News and thoughts:
Cell Stress, Vol. 1, No. 2, pp. 73 - 78; doi: 10.15698/cst2017.11.109
Tumor suppressive Ca2+ signaling is driven by IP3 receptor fitness

1 KU Leuven, Lab. Molecular and Cellular Signaling, Dep. Cellular and Molecular Medicine and Leuven Kanker Instituut (LKI), Campus Gasthuisberg O/N-I bus 802, Herestraat 49, BE-3000 Leuven, Belgium.
2 Department of Comparative Biomedical Sciences, Royal Veterinary College, NW1 0TU, London, United Kingdom.
3 University College London Consortium for Mitochondrial Research, University College London, WC1 6BT, London, United Kingdom.
Keywords: cancer, oncogenesis, tumor suppression, IP3R3, calcium signaling, PTEN, FBXL2, BAP1.
Received originally: 12/10/2017 Accepted: 20/10/2017
Published: 01/11/2017
Correspondence:
Geert Bultynck, KU Leuven, Lab. Molecular and Cellular Signaling, Dep. Cellular and Molecular Medicine and Leuven Kanker Instituut (LKI), Campus Gasthuisberg O/N-I bus 802, Herestraat 49, BE-3000 Leuven, Belgium geert.bultynck@kuleuven.be
Michelangelo Campanella, Hobday Building, RVC Camden Campus, Royal College Street, NW1 0TU, London, United Kingdom mcampanella@rvc.ac.uk
Conflict of interest statement: The authors declare no conflict of interest.
Please cite this article as: Geert Bultynck and Michelangelo Campanella (2017). Tumor suppressive Ca2+ signaling is driven by IP3 receptor fitness. Cell Stress 1(2): 73-78. doi: 10.15698/cst2017.11.109
Intracellular Ca2+ signals critically control a plethora of cellular functions, of which many impact cellular death and/or survival, processes often dysregulated in cancer [1]. In healthy cells, Ca2+ signaling is employed for normal cell physiology and survival functions [2]. Yet, when a cell is exposed to toxic stimuli or suffering from enduring cell stress, like irreparable DNA damage, the Ca2+-signaling toolbox can be rapidly switched from a “pro-survival” modus into a “pro-death” modus, thereby initiating demise pathways [3]. The highly dynamic nature of Ca2+ signaling allows cells to swiftly response to stress and damage, preventing the survival of damaged cells and malignant transformation that eventually results in tumor formation. Alterations in the expression, activity and regulation of Ca2+-transport systems both at the plasma membrane and at organelles like the endoplasmic reticulum (ER) and mitochondria have been implicated in oncogenesis and neoplasia [1][4]. These changes result in aberrant Ca2+-signaling events that could favor resistance to cell death, migration or senescence escape [5].
–
Over the last decade, we learnt that tight contacts and functional connections involving Ca2+ exchanges between the ER, the main intracellular Ca2+-storage organelle, and the mitochondria are pivotal for cell-fate decisions [3][6][7][8]. These contact sites contain chaperone-coupled Ca2+-flux systems: the IP3 receptors (IP3Rs) at the ER side and the voltage-dependent anion channels (VDACs) at the mitochondrial outer membrane side [9]. These are controlled/exploited by several cellular factors and regulatory proteins, including oncogenes and tumor suppressors [10][11][12]. Basal Ca2+ fluxes between ER and mitochondria sustain anabolic pathways for mitochondrial metabolism, ensuring proper cell cycle progression [13]. Yet, continued elevated ER-mitochondrial Ca2+ transfers result in loss of mitochondrial membrane integrity and release of apoptogenic factors [14]. Tuned ER-mitochondrial Ca2+ transfer is therefore key to cells’ response to pro-apoptotic stimuli: the failure of which results in cell death resistance, as often observed in cancer cells [15][16]. In fact, the efficacy of chemotherapeutic agents and photodynamic therapy depends on the ability of these agents to elicit ER-mitochondrial Ca2+ exchanges [15]. In the context of apoptosis, previous work proposed unique roles for the type 3 IP3R isoform (IP3R3) [17] and type 1 VDAC isoform (VDAC1) [18] even though other IP3R isoforms can contribute to the initiation of cell death programs [19][20].
–
In some cells, the role played by the IP3R3 in pro-apoptotic Ca2+ transfers from ER to mitochondria might relate to its ability to preferentially partner with the VDAC1 complex [18]. Notably, several tumor suppressors and oncogenes located at the ER membranes dodge ER Ca2+ homeostasis and dynamics [10][21]. In general, tumor suppressors increase ER-mitochondrial Ca2+ fluxes, whereas oncogenes suppress ER-mitochondrial Ca2+ fluxes. The actions of both tumor suppressors and oncogenes at the ER can involve changes of the steady-state ER Ca2+-filling state through modulation of sarco/endoplasmic reticulum Ca2+ ATPases (SERCA) and/or ER Ca2+-leak channels. For instance, during stress, the tumor suppressor p53 accumulates at the ER and enhances the Ca2+-pump activity SERCA, causing ER Ca2+ overload and the likelihood for pro-apoptotic ER-mitochondrial Ca2+ fluxes [22][23][24]. The anti-apoptotic protein Bcl-2 increases IP3R phosphorylation and its sensitivity to IP3, enhancing the passive Ca2+ leak from the ER, lowering ER Ca2+ levels and so pro-apoptotic ER-mitochondrial Ca2+ fluxes [25]. Several tumor suppressors and oncogenes have been identified as direct regulators of the IP3R, whereby tumor suppressors (like BRCA1, PTEN, PML) and oncogenes (like Bcl-2, PKB/Akt) that respectively promote and suppress the activity of IP3R channels by impacting their gating and consequently their open probability [10][21]. Besides this direct regulation of IP3R gating, it is clear that total IP3R-protein levels impact Ca2+ flux from the ER to the mitochondria and in turn cellular sensitivity to death as well. Insights are now available on IP3R degradation by ER-assisted and 26S proteasomal turnover after cell stimulation and IP3R activation [26]. In such conditions, IP3Rs become ubiquitinated due to recruitment of the erlin1/2 complex and RNF170, an E3 ubiquitin ligase [27][28][29].
–
Hitherto, not much was known about the molecular mechanisms impacting basal IP3R turn-over and controlling their steady-state in stressed cells; equally, whether dysregulation of these mechanisms was involved in oncogenesis and/or tumor progression. Nevertheless, it is clear that IP3R levels do impact apoptotic sensitivity [20][30][31][32], and hence cell death and survival proteins were found implicated in regulating IP3R levels [33][34].
–
Recent work from Kuchay et al. revealed an unexpected role for the tumor suppressor lipid/protein phosphatase PTEN, an allele frequently lost in cancer [35] and well-known negative regulator of PKB/Akt signaling, stabilizing IP3R channels by protecting them from proteasomal degradation [36] (Fig. 1). This novel function adds to its recently discovered presence at the mitochondria-associated membranes (MAMs), where it contributes to cell death sensitivity by suppressing IP3R3-mediated Ca2+ fluxes [37]. Independently of its catalytic activity, PTEN competes with the F-box protein FBXL2 (the receptor subunit of one of 69 human SCF (SKP1, CUL1, F-box protein) [38][39] to bind to IP3R3 channels, in particular to a region in the ligand-binding domain. Normal cells that express PTEN will have a low level of the IP3R3/FBXL2-complex formation, preventing the ubiquitination of IP3R3 channels and subsequent targeting to the proteasome. Consistently with previous observations [27][40], activation of cells with agonists increase FBXL2/IP3R3-complex formation and subsequent IP3R3 post-transcriptional regulation via ubiquitination similarly to RNF170-mediated ubiquitination of IP3Rs [27]. Interestingly, FBXL2 activity itself is Ca2+ dependent, but antagonized by calmodulin [41]. Hence, activation of IP3Rs may not only make these channels more susceptible for degradation by increased interaction with FBXL2 but release of Ca2+ itself through IP3Rs may trigger local activation of FBXL2 associated with IP3R3 leading to IP3R3 degradation. Notably, FBXL2 binding to its substrates (like cyclin D3) was found to occur via canonical calmodulin-binding motif thereby preventing ubiquitination. It is nonetheless likely that FBXL2 binding to IP3Rs does not occur by targeting its calmodulin-binding motif even though a role for calmodulin in regulating IP3R3/FBXL2-complex formation may not be ruled out. In any case, access to the degron region in the ligand-binding core of IP3R3 for FBXL2 is facilitated by deletion of the suppressor domain and loss of PTEN is associated with increased FBXL2 binding to IP3R3 and degradation of IP3R3, contributing to the apoptotic resistance of cells [36]. In cancer cells lacking PTEN, FBXL2 knockdown could therefore restore IP3R3 levels and apoptotic sensitivity.
–
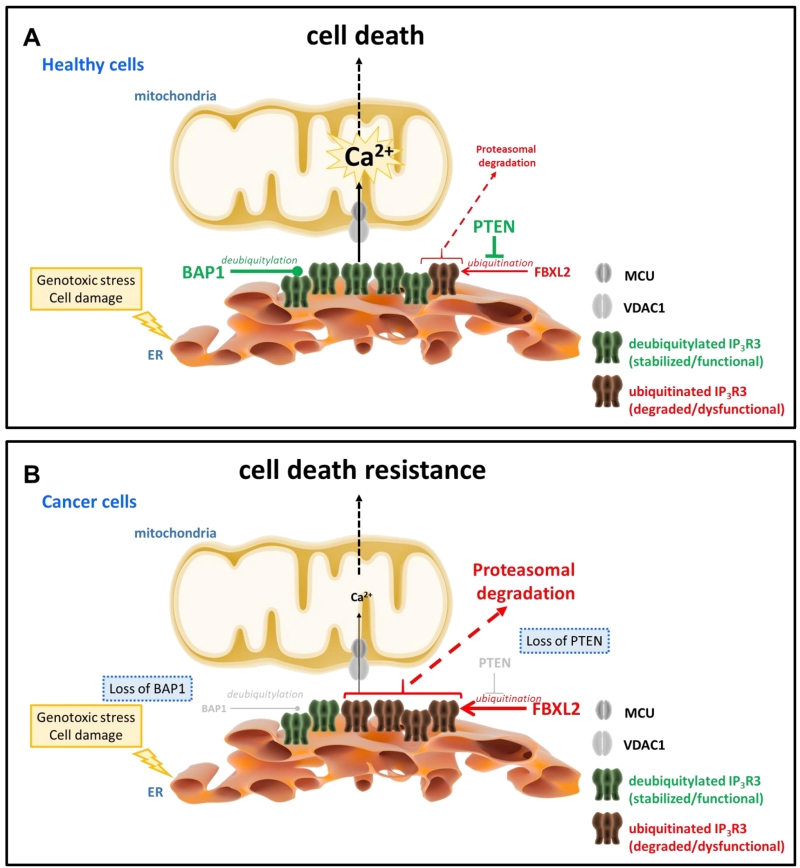 |
FIGURE 1: IP3R3 channels are targets for ubiquitination and deubiquitylation enzymes, whose activities are deregulated in cancer cells, thereby impacting IP3R3 stability and ultimately cell death sensitivity. (A) IP3Rs, located at the ER-mitochondrial interface, here particularly IP3R3, promote ER-mitochondrial Ca2+ fluxes, thereby enabling adequate cell death sensitivity in healthy cells undergoing genotoxic stress and cell damage, an important feature that prevents oncogenesis. The levels of IP3R3 channels are critical for this process and are dynamically regulated via ubiquitination. BAP1, a tumor suppressor with deubiquitylation enzyme activities and frequently mutated in cancers including mesothelioma, deubiquitylates IP3R3, preventing its proteasomal degradation and resulting in IP3R3 stabilization. In contrast, FBXL2, an F-Box protein with ubiquitin ligase activity, ubiquitinates IP3R3, directing it for proteasomal degradation and resulting in a decline in IP3R3 levels. Importantly, PTEN, another tumor suppressor that antagonizes Akt signaling and frequently mutated in a variety of cancers including prostate cancer, competes with FBXL2 for IP3R3 binding and thus antagonizes FBXL2-mediated ubiquitination of IP3R3, stabilizing IP3R3. In healthy cells, it is anticipated that the balance between deubiquitylation and ubiquitination of IP3R3 channels is favored towards deubiquitylated IP3R3, enabling sufficient IP3R3 channels to sustain adequate ER-mitochondrial Ca2+ fluxes in cells undergoing genotoxic stress and cell damage to engage cell death and eradicate pre-malignant cells with DNA damage. (B) Several cancer types display chromosomal aberrations with defects in the expression of several tumor suppressors, including a reduction in BAP1 levels and a loss in PTEN expression. As a consequence, both alterations will favor IP3R3 ubiquitination and degradation, since reduced BAP1 levels will lead to reduced deubiquitylation of IP3Rs and loss of PTEN will enable FBXL2 to mediate IP3R3 ubiquitination. As a consequence, the balance between ubiquitination and deubiquitylation of IP3R3s will shift towards ubiquitinated IP3R3, impairing ER-mitochondrial Ca2+ fluxes that are needed to engage cell death programs in response to genotoxic stress and cell damage. As such, cells will be able to withstand this cellular stress, resulting in their survival and proliferation despite accumulated DNA damage. This phenomenon is an early event in oncogenesis, enabling malignant cell formation and tumoral behavior. |
–
Notably, authors show that by mutating the degron region in the ligand-binding domain in IP3R3 the FBXL2 binding and IP3R3 degradation were both prevented. Using a knockin approach in which wild-type IP3R3 was altered into a non-degradable IP3R3 mutant version (IP3R3Q550A/F553A/R554A), the authors could restore the rise of Ca2+ induced by photodynamic therapy and apoptosis in PTEN-negative cancer cells. This correlation was eloquently observed in human tumor samples beyond the convincing in vivo xenograft models, in which tumor cells expressing a non-degradable IP3R3 version or treated with geranylgeranyltransferase inhibitor (that prevents FBXL2 accumulation at ER membranes and activity, which depends on its geranylgeranylation [42]) were greatly sensitized towards photodynamic therapy compared to tumor cells expressing wild-type (degradable) IP3R3 or untreated tumors.
–
It is important to note that FBXL2 has been previously implicated in cancer, but rather acting via the suppression of cell cycle progression and proliferation, as observed in lung tumors [43], leukemic cells [44], gastric cancer cells [45], and prevalently of tumor suppressive nature. Instead, in this work, the effects of FBXL2 on IP3R3 are tumor promoting by increasing IP3R3 degradation and making the cells more resistant to cell death [36]. These effects are neutralized by PTEN, which prevents FBLX2 binding to IP3R3 channels. This interference by PTEN is likely selective for IP3R3 and not for other FBXL2 targets regulating the cell cycle. Nevertheless, the anti-cancer properties of FBXL2 activators like the small molecule BC-1258 [46] will be adversely impacted by FBXL2-mediated IP3R3 degradation, likely limiting their application to PTEN-positive cancers. Vice versa, the previously discovered tumor suppressive properties of FBXL2 in cancer might be further boosted if FBXL2 could be selectively/subcellularly activated to pro-mote its cell-cycle targets while shielding IP3R3 for degradation.
–
In a separate study, Bononi et al. revealed a novel deubiquitylating enzyme that actually counteracts the level of IP3R3 ubiquitination, namely BRCA-associated protein 1 (BAP1) [47] (Fig. 1). BAP1 is a potent tumor suppressor, which protects against environmental stress and damage [48]. Loss of one BAP1 allele either inherited or acquired during life has been associated with environmental stress-induced carcinogenesis, like UV light for uveal melanoma and asbestos for mesothelioma. Germline mutations in BAP1 resulting in aberrant/loss of BAP1 expression were associated with a high incidence of familial mesothelioma and uveal melanoma, while somatic mutations in BAP1 were found in sporadic mesotheliomas [49]. Germline mutations in BAP1 greatly enhanced the sensitivity of mice to develop mesothelioma when exposed to asbestos [50]. Recently, it has been shown in patient fibroblasts that loss of BAP1 displayed a metabolic rewiring towards aerobic glycolysis and reduced mitochondrial respiration associated with malignancy and carcinogenesis [51]. BAP1 was therefore identified as a novel IP3R3-interacting protein that impacts its post-translational modification. BAP1 causes IP3R3 stabilization and prevents the channel to be degraded by the proteasome. Loss of only 1 allele in BAP1 is sufficient to protect cells from undergoing apoptotic cell death via suppressed Ca2+ release triggered by apoptotic stimuli like H2O2. An effect operated via the IP3R3 degradation due to a decreased BAP1-mediated deubiquitylation of IP3R3. As a consequence, exposure of cells being BAP1+/- to DNA-damaging conditions will result in a higher percentage of cells surviving despite having damaged DNA. Such cells bearing genomic aberrations are at high risk for neoplastic behavior and oncogenesis, resulting in malignant cell growth and tumor formation. However, restoring IP3R3 in these cells could be an attractive strategy to reinstate cell death sensitivity of BAP1+/- cells.
–
Based on these recent developments, it will be therefore important to examine the interplay between BAP1 and FBXL2. It is indeed not clear whether the increased IP3R3 ubiquitination observed when BAP1 levels are declined is mediated through FBXL2 and whether lack of BAP1 results in increased FBXL2 association with IP3R3 channels. In any case, by inhibiting FBXL2’s action on IP3R3 without inhibiting its action on cell cycle targets could be an attractive avenue to restore IP3R3 levels in these cancers. Another strategy can be boosting ER Ca2+-store loading in tumor cells by activating the SERCA pumps, thereby increasing the likelihood for ER-mitochondrial Ca2+ fluxes and restoring cellular sensitivity to apoptosis [22][52]. Tumor-selective SERCA modulation is challenging but not impossible though, as evidenced by the ability to locally release thapsigargin in the vicinity of pancreatic tumor cells using peptide-coupled prodrugs that are enzymatically cleaved by prostate-specific factors [53]. SERCA-activating approaches may be based on p53, recently proposed to activate SERCA activity in response to chemotherapy [22][23][24], although p53 is very frequently mutated in cancer. In wild-type p53 tumors, direct p53 activators might be of use. Other solace may come from SERCA-activating small molecules like CDN1163 [54], provided these agents can be delivered to tumor cells while sparing healthy cells [52].
–
These recent papers highlight that deregulation of IP3R3 ubiquitination homeostasis not only impacts death and survival of cells but also contributes to the oncogenic behavior of cells with dysfunctional tumor suppressors by either (i) lacking PTEN [36] or by (ii) displaying deficiencies in BAP1 [47]. They underpin the emerging role of altered Ca2+ signaling at the MAM level as a key event in apoptosis resistance that contributes early events associated with oncogenesis and tumor formation. Challenging will be the translation of these insights into anti-cancer therapies for which not only tumor-selective applications will be required but also further understanding in the selective targeting at the level of the IP3R3.
References
- G.R. Monteith, N. Prevarskaya, and S.J. Roberts-Thomson, "The calcium–cancer signalling nexus", Nature Reviews Cancer, vol. 17, pp. 373-380, 2017. http://dx.doi.org/10.1038/nrc.2017.18
- M.J. Berridge, M.D. Bootman, and H.L. Roderick, "Calcium signalling: dynamics, homeostasis and remodelling", Nature Reviews Molecular Cell Biology, vol. 4, pp. 517-529, 2003. http://dx.doi.org/10.1038/nrm1155
- S. Marchi, S. Patergnani, S. Missiroli, G. Morciano, A. Rimessi, M.R. Wieckowski, C. Giorgi, and P. Pinton, "Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death", Cell Calcium, vol. 69, pp. 62-72, 2018. http://dx.doi.org/10.1016/j.ceca.2017.05.003
- G. Bultynck, and J.B. Parys, "Ca2+ signaling and cell death: Focus on Ca2+-transport systems and their implication in cell death and survival", Cell Calcium, vol. 69, pp. 1-3, 2018. http://dx.doi.org/10.1016/j.ceca.2017.09.001
- H. Ivanova, M. Kerkhofs, R.M. La Rovere, and G. Bultynck, "Endoplasmic Reticulum–Mitochondrial Ca2+ Fluxes Underlying Cancer Cell Survival", Frontiers in Oncology, vol. 7, 2017. http://dx.doi.org/10.3389/fonc.2017.00070
- S. Marchi, S. Patergnani, and P. Pinton, "The endoplasmic reticulum–mitochondria connection: One touch, multiple functions", Biochimica et Biophysica Acta (BBA) - Bioenergetics, vol. 1837, pp. 461-469, 2014. http://dx.doi.org/10.1016/j.bbabio.2013.10.015
- S. Marchi, M. Bittremieux, S. Missiroli, C. Morganti, S. Patergnani, L. Sbano, A. Rimessi, M. Kerkhofs, J.B. Parys, G. Bultynck, C. Giorgi, and P. Pinton, "Endoplasmic Reticulum-Mitochondria Communication Through Ca2+ Signaling: The Importance of Mitochondria-Associated Membranes (MAMs)", Advances in Experimental Medicine and Biology, pp. 49-67, 2017. http://dx.doi.org/10.1007/978-981-10-4567-7_4
- R.M. La Rovere, G. Roest, G. Bultynck, and J.B. Parys, "Intracellular Ca 2+ signaling and Ca 2+ microdomains in the control of cell survival, apoptosis and autophagy", Cell Calcium, vol. 60, pp. 74-87, 2016. http://dx.doi.org/10.1016/j.ceca.2016.04.005
- C. Giorgi, S. Missiroli, S. Patergnani, J. Duszynski, M.R. Wieckowski, and P. Pinton, "Mitochondria-Associated Membranes: Composition, Molecular Mechanisms, and Physiopathological Implications", Antioxidants & Redox Signaling, vol. 22, pp. 995-1019, 2015. http://dx.doi.org/10.1089/ars.2014.6223
- M. Bittremieux, J.B. Parys, P. Pinton, and G. Bultynck, "ER functions of oncogenes and tumor suppressors: Modulators of intracellular Ca2+ signaling", Biochimica et Biophysica Acta (BBA) - Molecular Cell Research, vol. 1863, pp. 1364-1378, 2016. http://dx.doi.org/10.1016/j.bbamcr.2016.01.002
- T. Vervliet, E. Clerix, B. Seitaj, H. Ivanova, G. Monaco, and G. Bultynck, "Modulation of Ca2+ Signaling by Anti-apoptotic B-Cell Lymphoma 2 Proteins at the Endoplasmic Reticulum–Mitochondrial Interface", Frontiers in Oncology, vol. 7, 2017. http://dx.doi.org/10.3389/fonc.2017.00075
- S. Marchi, C. Giorgi, M. Oparka, J. Duszynski, M.R. Wieckowski, and P. Pinton, "Oncogenic and oncosuppressive signal transduction at mitochondria-associated endoplasmic reticulum membranes", Molecular & Cellular Oncology, vol. 1, pp. e956469, 2014. http://dx.doi.org/10.4161/23723548.2014.956469
- C. Cárdenas, and J.K. Foskett, "Mitochondrial Ca2+ signals in autophagy", Cell Calcium, vol. 52, pp. 44-51, 2012. http://dx.doi.org/10.1016/j.ceca.2012.03.001
- C. Giorgi, F. Baldassari, A. Bononi, M. Bonora, E. De Marchi, S. Marchi, S. Missiroli, S. Patergnani, A. Rimessi, J.M. Suski, M.R. Wieckowski, and P. Pinton, "Mitochondrial Ca2+ and apoptosis", Cell Calcium, vol. 52, pp. 36-43, 2012. http://dx.doi.org/10.1016/j.ceca.2012.02.008
- G. Pedriali, A. Rimessi, L. Sbano, C. Giorgi, M.R. Wieckowski, M. Previati, and P. Pinton, "Regulation of Endoplasmic Reticulum–Mitochondria Ca2+ Transfer and Its Importance for Anti-Cancer Therapies", Frontiers in Oncology, vol. 7, 2017. http://dx.doi.org/10.3389/fonc.2017.00180
- M. Kerkhofs, C. Giorgi, S. Marchi, B. Seitaj, J.B. Parys, P. Pinton, G. Bultynck, and M. Bittremieux, "Alterations in Ca2+ Signalling via ER-Mitochondria Contact Site Remodelling in Cancer", Advances in Experimental Medicine and Biology, pp. 225-254, 2017. http://dx.doi.org/10.1007/978-981-10-4567-7_17
- S. Marchi, M. Marinello, A. Bononi, M. Bonora, C. Giorgi, A. Rimessi, and P. Pinton, "Selective modulation of subtype III IP3R by Akt regulates ER Ca2+ release and apoptosis", Cell Death & Disease, vol. 3, pp. e304-e304, 2012. http://dx.doi.org/10.1038/cddis.2012.45
- D. De Stefani, A. Bononi, A. Romagnoli, A. Messina, V. De Pinto, P. Pinton, and R. Rizzuto, "VDAC1 selectively transfers apoptotic Ca2+ signals to mitochondria", Cell Death & Differentiation, vol. 19, pp. 267-273, 2011. http://dx.doi.org/10.1038/cdd.2011.92
- H. Ivanova, T. Vervliet, L. Missiaen, J.B. Parys, H. De Smedt, and G. Bultynck, "Inositol 1,4,5-trisphosphate receptor-isoform diversity in cell death and survival", Biochimica et Biophysica Acta (BBA) - Molecular Cell Research, vol. 1843, pp. 2164-2183, 2014. http://dx.doi.org/10.1016/j.bbamcr.2014.03.007
- T. Jayaraman, and A.R. Marks, "T cells deficient in inositol 1,4,5-trisphosphate receptor are resistant to apoptosis.", Molecular and cellular biology, 1997. http://www.ncbi.nlm.nih.gov/pubmed/9154798
- H. Akl, and G. Bultynck, "Altered Ca2+ signaling in cancer cells: Proto-oncogenes and tumor suppressors targeting IP3 receptors", Biochimica et Biophysica Acta (BBA) - Reviews on Cancer, vol. 1835, pp. 180-193, 2013. http://dx.doi.org/10.1016/j.bbcan.2012.12.001
- M. Bittremieux, and G. Bultynck, "p53 and Ca2+ signaling from the endoplasmic reticulum: partners in anti-cancer therapies", Oncoscience, vol. 2, pp. 233-238, 2015. http://dx.doi.org/10.18632/oncoscience.139
- C. Giorgi, M. Bonora, S. Missiroli, F. Poletti, F.G. Ramirez, G. Morciano, C. Morganti, P.P. Pandolfi, F. Mammano, and P. Pinton, "Intravital imaging reveals p53-dependent cancer cell death induced by phototherapy via calcium signaling", Oncotarget, vol. 6, pp. 1435-1445, 2014. http://dx.doi.org/10.18632/oncotarget.2935
- C. Giorgi, M. Bonora, G. Sorrentino, S. Missiroli, F. Poletti, J.M. Suski, F. Galindo Ramirez, R. Rizzuto, F. Di Virgilio, E. Zito, P.P. Pandolfi, M.R. Wieckowski, F. Mammano, G. Del Sal, and P. Pinton, "p53 at the endoplasmic reticulum regulates apoptosis in a Ca 2+ -dependent manner", Proceedings of the National Academy of Sciences, vol. 112, pp. 1779-1784, 2015. http://dx.doi.org/10.1073/pnas.1410723112
- S.A. Oakes, L. Scorrano, J.T. Opferman, M.C. Bassik, M. Nishino, T. Pozzan, and S.J. Korsmeyer, "Proapoptotic BAX and BAK regulate the type 1 inositol trisphosphate receptor and calcium leak from the endoplasmic reticulum", Proceedings of the National Academy of Sciences, vol. 102, pp. 105-110, 2004. http://dx.doi.org/10.1073/pnas.0408352102
- F. Wright, and R. Wojcikiewicz, "Chapter 4 - Inositol 1,4,5-Trisphosphate Receptor Ubiquitination", Progress in Molecular Biology and Translational Science, pp. 141-159, 2016. http://dx.doi.org/10.1016/bs.pmbts.2016.02.004
- J.P. Lu, Y. Wang, D.A. Sliter, M.M. Pearce, and R.J. Wojcikiewicz, "RNF170 Protein, an Endoplasmic Reticulum Membrane Ubiquitin Ligase, Mediates Inositol 1,4,5-Trisphosphate Receptor Ubiquitination and Degradation", Journal of Biological Chemistry, vol. 286, pp. 24426-24433, 2011. http://dx.doi.org/10.1074/jbc.M111.251983
- M.M. Pearce, D.B. Wormer, S. Wilkens, and R.J. Wojcikiewicz, "An Endoplasmic Reticulum (ER) Membrane Complex Composed of SPFH1 and SPFH2 Mediates the ER-associated Degradation of Inositol 1,4,5-Trisphosphate Receptors", Journal of Biological Chemistry, vol. 284, pp. 10433-10445, 2009. http://dx.doi.org/10.1074/jbc.M809801200
- F.A. Wright, J.P. Lu, D.A. Sliter, N. Dupré, G.A. Rouleau, and R.J. Wojcikiewicz, "A Point Mutation in the Ubiquitin Ligase RNF170 That Causes Autosomal Dominant Sensory Ataxia Destabilizes the Protein and Impairs Inositol 1,4,5-Trisphosphate Receptor-mediated Ca2+ Signaling", Journal of Biological Chemistry, vol. 290, pp. 13948-13957, 2015. http://dx.doi.org/10.1074/jbc.M115.655043
- M. Madesh, B.J. Hawkins, T. Milovanova, C.D. Bhanumathy, S.K. Joseph, S.P. RamachandraRao, K. Sharma, T. Kurosaki, and A.B. Fisher, "Selective role for superoxide in InsP3 receptor–mediated mitochondrial dysfunction and endothelial apoptosis", The Journal of Cell Biology, vol. 170, pp. 1079-1090, 2005. http://dx.doi.org/10.1083/jcb.200505022
- M.T. Khan, C.D. Bhanumathy, Z.T. Schug, and S.K. Joseph, "Role of Inositol 1,4,5-Trisphosphate Receptors in Apoptosis in DT40 Lymphocytes", Journal of Biological Chemistry, vol. 282, pp. 32983-32990, 2007. http://dx.doi.org/10.1074/jbc.M705183200
- Z. Assefa, G. Bultynck, K. Szlufcik, N. Nadif Kasri, E. Vermassen, J. Goris, L. Missiaen, G. Callewaert, J.B. Parys, and H. De Smedt, "Caspase-3-induced Truncation of Type 1 Inositol Trisphosphate Receptor Accelerates Apoptotic Cell Death and Induces Inositol Trisphosphate-independent Calcium Release during Apoptosis", Journal of Biological Chemistry, vol. 279, pp. 43227-43236, 2004. http://dx.doi.org/10.1074/jbc.M403872200
- J.J. Schulman, F.A. Wright, T. Kaufmann, and R.J. Wojcikiewicz, "The Bcl-2 Protein Family Member Bok Binds to the Coupling Domain of Inositol 1,4,5-Trisphosphate Receptors and Protects Them from Proteolytic Cleavage", Journal of Biological Chemistry, vol. 288, pp. 25340-25349, 2013. http://dx.doi.org/10.1074/jbc.M113.496570
- C. Pierro, S.J. Cook, T.C.F. Foets, M.D. Bootman, and H.L. Roderick, "Oncogenic K-Ras suppresses IP3-dependent Ca2+ release through remodeling of IP3Rs isoform composition and ER luminal Ca2+ levels in colorectal cancer cell lines", Journal of Cell Science, 2014. http://dx.doi.org/10.1242/jcs.141408
- R. Beroukhim, C.H. Mermel, D. Porter, G. Wei, S. Raychaudhuri, J. Donovan, J. Barretina, J.S. Boehm, J. Dobson, M. Urashima, K.T. Mc Henry, R.M. Pinchback, A.H. Ligon, Y. Cho, L. Haery, H. Greulich, M. Reich, W. Winckler, M.S. Lawrence, B.A. Weir, K.E. Tanaka, D.Y. Chiang, A.J. Bass, A. Loo, C. Hoffman, J. Prensner, T. Liefeld, Q. Gao, D. Yecies, S. Signoretti, E. Maher, F.J. Kaye, H. Sasaki, J.E. Tepper, J.A. Fletcher, J. Tabernero, J. Baselga, M. Tsao, F. Demichelis, M.A. Rubin, P.A. Janne, M.J. Daly, C. Nucera, R.L. Levine, B.L. Ebert, S. Gabriel, A.K. Rustgi, C.R. Antonescu, M. Ladanyi, A. Letai, L.A. Garraway, M. Loda, D.G. Beer, L.D. True, A. Okamoto, S.L. Pomeroy, S. Singer, T.R. Golub, E.S. Lander, G. Getz, W.R. Sellers, and M. Meyerson, "The landscape of somatic copy-number alteration across human cancers", Nature, vol. 463, pp. 899-905, 2010. http://dx.doi.org/10.1038/nature08822
- S. Kuchay, C. Giorgi, D. Simoneschi, J. Pagan, S. Missiroli, A. Saraf, L. Florens, M.P. Washburn, A. Collazo-Lorduy, M. Castillo-Martin, C. Cordon-Cardo, S.M. Sebti, P. Pinton, and M. Pagano, "PTEN counteracts FBXL2 to promote IP3R3- and Ca2+-mediated apoptosis limiting tumour growth", Nature, vol. 546, pp. 554-558, 2017. http://dx.doi.org/10.1038/nature22965
- A. Bononi, M. Bonora, S. Marchi, S. Missiroli, F. Poletti, C. Giorgi, P.P. Pandolfi, and P. Pinton, "Identification of PTEN at the ER and MAMs and its regulation of Ca2+ signaling and apoptosis in a protein phosphatase-dependent manner", Cell Death & Differentiation, vol. 20, pp. 1631-1643, 2013. http://dx.doi.org/10.1038/cdd.2013.77
- J.R. Skaar, J.K. Pagan, and M. Pagano, "SnapShot: F Box Proteins I", Cell, vol. 137, pp. 1160-1160.e1, 2009. http://dx.doi.org/10.1016/j.cell.2009.05.039
- G.P. Ilyin, M. Rialland, D. Glaise, and C. Guguen-Guillouzo, "Identification of a novel Skp2-like mammalian protein containing F-box and leucine-rich repeats.", FEBS letters, 1999. http://www.ncbi.nlm.nih.gov/pubmed/10508920
- C.C. Zhu, and R.J. Wojcikiewicz, "Ligand binding directly stimulates ubiquitination of the inositol 1, 4,5-trisphosphate receptor.", The Biochemical journal, 2000. http://www.ncbi.nlm.nih.gov/pubmed/10839985
- B.B. Chen, T.A. Coon, J.R. Glasser, and R.K. Mallampalli, "Calmodulin Antagonizes a Calcium-Activated SCF Ubiquitin E3 Ligase Subunit, FBXL2, To Regulate Surfactant Homeostasis", Molecular and Cellular Biology, vol. 31, pp. 1905-1920, 2011. http://dx.doi.org/10.1128/MCB.00723-10
- C. Wang, M. Gale, B.C. Keller, H. Huang, M.S. Brown, J.L. Goldstein, and J. Ye, "Identification of FBL2 As a Geranylgeranylated Cellular Protein Required for Hepatitis C Virus RNA Replication", Molecular Cell, vol. 18, pp. 425-434, 2005. http://dx.doi.org/10.1016/j.molcel.2005.04.004
- B.B. Chen, J.R. Glasser, T.A. Coon, and R.K. Mallampalli, "F-box protein FBXL2 exerts human lung tumor suppressor-like activity by ubiquitin-mediated degradation of cyclin D3 resulting in cell cycle arrest", Oncogene, vol. 31, pp. 2566-2579, 2011. http://dx.doi.org/10.1038/onc.2011.432
- B.B. Chen, J.R. Glasser, T.A. Coon, C. Zou, H.L. Miller, M. Fenton, J.F. McDyer, M. Boyiadzis, and R.K. Mallampalli, "F-box protein FBXL2 targets cyclin D2 for ubiquitination and degradation to inhibit leukemic cell proliferation", Blood, vol. 119, pp. 3132-3141, 2012. http://dx.doi.org/10.1182/blood-2011-06-358911
-
L. Li, D. Pan, H. Chen, L. Zhang, and W. Xie, "F‐box protein
FBXL 2 inhibits gastric cancer proliferation by ubiquitin‐mediated degradation of forkhead box M1", FEBS Letters, vol. 590, pp. 445-452, 2016. http://dx.doi.org/10.1002/1873-3468.12071 - B.B. Chen, J.R. Glasser, T.A. Coon, and R.K. Mallampalli, "Skp-cullin-F box E3 ligase component FBXL2 ubiquitinates Aurora B to inhibit tumorigenesis", Cell Death & Disease, vol. 4, pp. e759-e759, 2013. http://dx.doi.org/10.1038/cddis.2013.271
- A. Bononi, C. Giorgi, S. Patergnani, D. Larson, K. Verbruggen, M. Tanji, L. Pellegrini, V. Signorato, F. Olivetto, S. Pastorino, M. Nasu, A. Napolitano, G. Gaudino, P. Morris, G. Sakamoto, L.K. Ferris, A. Danese, A. Raimondi, C. Tacchetti, S. Kuchay, H.I. Pass, E.B. Affar, H. Yang, P. Pinton, and M. Carbone, "BAP1 regulates IP3R3-mediated Ca2+ flux to mitochondria suppressing cell transformation", Nature, vol. 546, pp. 549-553, 2017. http://dx.doi.org/10.1038/nature22798
- D.E. Jensen, M. Proctor, S.T. Marquis, H.P. Gardner, S.I. Ha, L.A. Chodosh, A.M. Ishov, N. Tommerup, H. Vissing, Y. Sekido, J. Minna, A. Borodovsky, D.C. Schultz, K.D. Wilkinson, G.G. Maul, N. Barlev, S.L. Berger, G.C. Prendergast, and F.J. Rauscher, "BAP1: a novel ubiquitin hydrolase which binds to the BRCA1 RING finger and enhances BRCA1-mediated cell growth suppression.", Oncogene, 1998. http://www.ncbi.nlm.nih.gov/pubmed/9528852
- J.R. Testa, M. Cheung, J. Pei, J.E. Below, Y. Tan, E. Sementino, N.J. Cox, A.U. Dogan, H.I. Pass, S. Trusa, M. Hesdorffer, M. Nasu, A. Powers, Z. Rivera, S. Comertpay, M. Tanji, G. Gaudino, H. Yang, and M. Carbone, "Germline BAP1 mutations predispose to malignant mesothelioma", Nature Genetics, vol. 43, pp. 1022-1025, 2011. http://dx.doi.org/10.1038/ng.912
- J. Xu, Y. Kadariya, M. Cheung, J. Pei, J. Talarchek, E. Sementino, Y. Tan, C.W. Menges, K.Q. Cai, S. Litwin, H. Peng, J. Karar, F.J. Rauscher, and J.R. Testa, "Germline Mutation of Bap1 Accelerates Development of Asbestos-Induced Malignant Mesothelioma", Cancer Research, vol. 74, pp. 4388-4397, 2014. http://dx.doi.org/10.1158/0008-5472.CAN-14-1328
- A. Bononi, H. Yang, C. Giorgi, S. Patergnani, L. Pellegrini, M. Su, G. Xie, V. Signorato, S. Pastorino, P. Morris, G. Sakamoto, S. Kuchay, G. Gaudino, H.I. Pass, A. Napolitano, P. Pinton, W. Jia, and M. Carbone, "Germline BAP1 mutations induce a Warburg effect", Cell Death & Differentiation, vol. 24, pp. 1694-1704, 2017. http://dx.doi.org/10.1038/cdd.2017.95
- E.R. Chemaly, L. Troncone, and D. Lebeche, "SERCA control of cell death and survival", Cell Calcium, vol. 69, pp. 46-61, 2018. http://dx.doi.org/10.1016/j.ceca.2017.07.001
- N.T.Q. Doan, E.S. Paulsen, P. Sehgal, J.V. Møller, P. Nissen, S.R. Denmeade, J.T. Isaacs, C.A. Dionne, and S.B. Christensen, "Targeting thapsigargin towards tumors", Steroids, vol. 97, pp. 2-7, 2015. http://dx.doi.org/10.1016/j.steroids.2014.07.009
- S. Kang, R. Dahl, W. Hsieh, A. Shin, K.M. Zsebo, C. Buettner, R.J. Hajjar, and D. Lebeche, "Small Molecular Allosteric Activator of the Sarco/Endoplasmic Reticulum Ca2+-ATPase (SERCA) Attenuates Diabetes and Metabolic Disorders", Journal of Biological Chemistry, vol. 291, pp. 5185-5198, 2016. http://dx.doi.org/10.1074/jbc.M115.705012
ACKNOWLEDGMENTS
The research activities led by M.C. are supported by the following funders, who are gratefully acknowledged: The Biotechnology and Biological Sciences Research Council; the London Interdisciplinary Doctoral Training Programme (LiDO); The Petplan Charitable Trust and the LAM-Bighi Grant Initiative. The research activities led by G.B. are supported by the following funders, who are gratefully acknowledged: Scientific Research – Flanders (FWO), Research Council – KU Leuven and Stichting Tegen Kanker.
COPYRIGHT
© 2017

Tumor suppressive Ca2+ signaling is driven by IP3 receptor fitness by Bultynck and Campanella is licensed under a Creative Commons Attribution 4.0 International License.


