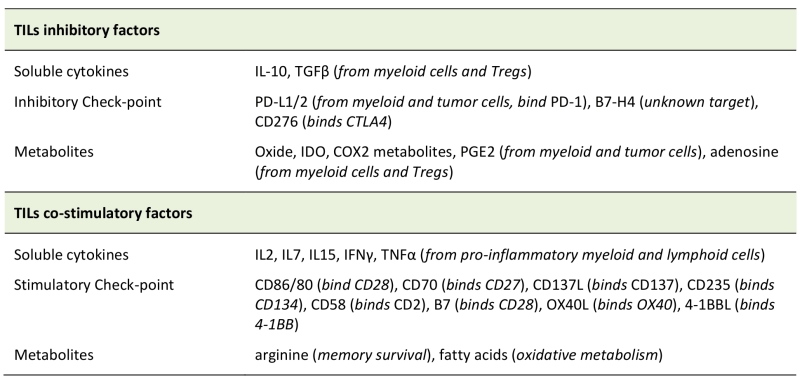 |
Abbreviations: TILs: tumor-infiltrating-T-lymphocytes; IDO: Indoleamine-2,3-dioxygenase; COX2: cyclo-oxigenase2; PGE2: Prostaglandin E2. |
–
Furthermore, the presence of a compact ECM increases the incapacity of T lymphocytes to recognise and kill tumour cells, since T lymphocytes need to actively infiltrate the TME to reach the neoplastic cells. Invasion is an active process in which secretion of particular enzymes is needed to degrade the ECM elements. Several groups, including our own one, reported how this invasion capacity is reduced in patients after high dose chemotherapy and after adoptive T cell transfer, in both cell and gene therapy settings [86]. In the last years, different researches focused on the optimization of chemotherapy regimens in combination with immunotherapies [87][88], such as Treg depletion [89][90], blockage of check-point inhibitors [91][92], anti-angiogenesis treatment [93][94], use of oncolytic virotherapy [95][96] and gene modification of T lymphocytes [86][97], in order to improve the penetration of T lymphocytes or other effector cells. This aspect will be addressed in the following paragraph.
PRE-CLINICAL AND CLINICAL DATA ON REDUCING T-CELL STRESS AND IMPROVING ANTI-TUMOUR ACTIVITY
In the last two to three decades, the development of a new field of research has focused on advanced anti-cancer immunotherapies. Strategies include monoclonal antibodies, adoptive cell transfer, check-point inhibitors, vaccine therapies, oncolytic therapies and gene modification of effector cells, all these approaches showing remarkable long-term efficacy in patients with various types of cancers [98][99][100][101][102][103]. Although conventional therapies, such as radiation and chemotherapy, induce positive responses in the majority of patients, relapse and resistance often occur in patients after prolonged treatment [104]. The strategies developed so far have focused their attention on implementation of tumour targeting. Recently, several pre-clinical and clinical studies have highlighted how most of the pitfalls observed during cancer treatment, in particular in the setting of solid tumours, are not only related to the presence of heterogeneous subpopulations of cancer cells, but also to the development and/or recruitment during tumorigenesis of particular cell types and chemical-physical barriers, which are instrumental to the pathologic manifestation of cancer [105][106].
–
Clinical trials underline how the presence of adequate and functional T cells within tumours correlates with favourable clinical outcome. Several studies in animal models have been carried out in order to fully understand how T-cell infiltration can be enhanced to promote tumour rejection, or to prevent recurrence [107][108][109]. The ever-deeper knowledge of the TME and its constitutive elements, and development of multidisciplinary approaches are laying the foundations for technologies that, one step at a time, are trying to tackle the problem of improving the invasive capacity of T lymphocytes, while improving both their ability to survive and their tumour specificity.
–
Immunotherapies targeted to counteract the mechanisms of tumour-induced T-cell dysfunction have successfully provided persistent clinical benefits in patients with advanced cancer. Most recently, they have focused on immune check-point blockade in order to block the activation of co-inhibitory receptors, or to reduce their levels on the surface of exhausted T cells in cancer patients. Most of these therapies proved to be successful by increasing T-cell functionality inside the TME by restoring their cytokine production and/or cytotoxic activity. Up to now, several check-point inhibitors are currently tested in clinical trials [110] including: combinations of PD1 or of its ligand PDL1, CTLA4 and LAG3 signalling inhibitors [100][101][111][112][113], or agents promoting the immune response with CD40/CD40L, CD137, OX40 and GITR stimulation/engagement [114].
–
Up to now several pre-clinical and clinical studies have shown the feasibility to redirect T lymphocytes on cancer cells through chimeric antigen receptors (CARs) in order to guarantee the specificity of T lymphocytes, overcoming the problem of human MHC protein down-regulation and the lack of costimulatory molecules [115]. A typical CAR consists of a single-chain variable fragment (scFv) linked to an intracellular signalling domain, derived normally from T cells, and more recently also from NK cells [116][117]. A more advanced generation of these molecules is being engineered to recapitulate the costimulatory events that occur upon TCR triggering to fully activate T lymphocytes. Signaling domains derived from T-cell costimulatory receptors are thus directly incorporated in tandem with the TCR co-receptor CD3ζ chain. Intra-cytoplasmic signaling domains of CD28, CD134 (OX40), CD137 (4-1BB), inducible costimulator (ICOS), CD27, DAP10 or CD244 (2B4) in various combinations have been used to construct second and third-generation CARs [118]. After binding to tumour antigen through the scFv, the CAR activates T cells in an antigen-specific and MHC-independent manner, inducing lysis of the engaged target cells through granzyme-B and perforin pathways. Clinical trials with CAR T therapy have shown incredible efficacy in patients with acute lymphoblastic leukaemia, with a complete response rate of nearly 90% un patients who had already failed several lines of conventional therapies, including allogeneic hematopoietic stem cell transplantation [119]. However, attempts to apply CAR T therapy to solid tumours has been less successful [120], and extensive efforts have been devoted to increasing CAR T-cell activity inside solid tumours, as well as their target specificity. For example, engineering CAR T cell to produce cytokines such as IL-7 and CCL19 have proved to increase their infiltration into solid tumour mass, leading to complete regression of pre-established tumours and prolonging survival in mice [121].
–
Several pre-clinical studies brought forward new strategies that can be applied to increase the infiltration of T cells, taking advantage of the negative feedback present in the TME, and producing particular factors capable to boost their persistence, as well as recruiting the innate immune system and inflammatory components. We proved that the T lymphocytes overexpression of heparanase, one of the enzymes involved in the ECM modelling, significantly increased the capacity to degrade the ECM, thus resulting in enhanced tumour infiltration and antitumor activity [86]. Another updated strategy to implement T-cell infiltration is that based on the use of oncolytic viruses, which are able to infect and kill only tumour cells. Unfortunately, although highly promising in vitro [107], it did not show the expected results in clinical trials. Indeed, our group proposed the possibility to further arm the oncolytic therapy with chemokines and cytokines and to combine it with a CAR T-cell approach. The results of this strategy underline how this combinatory therapy is able to improve significantly tumour eradication and T-cell persistence [96]. Based on this data, a growing number of clinical trials proposed to evaluate the clinical efficacy of: check-point inhibitors of soluble mediators (IDO, A2aR, CSF1R, IL-10 or TGFβ), agonistic antibodies targeting and activating receptors on T cells, anti-tumour vaccines [122][123] and adoptive transfer of CAR T cells [124][125].
–
Furthermore, different groups investigate also the possibility to manipulate the TME chemokine profile in order to recruit sufficient numbers of effector cells into the tumour sites. In this regard, interesting data were produced with T,cell chemo-attractants, such as CCL4, CCL5, CCL21, CXCL10, TNFα, IFNβ and TNFSF14 [96][126][127][128][129]. Nevertheless, alternative strategies increasing T cell infiltration into the tumour mass is one of the main challenges that researchers will still have to face in the future. In this way, unconventional and unexpected regulators of these processes, such as mitochondria-dependent myosin fuelling of T-cell migration (see next paragraph for further details) [130][131], could unmask additional therapeutic opportunities to be exploited. In addition, new strategies, to be developed in future, could consider acting on satisfying the metabolic requirements of T cells inside the TME, because of the frequent low nutrient availability and hypoxic conditions. According to this hypothesis, several encouraging results have been recently obtained, such as for example, forcing T cells to use metabolites alternative to glucose, as fatty acids [132] (Fig. 1).
–
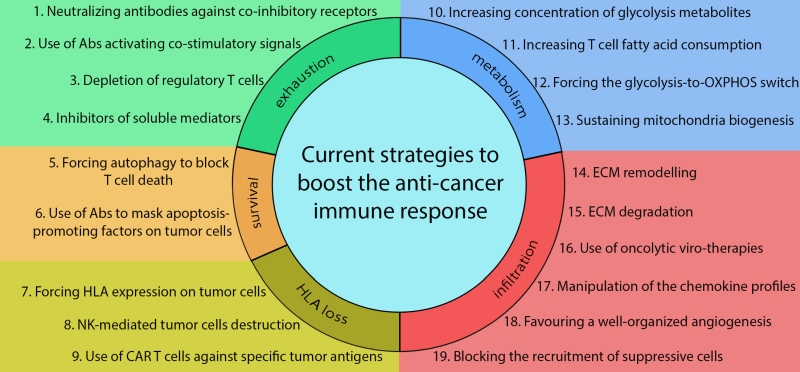 |
FIGURE 1: Current strategies to increase anti-cancer immune responses. The panel summarizes current strategies adopted in clinical trials or in basic research to increase the efficacy of immune-therapies against solid malignancies. Acting on T-cell exhaustion, researchers are currently testing the possibility to use soluble antibodies to neutralize co-inhibitory receptors (1) or to activate co-stimulatory signals on T cells (2). Other approaches involve depletion of immune suppressive Tregs (3) or the inhibition of soluble mediators produced by immune suppressive populations (4). Moreover, strategies could be adopted to prevent T-cell death inside the TME, by forcing autophagy in T cells (5) or by inhibiting the interaction between apoptosis-promoting factors on the tumour cell surface with apoptosis receptors on T cells using soluble neutralizing antibodies (6). In those cases, in which tumor cells evade the immune response due to a lack of antigen presentation, it is possible to force HLA expression on tumor cells (7) or to exploit the ability of NK cells to recognize and kill HLA-negative cells (8). Very recently, engineered CAR T cells have been used for their ability to kill tumour cells expressing specific antigens (10). Moreover, T cell functionality inside the TME may be increased through the modulation of its metabolism by increasing glycolysis metabolite concentration in the TME (10), by forcing T cells to utilize alternative substrates, such as fatty acids (11) or OXPHOS substrates (12), or by directly sustaining their mitochondria biogenesis and oxidative metabolism (13). Finally, several approaches are currently adopted to increase T-cell infiltration into the TME by forcing the expression of specific molecules by T cells, which can thus remodel (14) or completely degrade (15) the extracellular matrix (ECM), also by means of oncolytic-viruses (16). In addition, T-cell infiltration can be increased by modifying the chemokines expressed into the TME (17) or favouring a well-organized angiogenesis (18), which increases the ability of T cells to invade the tumour-surrounding stroma. By contrast, modulation of the chemokine profile could be used to prevent recruitment of suppressive immune populations into the TME (19). See text for details. |
–
In recent years, several studies have highlighted the role of mitochondria in regulating several key processes in T cells. It has long been recognized that, while effector T cells upregulate glycolysis to quickly produce adenosine triphosphate and to generate precursors for biosynthesis of macromolecules [133], memory T cells mainly rely on mitochondria-based oxidative metabolism, sustained at least in part by fatty acid oxidation [134]. Interestingly, the morphology of the mitochondrial network is tightly linked to the cell metabolic status and it can actively control it. Indeed, pharmacological manipulations favouring mitochondria elongation and OXPHOS activity, on in vitro isolated T cells, have been shown to reprogram T cells towards a memory phenotype, thus favouring their long-term survival and increasing their anti-tumour function [135]. In addition, forcing T cells to utilize alternative pathways, instead of glycolysis, may favour their survival in glucose-deprived TME. In this way, increasing mitochondria-based fatty acid utilization could increase T-cell functionality inside the tumour [132]. In addition, a recent paper shows that TILs undergo downregulation of mitochondrial mass inside the TME [136]. Interestingly, the chronic antigen stimulation inside the TME leads to upregulated Akt levels, which, in turns, repress the activity of peroxisome proliferator-activated receptor gamma coactivator 1α, (PGC-1α) the master regulator of mitochondrial biogenesis. Therefore, dysregulation of mitochondrial oxidative metabolism, shut-down by hypoxia, has a strong negative effect on TILs functionality [136]. Besides regulating T-cell metabolism, mitochondria have also been implicated in T-cell migration, proliferation and apoptosis, all key aspects for an optimal T-cell anti-tumour response. We demonstrated that dynamin-related protein 1 (Drp1)-dependent mitochondria remodelling is crucial to sustain T-cell chemotaxis [130] and we have clues that it also controls extravasation towards a solid tumour mass. Moreover, Drp1-mediated fission of mitochondrial network is essential for the redistribution of these organelles to daughter cells during cell division [137] and, in the absence of this process, the clonal expansion of T cells upon activation is strongly impaired (unpublished personal results). Therefore, modulation of mitochondrial dynamics may represent an important tool, in the future, to increase T-cell invasiveness and expansion into solid malignancies, in addition to modulate their energy utilization. This could become highly interesting, particularly for those “immune-excluded” tumours, where immune cells do not efficiently infiltrate into the tumour mass.
–
Mitochondria are also responsible for the release of cytochrome-C (cyt-C) in the cytosol during apoptosis. Although this process has long been investigated in cancer cells [138], we recently demonstrated that the morphology of the mitochondrial network tightly regulates also the physiological T-cell Activation-Induced Cell Death (AICD), a process normally involved in the shut-down of the immune response and exploited by tumours to kill them in the TME [139], by promoting cyt-C release [140]. Indeed, the high rate of effector T-cell apoptosis inside the TME, due to chronic antigen stimulation, is one of the main obstacles for an efficient immune-response against solid malignancies. Again, engineering in vitro manipulated T cells to overcome this mitochondria-based processes could represent a novel approach to increase T-cell survival in the TME, and promising results have been recently obtained in this direction [141][142]. At last, during T-cell stimulation, the autophagic machinery has important roles for an optimal T cell functionality. For example, it is essential to sustain T-cell survival and proliferation and it also controls the generation of long-lived memory T cells (see [143] for a review). Interestingly, we recently demonstrated that autophagy inhibition is necessary and strictly regulated to allow the onset of AICD, while forcing its activation prevents T-cell death [140]. Whether modulation of autophagy could be exploited to overcome the high rate of apoptosis in TME infiltrating T cells is a still an unexplored field, but it may represent another promising tool for future therapeutic purposes.
–
Overall, this large mass of data suggests that additional and completely new targets in T cells have been unmasked in crucial cellular processes and organelles, which could be exploited in the future to boost anti-cancer T cell response.
References
- G.P. Dunn, A.T. Bruce, H. Ikeda, L.J. Old, and R.D. Schreiber, "Cancer immunoediting: from immunosurveillance to tumor escape", Nature Immunology, vol. 3, pp. 991-998, 2002. http://dx.doi.org/10.1038/ni1102-991
- E.J. Wherry, and M. Kurachi, "Molecular and cellular insights into T cell exhaustion", Nature Reviews Immunology, vol. 15, pp. 486-499, 2015. http://dx.doi.org/10.1038/nri3862
- S. Hadrup, M. Donia, and P. thor Straten, "Effector CD4 and CD8 T Cells and Their Role in the Tumor Microenvironment", Cancer Microenvironment, vol. 6, pp. 123-133, 2012. http://dx.doi.org/10.1007/s12307-012-0127-6
- Y. Takeuchi, and H. Nishikawa, "Roles of regulatory T cells in cancer immunity", International Immunology, vol. 28, pp. 401-409, 2016. http://dx.doi.org/10.1093/intimm/dxw025
- G.J. Yuen, E. Demissie, and S. Pillai, "B Lymphocytes and Cancer: A Love–Hate Relationship", Trends in Cancer, vol. 2, pp. 747-757, 2016. http://dx.doi.org/10.1016/j.trecan.2016.10.010
- A. Gardner, and B. Ruffell, "Dendritic Cells and Cancer Immunity", Trends in Immunology, vol. 37, pp. 855-865, 2016. http://dx.doi.org/10.1016/j.it.2016.09.006
- B. Vallentin, V. Barlogis, C. Piperoglou, S. Cypowyj, N. Zucchini, M. Chéné, F. Navarro, C. Farnarier, E. Vivier, and F. Vély, "Innate Lymphoid Cells in Cancer", Cancer Immunology Research, vol. 3, pp. 1109-1114, 2015. http://dx.doi.org/10.1158/2326-6066.CIR-15-0222
- M.G. Morvan, and L.L. Lanier, "NK cells and cancer: you can teach innate cells new tricks", Nature Reviews Cancer, vol. 16, pp. 7-19, 2015. http://dx.doi.org/10.1038/nrc.2015.5
- M. Lawand, J. Déchanet-Merville, and M. Dieu-Nosjean, "Key Features of Gamma-Delta T-Cell Subsets in Human Diseases and Their Immunotherapeutic Implications", Frontiers in Immunology, vol. 8, 2017. http://dx.doi.org/10.3389/fimmu.2017.00761
- A. Capsomidis, G. Benthall, H.H. Van Acker, J. Fisher, A.M. Kramer, Z. Abeln, Y. Majani, T. Gileadi, R. Wallace, K. Gustafsson, B. Flutter, and J. Anderson, "Chimeric Antigen Receptor-Engineered Human Gamma Delta T Cells: Enhanced Cytotoxicity with Retention of Cross Presentation", Molecular Therapy, vol. 26, pp. 354-365, 2018. http://dx.doi.org/10.1016/j.ymthe.2017.12.001
- V. Umansky, C. Blattner, C. Gebhardt, and J. Utikal, "The Role of Myeloid-Derived Suppressor Cells (MDSC) in Cancer Progression", Vaccines, vol. 4, pp. 36, 2016. http://dx.doi.org/10.3390/vaccines4040036
- R. Noy, and J. Pollard, "Tumor-Associated Macrophages: From Mechanisms to Therapy", Immunity, vol. 41, pp. 49-61, 2014. http://dx.doi.org/10.1016/j.immuni.2014.06.010
- D.S. Chen, and I. Mellman, "Elements of cancer immunity and the cancer–immune set point", Nature, vol. 541, pp. 321-330, 2017. http://dx.doi.org/10.1038/nature21349
- F. Balkwill, and A. Mantovani, "Inflammation and cancer: back to Virchow?", The Lancet, vol. 357, pp. 539-545, 2001. http://dx.doi.org/10.1016/S0140-6736(00)04046-0
- M. Egeblad, E.S. Nakasone, and Z. Werb, "Tumors as Organs: Complex Tissues that Interface with the Entire Organism", Developmental Cell, vol. 18, pp. 884-901, 2010. http://dx.doi.org/10.1016/j.devcel.2010.05.012
- D.E. Discher, P. Janmey, and Y. Wang, "Tissue Cells Feel and Respond to the Stiffness of Their Substrate", Science, vol. 310, pp. 1139-1143, 2005. http://dx.doi.org/10.1126/science.1116995
- C. Bonnans, J. Chou, and Z. Werb, "Remodelling the extracellular matrix in development and disease", Nature Reviews Molecular Cell Biology, vol. 15, pp. 786-801, 2014. http://dx.doi.org/10.1038/nrm3904
- D.A. Stewart, C.R. Cooper, and R.A. Sikes, "Array", Reproductive Biology and Endocrinology, vol. 2, pp. 2, 2004. http://dx.doi.org/10.1186/1477-7827-2-2
- P. Lu, V.M. Weaver, and Z. Werb, "The extracellular matrix: A dynamic niche in cancer progression", Journal of Cell Biology, vol. 196, pp. 395-406, 2012. http://dx.doi.org/10.1083/jcb.201102147
- D. Hanahan, and R.A. Weinberg, "The Hallmarks of Cancer", Cell, vol. 100, pp. 57-70, 2000. http://dx.doi.org/10.1016/s0092-8674(00)81683-9
- E. Reeves, and E. James, "Antigen processing and immune regulation in the response to tumours", Immunology, vol. 150, pp. 16-24, 2016. http://dx.doi.org/10.1111/imm.12675
- A. Bhatia, and Y. Kumar, "Cellular and molecular mechanisms in cancer immune escape: a comprehensive review", Expert Review of Clinical Immunology, vol. 10, pp. 41-62, 2013. http://dx.doi.org/10.1586/1744666X.2014.865519
- D. Hanahan, and R. Weinberg, "Hallmarks of Cancer: The Next Generation", Cell, vol. 144, pp. 646-674, 2011. http://dx.doi.org/10.1016/j.cell.2011.02.013
- G.C. Prendergast, C. Smith, S. Thomas, L. Mandik-Nayak, L. Laury-Kleintop, R. Metz, and A.J. Muller, "Indoleamine 2,3-dioxygenase pathways of pathogenic inflammation and immune escape in cancer", Cancer Immunology, Immunotherapy, vol. 63, pp. 721-735, 2014. http://dx.doi.org/10.1007/s00262-014-1549-4
- A. Banh, J. Zhang, H. Cao, D.M. Bouley, S. Kwok, C. Kong, A.J. Giaccia, A.C. Koong, and Q. Le, "Tumor Galectin-1 Mediates Tumor Growth and Metastasis through Regulation of T-Cell Apoptosis", Cancer Research, vol. 71, pp. 4423-4431, 2011. http://dx.doi.org/10.1158/0008-5472.CAN-10-4157
- M. Grimm, M. Kim, A. Rosenwald, B. von Rahden, I. Tsaur, E. Meier, U. Heemann, C. Germer, M. Gasser, and A.M. Waaga-Gasser, "Tumour-mediated TRAIL-Receptor expression indicates effective apoptotic depletion of infiltrating CD8+ immune cells in clinical colorectal cancer", European Journal of Cancer, vol. 46, pp. 2314-2323, 2010. http://dx.doi.org/10.1016/j.ejca.2010.05.025
- J. Zhu, C.G. Powis de Tenbossche, S. Cané, D. Colau, N. van Baren, C. Lurquin, A. Schmitt-Verhulst, P. Liljeström, C. Uyttenhove, and B.J. Van den Eynde, "Resistance to cancer immunotherapy mediated by apoptosis of tumor-infiltrating lymphocytes", Nature Communications, vol. 8, 2017. http://dx.doi.org/10.1038/s41467-017-00784-1
- A. Viale, P. Pettazzoni, C.A. Lyssiotis, H. Ying, N. Sánchez, M. Marchesini, A. Carugo, T. Green, S. Seth, V. Giuliani, M. Kost-Alimova, F. Muller, S. Colla, L. Nezi, G. Genovese, A.K. Deem, A. Kapoor, W. Yao, E. Brunetto, Y. Kang, M. Yuan, J.M. Asara, Y.A. Wang, T.P. Heffernan, A.C. Kimmelman, H. Wang, J.B. Fleming, L.C. Cantley, R.A. DePinho, and G.F. Draetta, "Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function", Nature, vol. 514, pp. 628-632, 2014. http://dx.doi.org/10.1038/nature13611
- O. Warburg, "On the Origin of Cancer Cells", Science, vol. 123, pp. 309-314, 1956. http://dx.doi.org/10.1126/science.123.3191.309
- R. Wang, C. Dillon, L. Shi, S. Milasta, R. Carter, D. Finkelstein, L. McCormick, P. Fitzgerald, H. Chi, J. Munger, and D. Green, "The Transcription Factor Myc Controls Metabolic Reprogramming upon T Lymphocyte Activation", Immunity, vol. 35, pp. 871-882, 2011. http://dx.doi.org/10.1016/j.immuni.2011.09.021
- C. Chang, J. Qiu, D. O’Sullivan, M. Buck, T. Noguchi, J. Curtis, Q. Chen, M. Gindin, M. Gubin, G. van der Windt, E. Tonc, R. Schreiber, E. Pearce, and E. Pearce, "Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression", Cell, vol. 162, pp. 1229-1241, 2015. http://dx.doi.org/10.1016/j.cell.2015.08.016
- K. Fischer, P. Hoffmann, S. Voelkl, N. Meidenbauer, J. Ammer, M. Edinger, E. Gottfried, S. Schwarz, G. Rothe, S. Hoves, K. Renner, B. Timischl, A. Mackensen, L. Kunz-Schughart, R. Andreesen, S.W. Krause, and M. Kreutz, "Inhibitory effect of tumor cell–derived lactic acid on human T cells", Blood, vol. 109, pp. 3812-3819, 2007. http://dx.doi.org/10.1182/blood-2006-07-035972
- C. Chang, J. Curtis, L. Maggi, B. Faubert, A. Villarino, D. O’Sullivan, S. Huang, G. van der Windt, J. Blagih, J. Qiu, J. Weber, E. Pearce, R. Jones, and E. Pearce, "Posttranscriptional Control of T Cell Effector Function by Aerobic Glycolysis", Cell, vol. 153, pp. 1239-1251, 2013. http://dx.doi.org/10.1016/j.cell.2013.05.016
- H.O. Alsaab, S. Sau, R. Alzhrani, K. Tatiparti, K. Bhise, S.K. Kashaw, and A.K. Iyer, "PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome", Frontiers in Pharmacology, vol. 8, 2017. http://dx.doi.org/10.3389/fphar.2017.00561
- E.N. Rozali, S.V. Hato, B.W. Robinson, R.A. Lake, and W.J. Lesterhuis, "Programmed Death Ligand 2 in Cancer-Induced Immune Suppression", Clinical and Developmental Immunology, vol. 2012, pp. 1-8, 2012. http://dx.doi.org/10.1155/2012/656340
- R.V. Parry, J.M. Chemnitz, K.A. Frauwirth, A.R. Lanfranco, I. Braunstein, S.V. Kobayashi, P.S. Linsley, C.B. Thompson, and J.L. Riley, "CTLA-4 and PD-1 Receptors Inhibit T-Cell Activation by Distinct Mechanisms", Molecular and Cellular Biology, vol. 25, pp. 9543-9553, 2005. http://dx.doi.org/10.1128/MCB.25.21.9543-9553.2005
- R.D. Michalek, V.A. Gerriets, S.R. Jacobs, A.N. Macintyre, N.J. MacIver, E.F. Mason, S.A. Sullivan, A.G. Nichols, and J.C. Rathmell, "Cutting Edge: Distinct Glycolytic and Lipid Oxidative Metabolic Programs Are Essential for Effector and Regulatory CD4+ T Cell Subsets", The Journal of Immunology, vol. 186, pp. 3299-3303, 2011. http://dx.doi.org/10.4049/jimmunol.1003613
- C. Procaccini, F. Carbone, D. Di Silvestre, F. Brambilla, V. De Rosa, M. Galgani, D. Faicchia, G. Marone, D. Tramontano, M. Corona, C. Alviggi, A. Porcellini, A. La Cava, P. Mauri, and G. Matarese, "The Proteomic Landscape of Human Ex Vivo Regulatory and Conventional T Cells Reveals Specific Metabolic Requirements", Immunity, vol. 44, pp. 712, 2016. http://dx.doi.org/10.1016/j.immuni.2016.02.022
- A. Angelin, L. Gil-de-Gómez, S. Dahiya, J. Jiao, L. Guo, M.H. Levine, Z. Wang, W.J. Quinn, P.K. Kopinski, L. Wang, T. Akimova, Y. Liu, T.R. Bhatti, R. Han, B.L. Laskin, J.A. Baur, I.A. Blair, D.C. Wallace, W.W. Hancock, and U.H. Beier, "Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments", Cell Metabolism, vol. 25, pp. 1282-1293.e7, 2017. http://dx.doi.org/10.1016/j.cmet.2016.12.018
- G.T. Motz, S.P. Santoro, L. Wang, T. Garrabrant, R.R. Lastra, I.S. Hagemann, P. Lal, M.D. Feldman, F. Benencia, and G. Coukos, "Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors", Nature Medicine, vol. 20, pp. 607-615, 2014. http://dx.doi.org/10.1038/nm.3541
- K.L. Singel, and B.H. Segal, "Neutrophils in the tumor microenvironment: trying to heal the wound that cannot heal", Immunological Reviews, vol. 273, pp. 329-343, 2016. http://dx.doi.org/10.1111/imr.12459
- R. Carretero, I.M. Sektioglu, N. Garbi, O.C. Salgado, P. Beckhove, and G.J. Hämmerling, "Eosinophils orchestrate cancer rejection by normalizing tumor vessels and enhancing infiltration of CD8+ T cells", Nature Immunology, vol. 16, pp. 609-617, 2015. http://dx.doi.org/10.1038/ni.3159
- I.M. Sektioglu, R. Carretero, N. Bulbuc, T. Bald, T. Tüting, A.Y. Rudensky, and G.J. Hämmerling, "Basophils Promote Tumor Rejection via Chemotaxis and Infiltration of CD8+ T Cells", Cancer Research, vol. 77, pp. 291-302, 2017. http://dx.doi.org/10.1158/0008-5472.CAN-16-0993
- S. Ugel, F. De Sanctis, S. Mandruzzato, and V. Bronte, "Tumor-induced myeloid deviation: when myeloid-derived suppressor cells meet tumor-associated macrophages", Journal of Clinical Investigation, vol. 125, pp. 3365-3376, 2015. http://dx.doi.org/10.1172/JCI80006
- R. Geiger, J.C. Rieckmann, T. Wolf, C. Basso, Y. Feng, T. Fuhrer, M. Kogadeeva, P. Picotti, F. Meissner, M. Mann, N. Zamboni, F. Sallusto, and A. Lanzavecchia, "L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity", Cell, vol. 167, pp. 829-842.e13, 2016. http://dx.doi.org/10.1016/j.cell.2016.09.031
- S. Spranger, R.M. Spaapen, Y. Zha, J. Williams, Y. Meng, T.T. Ha, and T.F. Gajewski, "Up-Regulation of PD-L1, IDO, and T regs in the Melanoma Tumor Microenvironment Is Driven by CD8 + T Cells", Science Translational Medicine, vol. 5, 2013. http://dx.doi.org/10.1126/scitranslmed.3006504
- K. Sakuishi, P. Jayaraman, S.M. Behar, A.C. Anderson, and V.K. Kuchroo, "Emerging Tim-3 functions in antimicrobial and tumor immunity", Trends in Immunology, vol. 32, pp. 345-349, 2011. http://dx.doi.org/10.1016/j.it.2011.05.003
- K. De Bock, S. Cauwenberghs, and P. Carmeliet, "Vessel abnormalization: another hallmark of cancer?Molecular mechanisms and therapeutic implications", Current Opinion in Genetics & Development, vol. 21, pp. 73-79, 2011. http://dx.doi.org/10.1016/j.gde.2010.10.008
- D. Hanahan, and J. Folkman, "Patterns and Emerging Mechanisms of the Angiogenic Switch during Tumorigenesis", Cell, vol. 86, pp. 353-364, 1996. http://dx.doi.org/10.1016/s0092-8674(00)80108-7
- N. Koshikawa, A. Iyozumi, M. Gassmann, and K. Takenaga, "Constitutive upregulation of hypoxia-inducible factor-1α mRNA occurring in highly metastatic lung carcinoma cells leads to vascular endothelial growth factor overexpression upon hypoxic exposure", Oncogene, vol. 22, pp. 6717-6724, 2003. http://dx.doi.org/10.1038/sj.onc.1206765
- F. Guillaumond, J. Leca, O. Olivares, M. Lavaut, N. Vidal, P. Berthezène, N.J. Dusetti, C. Loncle, E. Calvo, O. Turrini, J.L. Iovanna, R. Tomasini, and S. Vasseur, "Strengthened glycolysis under hypoxia supports tumor symbiosis and hexosamine biosynthesis in pancreatic adenocarcinoma", Proceedings of the National Academy of Sciences, vol. 110, pp. 3919-3924, 2013. http://dx.doi.org/10.1073/pnas.1219555110
- J.D. Shields, M. Borsetti, H. Rigby, S.J. Harper, P.S. Mortimer, J.R. Levick, A. Orlando, and D.O. Bates, "Lymphatic density and metastatic spread in human malignant melanoma", British Journal of Cancer, vol. 90, pp. 693-700, 2004. http://dx.doi.org/10.1038/sj.bjc.6601571
- R.H. Farnsworth, T. Karnezis, R. Shayan, M. Matsumoto, C.J. Nowell, M.G. Achen, and S.A. Stacker, "A Role for Bone Morphogenetic Protein-4 in Lymph Node Vascular Remodeling and Primary Tumor Growth", Cancer Research, vol. 71, pp. 6547-6557, 2011. http://dx.doi.org/10.1158/0008-5472.CAN-11-0200
- C.Y. Slaney, M.H. Kershaw, and P.K. Darcy, "Trafficking of T Cells into Tumors", Cancer Research, vol. 74, pp. 7168-7174, 2014. http://dx.doi.org/10.1158/0008-5472.CAN-14-2458
- I.S. Mauldin, N.A. Wages, A.M. Stowman, E. Wang, M.E. Smolkin, W.C. Olson, D.H. Deacon, K.T. Smith, N.V. Galeassi, K.A. Chianese‐Bullock, L.T. Dengel, F.M. Marincola, G.R. Petroni, D.W. Mullins, and C.L. Slingluff, "Intratumoral interferon-gamma increases chemokine production but fails to increase T cell infiltration of human melanoma metastases", Cancer Immunology, Immunotherapy, vol. 65, pp. 1189-1199, 2016. http://dx.doi.org/10.1007/s00262-016-1881-y
- A.K. Neumann, J. Yang, M.P. Biju, S.K. Joseph, R.S. Johnson, V.H. Haase, B.D. Freedman, and L.A. Turka, "Hypoxia inducible factor 1α regulates T cell receptor signal transduction", Proceedings of the National Academy of Sciences, vol. 102, pp. 17071-17076, 2005. http://dx.doi.org/10.1073/pnas.0506070102
- M. Kondoh, N. Ohga, K. Akiyama, Y. Hida, N. Maishi, A.M. Towfik, N. Inoue, M. Shindoh, and K. Hida, "Hypoxia-Induced Reactive Oxygen Species Cause Chromosomal Abnormalities in Endothelial Cells in the Tumor Microenvironment", PLoS ONE, vol. 8, pp. e80349, 2013. http://dx.doi.org/10.1371/journal.pone.0080349
- I.B. Barsoum, C.A. Smallwood, D.R. Siemens, and C.H. Graham, "A Mechanism of Hypoxia-Mediated Escape from Adaptive Immunity in Cancer Cells", Cancer Research, vol. 74, pp. 665-674, 2014. http://dx.doi.org/10.1158/0008-5472.CAN-13-0992
- H.E. Barker, J. Chang, T.R. Cox, G. Lang, D. Bird, M. Nicolau, H.R. Evans, A. Gartland, and J.T. Erler, "LOXL2-Mediated Matrix Remodeling in Metastasis and Mammary Gland Involution", Cancer Research, vol. 71, pp. 1561-1572, 2011. http://dx.doi.org/10.1158/0008-5472.CAN-10-2868
- J. Zhu, L. Liang, Y. Jiao, L. Liu, and . , "Enhanced Invasion of Metastatic Cancer Cells via Extracellular Matrix Interface", PLOS ONE, vol. 10, pp. e0118058, 2015. http://dx.doi.org/10.1371/journal.pone.0118058
- F. Del Bufalo, T. Manzo, V. Hoyos, S. Yagyu, I. Caruana, J. Jacot, O. Benavides, D. Rosen, and M.K. Brenner, "3D modeling of human cancer: A PEG-fibrin hydrogel system to study the role of tumor microenvironment and recapitulate the in vivo effect of oncolytic adenovirus", Biomaterials, vol. 84, pp. 76-85, 2016. http://dx.doi.org/10.1016/j.biomaterials.2016.01.030
- K.R. Levental, H. Yu, L. Kass, J.N. Lakins, M. Egeblad, J.T. Erler, S.F. Fong, K. Csiszar, A. Giaccia, W. Weninger, M. Yamauchi, D.L. Gasser, and V.M. Weaver, "Matrix Crosslinking Forces Tumor Progression by Enhancing Integrin Signaling", Cell, vol. 139, pp. 891-906, 2009. http://dx.doi.org/10.1016/j.cell.2009.10.027
- N. Murakami, and L.V. Riella, "Co-Inhibitory Pathways and Their Importance in Immune Regulation", Transplantation, vol. 98, pp. 3-14, 2014. http://dx.doi.org/10.1097/TP.0000000000000169
- T. Maj, S. Wei, T. Welling, and W. Zou, "T Cells and Costimulation in Cancer", The Cancer Journal, vol. 19, pp. 473-482, 2013. http://dx.doi.org/10.1097/PPO.0000000000000002
- P.S. Kim, and R. Ahmed, "Features of responding T cells in cancer and chronic infection", Current Opinion in Immunology, vol. 22, pp. 223-230, 2010. http://dx.doi.org/10.1016/j.coi.2010.02.005
- L. Baitsch, S.A. Fuertes-Marraco, A. Legat, C. Meyer, and D.E. Speiser, "The three main stumbling blocks for anticancer T cells", Trends in Immunology, vol. 33, pp. 364-372, 2012. http://dx.doi.org/10.1016/j.it.2012.02.006
- G. Driessens, J. Kline, and T.F. Gajewski, "Costimulatory and coinhibitory receptors in anti‐tumor immunity", Immunological Reviews, vol. 229, pp. 126-144, 2009. http://dx.doi.org/10.1111/j.1600-065X.2009.00771.x
- J. Fourcade, Z. Sun, M. Benallaoua, P. Guillaume, I.F. Luescher, C. Sander, J.M. Kirkwood, V. Kuchroo, and H.M. Zarour, "Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen–specific CD8+ T cell dysfunction in melanoma patients", Journal of Experimental Medicine, vol. 207, pp. 2175-2186, 2010. http://dx.doi.org/10.1084/jem.20100637
- J. Fourcade, Z. Sun, O. Pagliano, P. Guillaume, I.F. Luescher, C. Sander, J.M. Kirkwood, D. Olive, V. Kuchroo, and H.M. Zarour, "CD8+ T Cells Specific for Tumor Antigens Can Be Rendered Dysfunctional by the Tumor Microenvironment through Upregulation of the Inhibitory Receptors BTLA and PD-1", Cancer Research, vol. 72, pp. 887-896, 2012. http://dx.doi.org/10.1158/0008-5472.CAN-11-2637
- J. Chauvin, O. Pagliano, J. Fourcade, Z. Sun, H. Wang, C. Sander, J.M. Kirkwood, T.T. Chen, M. Maurer, A.J. Korman, and H.M. Zarour, "TIGIT and PD-1 impair tumor antigen–specific CD8+ T cells in melanoma patients", Journal of Clinical Investigation, vol. 125, pp. 2046-2058, 2015. http://dx.doi.org/10.1172/JCI80445
- E.J. Wherry, S. Ha, S.M. Kaech, W.N. Haining, S. Sarkar, V. Kalia, S. Subramaniam, J.N. Blattman, D.L. Barber, and R. Ahmed, "Molecular Signature of CD8+ T Cell Exhaustion during Chronic Viral Infection", Immunity, vol. 27, pp. 670-684, 2007. http://dx.doi.org/10.1016/j.immuni.2007.09.006
- J.M. Angelosanto, S.D. Blackburn, A. Crawford, and E.J. Wherry, "Progressive Loss of Memory T Cell Potential and Commitment to Exhaustion during Chronic Viral Infection", Journal of Virology, vol. 86, pp. 8161-8170, 2012. http://dx.doi.org/10.1128/JVI.00889-12
- S. Zhu, J. Lin, G. Qiao, X. Wang, and Y. Xu, "Tim-3 identifies exhausted follicular helper T cells in breast cancer patients", Immunobiology, vol. 221, pp. 986-993, 2016. http://dx.doi.org/10.1016/j.imbio.2016.04.005
-
M. Giordano, C. Henin, J. Maurizio, C. Imbratta, P. Bourdely, M. Buferne, L. Baitsch, L. Vanhille, M.H. Sieweke, D.E. Speiser, N. Auphan‐Anezin, A. Schmitt‐Verhulst, and G. Verdeil, "Molecular profiling of
CD 8 T cells in autochthonous melanoma identifies Maf as driver of exhaustion", The EMBO Journal, vol. 34, pp. 2042-2058, 2015. http://dx.doi.org/10.15252/embj.201490786 - A.S. Japp, M.A. Kursunel, S. Meier, J.N. Mälzer, X. Li, N.A. Rahman, W. Jekabsons, H. Krause, A. Magheli, C. Klopf, A. Thiel, and M. Frentsch, "Dysfunction of PSA-specific CD8+ T cells in prostate cancer patients correlates with CD38 and Tim-3 expression", Cancer Immunology, Immunotherapy, vol. 64, pp. 1487-1494, 2015. http://dx.doi.org/10.1007/s00262-015-1752-y
- C. Goh, S. Narayanan, and Y.S. Hahn, "Myeloid‐derived suppressor cells: the dark knight or the joker in viral infections?", Immunological Reviews, vol. 255, pp. 210-221, 2013. http://dx.doi.org/10.1111/imr.12084
- T.A.W. Holderried, P.A. Lang, H. Kim, and H. Cantor, "Genetic disruption of CD8 + Treg activity enhances the immune response to viral infection", Proceedings of the National Academy of Sciences, vol. 110, pp. 21089-21094, 2013. http://dx.doi.org/10.1073/pnas.1320999110
- S. Lee, and K. Margolin, "Cytokines in Cancer Immunotherapy", Cancers, vol. 3, pp. 3856-3893, 2011. http://dx.doi.org/10.3390/cancers3043856
- R. Eil, S.K. Vodnala, D. Clever, C.A. Klebanoff, M. Sukumar, J.H. Pan, D.C. Palmer, A. Gros, T.N. Yamamoto, S.J. Patel, G.C. Guittard, Z. Yu, V. Carbonaro, K. Okkenhaug, D.S. Schrump, W.M. Linehan, R. Roychoudhuri, and N.P. Restifo, "Ionic immune suppression within the tumour microenvironment limits T cell effector function", Nature, vol. 537, pp. 539-543, 2016. http://dx.doi.org/10.1038/nature19364
- A.A. Chimote, P. Hajdu, A.M. Sfyris, B.N. Gleich, T. Wise-Draper, K.A. Casper, and L. Conforti, "Kv1.3 Channels Mark Functionally Competent CD8+ Tumor-Infiltrating Lymphocytes in Head and Neck Cancer", Cancer Research, vol. 77, pp. 53-61, 2017. http://dx.doi.org/10.1158/0008-5472.CAN-16-2372
- A. Zlotnik, "Chemokines in neoplastic progression", Seminars in Cancer Biology, vol. 14, pp. 181-185, 2004. http://dx.doi.org/10.1016/j.semcancer.2003.10.004
- A. Viola, A. Sarukhan, V. Bronte, and B. Molon, "The pros and cons of chemokines in tumor immunology", Trends in Immunology, vol. 33, pp. 496-504, 2012. http://dx.doi.org/10.1016/j.it.2012.05.007
- A.V. Gorbachev, and R.L. Fairchild, "Regulation of Chemokine Expression in the Tumor Microenvironment", Critical Reviews in Immunology, vol. 34, pp. 103-120, 2014. http://dx.doi.org/10.1615/critrevimmunol.2014010062
- C. Feig, J.O. Jones, M. Kraman, R.J.B. Wells, A. Deonarine, D.S. Chan, C.M. Connell, E.W. Roberts, Q. Zhao, O.L. Caballero, S.A. Teichmann, T. Janowitz, D.I. Jodrell, D.A. Tuveson, and D.T. Fearon, "Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti–PD-L1 immunotherapy in pancreatic cancer", Proceedings of the National Academy of Sciences, vol. 110, pp. 20212-20217, 2013. http://dx.doi.org/10.1073/pnas.1320318110
- B. Molon, S. Ugel, F. Del Pozzo, C. Soldani, S. Zilio, D. Avella, A. De Palma, P. Mauri, A. Monegal, M. Rescigno, B. Savino, P. Colombo, N. Jonjic, S. Pecanic, L. Lazzarato, R. Fruttero, A. Gasco, V. Bronte, and A. Viola, "Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells", Journal of Experimental Medicine, vol. 208, pp. 1949-1962, 2011. http://dx.doi.org/10.1084/jem.20101956
- I. Caruana, B. Savoldo, V. Hoyos, G. Weber, H. Liu, E.S. Kim, M.M. Ittmann, D. Marchetti, and G. Dotti, "Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes", Nature Medicine, vol. 21, pp. 524-529, 2015. http://dx.doi.org/10.1038/nm.3833
- D. Alizadeh, M. Trad, N.T. Hanke, C.B. Larmonier, N. Janikashvili, B. Bonnotte, E. Katsanis, and N. Larmonier, "Doxorubicin Eliminates Myeloid-Derived Suppressor Cells and Enhances the Efficacy of Adoptive T-Cell Transfer in Breast Cancer", Cancer Research, vol. 74, pp. 104-118, 2014. http://dx.doi.org/10.1158/0008-5472.CAN-13-1545
- J.P. Machiels, R.T. Reilly, L.A. Emens, A.M. Ercolini, R.Y. Lei, D. Weintraub, F.I. Okoye, and E.M. Jaffee, "Cyclophosphamide, doxorubicin, and paclitaxel enhance the antitumor immune response of granulocyte/macrophage-colony stimulating factor-secreting whole-cell vaccines in HER-2/neu tolerized mice.", Cancer research, 2001. http://www.ncbi.nlm.nih.gov/pubmed/11325840
- J.L. Soriano, N. Batista, E. Santiesteban, M. Lima, J. González, R. García, Y. Zarza, M.V. López, M. Rodríguez, J.L. Loys, N. Montejo, F. Aguirre, A. Macías, and A.M. Vázquez, "Metronomic Cyclophosphamide and Methotrexate Chemotherapy Combined with 1E10 Anti-Idiotype Vaccine in Metastatic Breast Cancer", International Journal of Breast Cancer, vol. 2011, pp. 1-6, 2011. http://dx.doi.org/10.4061/2011/710292
- Z. Wang, S.G. Pratts, H. Zhang, P.J. Spencer, R. Yu, M. Tonsho, J.A. Shah, T. Tanabe, H.R. Powell, C.A. Huang, J.C. Madsen, D.H. Sachs, and Z. Wang, "Treg depletion in non‐human primates using a novel diphtheria toxin‐based anti‐human CCR4 immunotoxin", Molecular Oncology, vol. 10, pp. 553-565, 2015. http://dx.doi.org/10.1016/j.molonc.2015.11.008
- F.S. Hodi, S.J. O'Day, D.F. McDermott, R.W. Weber, J.A. Sosman, J.B. Haanen, R. Gonzalez, C. Robert, D. Schadendorf, J.C. Hassel, W. Akerley, A.J. van den Eertwegh, J. Lutzky, P. Lorigan, J.M. Vaubel, G.P. Linette, D. Hogg, C.H. Ottensmeier, C. Lebbé, C. Peschel, I. Quirt, J.I. Clark, J.D. Wolchok, J.S. Weber, J. Tian, M.J. Yellin, G.M. Nichol, A. Hoos, and W.J. Urba, "Improved Survival with Ipilimumab in Patients with Metastatic Melanoma", New England Journal of Medicine, vol. 363, pp. 711-723, 2010. http://dx.doi.org/10.1056/NEJMoa1003466
- L. Wu, Z. Yun, T. Tagawa, K. Rey-McIntyre, and M. de Perrot, "CTLA-4 Blockade Expands Infiltrating T Cells and Inhibits Cancer Cell Repopulation during the Intervals of Chemotherapy in Murine Mesothelioma", Molecular Cancer Therapeutics, vol. 11, pp. 1809-1819, 2012. http://dx.doi.org/10.1158/1535-7163.MCT-11-1014
- A.W. Griffioen, "Anti-angiogenesis: making the tumor vulnerable to the immune system", Cancer Immunology, Immunotherapy, vol. 57, pp. 1553-1558, 2008. http://dx.doi.org/10.1007/s00262-008-0524-3
- P. Bocca, E. Di Carlo, I. Caruana, L. Emionite, M. Cilli, B. De Angelis, C. Quintarelli, A. Pezzolo, L. Raffaghello, F. Morandi, F. Locatelli, V. Pistoia, and I. Prigione, "Bevacizumab-mediated tumor vasculature remodelling improves tumor infiltration and antitumor efficacy of GD2-CAR T cells in a human neuroblastoma preclinical model", OncoImmunology, vol. 7, pp. e1378843, 2017. http://dx.doi.org/10.1080/2162402X.2017.1378843
- V. Hoyos, F. Del Bufalo, S. Yagyu, M. Ando, G. Dotti, M. Suzuki, L. Bouchier-Hayes, R. Alemany, and M.K. Brenner, "Mesenchymal Stromal Cells for Linked Delivery of Oncolytic and Apoptotic Adenoviruses to Non-small-cell Lung Cancers", Molecular Therapy, vol. 23, pp. 1497-1506, 2015. http://dx.doi.org/10.1038/mt.2015.110
- N. Nishio, I. Diaconu, H. Liu, V. Cerullo, I. Caruana, V. Hoyos, L. Bouchier-Hayes, B. Savoldo, and G. Dotti, "Armed Oncolytic Virus Enhances Immune Functions of Chimeric Antigen Receptor–Modified T Cells in Solid Tumors", Cancer Research, vol. 74, pp. 5195-5205, 2014. http://dx.doi.org/10.1158/0008-5472.CAN-14-0697
- I. Caruana, G. Weber, B.C. Ballard, M.S. Wood, B. Savoldo, and G. Dotti, "K562-Derived Whole-Cell Vaccine Enhances Antitumor Responses of CAR-Redirected Virus-Specific Cytotoxic T Lymphocytes In Vivo", Clinical Cancer Research, vol. 21, pp. 2952-2962, 2015. http://dx.doi.org/10.1158/1078-0432.CCR-14-2998
- J. Couzin-Frankel, "Cancer Immunotherapy", Science, vol. 342, pp. 1432-1433, 2013. http://dx.doi.org/10.1126/science.342.6165.1432
- D.L. Porter, B.L. Levine, M. Kalos, A. Bagg, and C.H. June, "Chimeric Antigen Receptor–Modified T Cells in Chronic Lymphoid Leukemia", New England Journal of Medicine, vol. 365, pp. 725-733, 2011. http://dx.doi.org/10.1056/NEJMoa1103849
- S.L. Topalian, F.S. Hodi, J.R. Brahmer, S.N. Gettinger, D.C. Smith, D.F. McDermott, J.D. Powderly, R.D. Carvajal, J.A. Sosman, M.B. Atkins, P.D. Leming, D.R. Spigel, S.J. Antonia, L. Horn, C.G. Drake, D.M. Pardoll, L. Chen, W.H. Sharfman, R.A. Anders, J.M. Taube, T.L. McMiller, H. Xu, A.J. Korman, M. Jure-Kunkel, S. Agrawal, D. McDonald, G.D. Kollia, A. Gupta, J.M. Wigginton, and M. Sznol, "Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer", New England Journal of Medicine, vol. 366, pp. 2443-2454, 2012. http://dx.doi.org/10.1056/NEJMoa1200690
- J.R. Brahmer, S.S. Tykodi, L.Q. Chow, W. Hwu, S.L. Topalian, P. Hwu, C.G. Drake, L.H. Camacho, J. Kauh, K. Odunsi, H.C. Pitot, O. Hamid, S. Bhatia, R. Martins, K. Eaton, S. Chen, T.M. Salay, S. Alaparthy, J.F. Grosso, A.J. Korman, S.M. Parker, S. Agrawal, S.M. Goldberg, D.M. Pardoll, A. Gupta, and J.M. Wigginton, "Safety and Activity of Anti–PD-L1 Antibody in Patients with Advanced Cancer", New England Journal of Medicine, vol. 366, pp. 2455-2465, 2012. http://dx.doi.org/10.1056/NEJMoa1200694
- G. Weber, I. Caruana, R.H. Rouce, A.J. Barrett, U. Gerdemann, A.M. Leen, K.R. Rabin, and C.M. Bollard, "Generation of Tumor Antigen-Specific T Cell Lines from Pediatric Patients with Acute Lymphoblastic Leukemia—Implications for Immunotherapy", Clinical Cancer Research, vol. 19, pp. 5079-5091, 2013. http://dx.doi.org/10.1158/1078-0432.CCR-13-0955
- G. Weber, U. Gerdemann, I. Caruana, B. Savoldo, N.F. Hensel, K.R. Rabin, E.J. Shpall, J.J. Melenhorst, A.M. Leen, A.J. Barrett, and C.M. Bollard, "Generation of multi-leukemia antigen-specific T cells to enhance the graft-versus-leukemia effect after allogeneic stem cell transplant", Leukemia, vol. 27, pp. 1538-1547, 2013. http://dx.doi.org/10.1038/leu.2013.66
- C. Holohan, S. Van Schaeybroeck, D.B. Longley, and P.G. Johnston, "Cancer drug resistance: an evolving paradigm", Nature Reviews Cancer, vol. 13, pp. 714-726, 2013. http://dx.doi.org/10.1038/nrc3599
- F. Klemm, and J.A. Joyce, "Microenvironmental regulation of therapeutic response in cancer", Trends in Cell Biology, vol. 25, pp. 198-213, 2015. http://dx.doi.org/10.1016/j.tcb.2014.11.006
- C. Devaud, L.B. John, J.A. Westwood, P.K. Darcy, and M.H. Kershaw, "Immune modulation of the tumor microenvironment for enhancing cancer immunotherapy", OncoImmunology, vol. 2, pp. e25961, 2013. http://dx.doi.org/10.4161/onci.25961
- C. Oelkrug, and J.M. Ramage, "Enhancement of T cell recruitment and infiltration into tumours", Clinical and Experimental Immunology, vol. 178, pp. 1-8, 2014. http://dx.doi.org/10.1111/cei.12382
- H. Tang, Y. Wang, L. Chlewicki, Y. Zhang, J. Guo, W. Liang, J. Wang, X. Wang, and Y. Fu, "Facilitating T Cell Infiltration in Tumor Microenvironment Overcomes Resistance to PD-L1 Blockade", Cancer Cell, vol. 30, pp. 500, 2016. http://dx.doi.org/10.1016/j.ccell.2016.08.011
- F. Allen, I.D. Bobanga, P. Rauhe, D. Barkauskas, N. Teich, C. Tong, J. Myers, and A.Y. Huang, "CCL3 augments tumor rejection and enhances CD8+T cell infiltration through NK and CD103+dendritic cell recruitment via IFNγ", OncoImmunology, vol. 7, pp. e1393598, 2017. http://dx.doi.org/10.1080/2162402X.2017.1393598
- J.D. Wolchok, H. Kluger, M.K. Callahan, M.A. Postow, N.A. Rizvi, A.M. Lesokhin, N.H. Segal, C.E. Ariyan, R. Gordon, K. Reed, M.M. Burke, A. Caldwell, S.A. Kronenberg, B.U. Agunwamba, X. Zhang, I. Lowy, H.D. Inzunza, W. Feely, C.E. Horak, Q. Hong, A.J. Korman, J.M. Wigginton, A. Gupta, and M. Sznol, "Nivolumab plus Ipilimumab in Advanced Melanoma", New England Journal of Medicine, vol. 369, pp. 122-133, 2013. http://dx.doi.org/10.1056/NEJMoa1302369
- K. Catakovic, E. Klieser, D. Neureiter, and R. Geisberger, "T cell exhaustion: from pathophysiological basics to tumor immunotherapy", Cell Communication and Signaling, vol. 15, 2017. http://dx.doi.org/10.1186/s12964-016-0160-z
- E. Cha, M. Klinger, Y. Hou, C. Cummings, A. Ribas, M. Faham, and L. Fong, "Improved Survival with T Cell Clonotype Stability After Anti–CTLA-4 Treatment in Cancer Patients", Science Translational Medicine, vol. 6, 2014. http://dx.doi.org/10.1126/scitranslmed.3008211
- M.V. Goldberg, and C.G. Drake, "LAG-3 in Cancer Immunotherapy", Current Topics in Microbiology and Immunology, pp. 269-278, 2010. http://dx.doi.org/10.1007/82_2010_114
- I. Melero, S. Hervas-Stubbs, M. Glennie, D.M. Pardoll, and L. Chen, "Immunostimulatory monoclonal antibodies for cancer therapy", Nature Reviews Cancer, vol. 7, pp. 95-106, 2007. http://dx.doi.org/10.1038/nrc2051
- I. Caruana, I. Diaconu, and G. Dotti, "From Monoclonal Antibodies to Chimeric Antigen Receptors for the Treatment of Human Malignancies", Seminars in Oncology, vol. 41, pp. 661-666, 2014. http://dx.doi.org/10.1053/j.seminoncol.2014.08.005
- K. Töpfer, M. Cartellieri, S. Michen, R. Wiedemuth, N. Müller, D. Lindemann, M. Bachmann, M. Füssel, G. Schackert, and A. Temme, "DAP12-Based Activating Chimeric Antigen Receptor for NK Cell Tumor Immunotherapy", The Journal of Immunology, vol. 194, pp. 3201-3212, 2015. http://dx.doi.org/10.4049/jimmunol.1400330
- B. Demoulin, W.J. Cook, J. Murad, D.J. Graber, M. Sentman, C. Lonez, D.E. Gilham, C.L. Sentman, and S. Agaugue, "Exploiting natural killer group 2D receptors for CAR T-cell therapy", Future Oncology, vol. 13, pp. 1593-1605, 2017. http://dx.doi.org/10.2217/fon-2017-0102
- C. Quintarelli, D. Orlando, I. Boffa, M. Guercio, V.A. Polito, A. Petretto, C. Lavarello, M. Sinibaldi, G. Weber, F. Del Bufalo, E. Giorda, M. Scarsella, S. Petrini, D. Pagliara, F. Locatelli, B. De Angelis, and I. Caruana, "Choice of costimulatory domains and of cytokines determines CAR T-cell activity in neuroblastoma", OncoImmunology, vol. 7, 2018. http://dx.doi.org/10.1080/2162402X.2018.1433518
- S.A. Grupp, M. Kalos, D. Barrett, R. Aplenc, D.L. Porter, S.R. Rheingold, D.T. Teachey, A. Chew, B. Hauck, J.F. Wright, M.C. Milone, B.L. Levine, and C.H. June, "Chimeric Antigen Receptor–Modified T Cells for Acute Lymphoid Leukemia", New England Journal of Medicine, vol. 368, pp. 1509-1518, 2013. http://dx.doi.org/10.1056/NEJMoa1215134
- M.A. Pule, B. Savoldo, G.D. Myers, C. Rossig, H.V. Russell, G. Dotti, M.H. Huls, E. Liu, A.P. Gee, Z. Mei, E. Yvon, H.L. Weiss, H. Liu, C.M. Rooney, H.E. Heslop, and M.K. Brenner, "Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma", Nature Medicine, vol. 14, pp. 1264-1270, 2008. http://dx.doi.org/10.1038/nm.1882
- K. Adachi, Y. Kano, T. Nagai, N. Okuyama, Y. Sakoda, and K. Tamada, "IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor", Nature Biotechnology, vol. 36, pp. 346-351, 2018. http://dx.doi.org/10.1038/nbt.4086
- K.F. Bol, G. Schreibelt, W.R. Gerritsen, I.J.M. de Vries, and C.G. Figdor, "Dendritic Cell–Based Immunotherapy: State of the Art and Beyond", Clinical Cancer Research, vol. 22, pp. 1897-1906, 2016. http://dx.doi.org/10.1158/1078-0432.CCR-15-1399
- �. Türeci, M. Vormehr, M. Diken, S. Kreiter, C. Huber, and U. Sahin, "Targeting the Heterogeneity of Cancer with Individualized Neoepitope Vaccines", Clinical Cancer Research, vol. 22, pp. 1885-1896, 2016. http://dx.doi.org/10.1158/1078-0432.CCR-15-1509
- M.V. Maus, and C.H. June, "Making Better Chimeric Antigen Receptors for Adoptive T-cell Therapy", Clinical Cancer Research, vol. 22, pp. 1875-1884, 2016. http://dx.doi.org/10.1158/1078-0432.CCR-15-1433
- A. Heczey, C.U. Louis, B. Savoldo, O. Dakhova, A. Durett, B. Grilley, H. Liu, M.F. Wu, Z. Mei, A. Gee, B. Mehta, H. Zhang, N. Mahmood, H. Tashiro, H.E. Heslop, G. Dotti, C.M. Rooney, and M.K. Brenner, "CAR T Cells Administered in Combination with Lymphodepletion and PD-1 Inhibition to Patients with Neuroblastoma", Molecular Therapy, vol. 25, pp. 2214-2224, 2017. http://dx.doi.org/10.1016/j.ymthe.2017.05.012
- S. Yang, S. Hillinger, K. Riedl, L. Zhang, L. Zhu, M. Huang, K. Atianzar, B.Y. Kuo, B. Gardner, R.K. Batra, R.M. Strieter, S.M. Dubinett, and S. Sharma, "Intratumoral Administration of Dendritic Cells Overexpressing CCL21 Generates Systemic Antitumor Responses and Confers Tumor Immunity", Clinical Cancer Research, vol. 10, pp. 2891-2901, 2004. http://dx.doi.org/10.1158/1078-0432.ccr-03-0380
- J.D. Shields, I.C. Kourtis, A.A. Tomei, J.M. Roberts, and M.A. Swartz, "Induction of Lymphoidlike Stroma and Immune Escape by Tumors That Express the Chemokine CCL21", Science, vol. 328, pp. 749-752, 2010. http://dx.doi.org/10.1126/science.1185837
- R. Barreira da Silva, M.E. Laird, N. Yatim, L. Fiette, M.A. Ingersoll, and M.L. Albert, "Dipeptidylpeptidase 4 inhibition enhances lymphocyte trafficking, improving both naturally occurring tumor immunity and immunotherapy", Nature Immunology, vol. 16, pp. 850-858, 2015. http://dx.doi.org/10.1038/ni.3201
- A. Di Stasi, B. De Angelis, C.M. Rooney, L. Zhang, A. Mahendravada, A.E. Foster, H.E. Heslop, M.K. Brenner, G. Dotti, and B. Savoldo, "T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model", Blood, vol. 113, pp. 6392-6402, 2009. http://dx.doi.org/10.1182/blood-2009-03-209650
- S. Campello, R.A. Lacalle, M. Bettella, S. Mañes, L. Scorrano, and A. Viola, "Orchestration of lymphocyte chemotaxis by mitochondrial dynamics", The Journal of Experimental Medicine, vol. 203, pp. 2879-2886, 2006. http://dx.doi.org/10.1084/jem.20061877
- J. Jacobelli, M. Estin Matthews, S. Chen, and M.F. Krummel, "Activated T Cell Trans-Endothelial Migration Relies on Myosin-IIA Contractility for Squeezing the Cell Nucleus through Endothelial Cell Barriers", PLoS ONE, vol. 8, pp. e75151, 2013. http://dx.doi.org/10.1371/journal.pone.0075151
- Y. Zhang, R. Kurupati, L. Liu, X.Y. Zhou, G. Zhang, A. Hudaihed, F. Filisio, W. Giles-Davis, X. Xu, G.C. Karakousis, L.M. Schuchter, W. Xu, R. Amaravadi, M. Xiao, N. Sadek, C. Krepler, M. Herlyn, G.J. Freeman, J.D. Rabinowitz, and H.C. Ertl, "Enhancing CD8+ T Cell Fatty Acid Catabolism within a Metabolically Challenging Tumor Microenvironment Increases the Efficacy of Melanoma Immunotherapy", Cancer Cell, vol. 32, pp. 377-391.e9, 2017. http://dx.doi.org/10.1016/j.ccell.2017.08.004
- C.S. Palmer, M. Ostrowski, B. Balderson, N. Christian, and S.M. Crowe, "Glucose Metabolism Regulates T Cell Activation, Differentiation, and Functions", Frontiers in Immunology, vol. 6, 2015. http://dx.doi.org/10.3389/fimmu.2015.00001
- G. van der Windt, B. Everts, C. Chang, J. Curtis, T. Freitas, E. Amiel, E. Pearce, and E. Pearce, "Mitochondrial Respiratory Capacity Is a Critical Regulator of CD8+ T Cell Memory Development", Immunity, vol. 36, pp. 68-78, 2012. http://dx.doi.org/10.1016/j.immuni.2011.12.007
- M. Buck, D. O’Sullivan, R. Klein Geltink, J. Curtis, C. Chang, D. Sanin, J. Qiu, O. Kretz, D. Braas, G. van der Windt, Q. Chen, S. Huang, C. O’Neill, B. Edelson, E. Pearce, H. Sesaki, T. Huber, A. Rambold, and E. Pearce, "Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming", Cell, vol. 166, pp. 63-76, 2016. http://dx.doi.org/10.1016/j.cell.2016.05.035
- N. Scharping, A. Menk, R. Moreci, R. Whetstone, R. Dadey, S. Watkins, R. Ferris, and G. Delgoffe, "The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction", Immunity, vol. 45, pp. 701-703, 2016. http://dx.doi.org/10.1016/j.immuni.2016.08.009
- W. Qian, S. Choi, G.A. Gibson, S.C. Watkins, C.J. Bakkenist, and B. Van Houten, "Mitochondrial hyperfusion induced by loss of the fission protein Drp1 causes ATM-dependent G2/M arrest and aneuploidy through DNA replication stress", Journal of Cell Science, vol. 125, pp. 5745-5757, 2012. http://dx.doi.org/10.1242/jcs.109769
- L. Simula, F. Nazio, and S. Campello, "The mitochondrial dynamics in cancer and immune-surveillance", Seminars in Cancer Biology, vol. 47, pp. 29-42, 2017. http://dx.doi.org/10.1016/j.semcancer.2017.06.007
- J.H. Russell, C.L. White, D.Y. Loh, and P. Meleedy-Rey, "Receptor-stimulated death pathway is opened by antigen in mature T cells.", Proceedings of the National Academy of Sciences, vol. 88, pp. 2151-2155, 1991. http://dx.doi.org/10.1073/pnas.88.6.2151
- M. Corrado, F.R. Mariotti, L. Trapani, L. Taraborrelli, F. Nazio, V. Cianfanelli, M.E. Soriano, E. Schrepfer, F. Cecconi, L. Scorrano, and S. Campello, "Macroautophagy inhibition maintains fragmented mitochondria to foster T cell receptor‐dependent apoptosis", The EMBO Journal, vol. 35, pp. 1793-1809, 2016. http://dx.doi.org/10.15252/embj.201593727
- B.L. Horton, J.B. Williams, A. Cabanov, S. Spranger, and T.F. Gajewski, "Intratumoral CD8+ T-cell Apoptosis Is a Major Component of T-cell Dysfunction and Impedes Antitumor Immunity", Cancer Immunology Research, vol. 6, pp. 14-24, 2018. http://dx.doi.org/10.1158/2326-6066.CIR-17-0249
- P.J. Ebert, J. Cheung, Y. Yang, E. McNamara, R. Hong, M. Moskalenko, S.E. Gould, H. Maecker, B.A. Irving, J.M. Kim, M. Belvin, and I. Mellman, "MAP Kinase Inhibition Promotes T Cell and Anti-tumor Activity in Combination with PD-L1 Checkpoint Blockade", Immunity, vol. 44, pp. 609-621, 2016. http://dx.doi.org/10.1016/j.immuni.2016.01.024
- S.D. Dowling, and F. Macian, "Autophagy and T cell metabolism", Cancer Letters, vol. 419, pp. 20-26, 2018. http://dx.doi.org/10.1016/j.canlet.2018.01.033
ACKNOWLEDGMENTS
This work was funded by AIRC Founda-tion ("MoTIs" IG 19826 grant) to SC.
COPYRIGHT
© 2018

T lymphocytes against solid malignancies: winning ways to defeat tumours by Caruana et al. is licensed under a Creative Commons Attribution 4.0 International License.


