Reviews:
Cell Stress, Vol. 1, No. 3, pp. 115 - 133; doi: 10.15698/cst2017.12.114
A game of substrates: replication fork remodeling and its roles in genome stability and chemo-resistance

1 Department of Pathology, University of Washington, Seattle, Washington, USA.
Keywords: DNA, replication fork, S phase, end resection, human, vertebrate, chemotherapy.
Abbreviations:
ds – double-stranded
DSB – double strand break,
HR – homologous recombination,
HU – hydroxyurea,
IR – ionizing radiation,
NHEJ – non-homologous end joining,
ss – single-stranded.
Received originally: 18/09/2017 Received in revised form: 27/11/2017
Accepted: 29/11/2017
Published: 05/12/2017
Correspondence:
Julia Sidorova, Department of Pathology, P.O. Box 357705, 1959 NE Pacific St., Seattle, WA 98155; phone (206) 221-4237 julias@uw.edu
Conflict of interest statement: The author declares no conflict of interest related to the writing of this review.
Please cite this article as: Julia Sidorova (2017). A game of substrates: replication fork remodeling and its roles in genome stability and chemo-resistance. Cell Stress 1(3): 115-133. doi: 10.15698/cst2017.12.114
Abstract
During the hours that human cells spend in the DNA synthesis (S) phase of the cell cycle, they may encounter adversities such as DNA damage or shortage of nucleotides. Under these stresses, replication forks in DNA may experience slowing, stalling, and breakage. Fork remodeling mechanisms, which stabilize slow or stalled replication forks and ensure their ability to continue or resume replication, protect cells from genomic instability and carcinogenesis. Fork remodeling includes DNA strand exchanges that result in annealing of newly synthesized strands (fork reversal), controlled DNA resection, and cleavage of DNA strands. Defects in major tumor suppressor genes BRCA1 and BRCA2, and a subset of the Fanconi Anemia genes have been shown to result in deregulation in fork remodeling, and most prominently, loss of kilobases of nascent DNA from stalled replication forks. This phenomenon has recently gained spotlight as a potential marker and mediator of chemo-sensitivity in cancer cells and, conversely, its suppression – as a hallmark of acquired chemo-resistance. Moreover, nascent strand degradation at forks is now known to also trigger innate immune response to self-DNA. An increasingly sophisticated molecular description of these events now points at a combination of unbalanced fork reversal and end-resection as a root cause, yet also reveals the multi-layered complexity and heterogeneity of the underlying processes in normal and cancer cells.
INTRODUCTION
The three billion base pairs of the human genome are replicated in about six to eight hours of the S phase of the cell cycle. At the height of S phase, there are hundreds of replication forks running through chromosomes, each carrying on its three-way DNA junction anywhere from dozens to perhaps low hundreds of proteins that service every aspect of chromatin duplication. When replication forks experience disruptions to their normal mode of operation, which manifests as forks’ stalling or slowing, the condition is referred to as replication (or replicative) stress. Among classic causes of replication stress are DNA damage during S phase and unbalanced or reduced levels of intracellular nucleotide pools, both of which can be elicited by exogenous agents such as chemotherapy drugs. Other recognized causes include conflicts with transcription, epigenetic abnormalities, and defective cell cycle regulation. Prolonged stalling of forks during stress can cause fork collapse, i.e. DNA damage and loss of protein machinery of the fork, which can jeopardize complete duplication of the genome. Replication stress is considered one of the major drivers of genomic instability as well as premature senescence and carcinogenesis. For in-depth review of these topics, see [1][2][3][4][5][6][7][8][9].
–
Eukaryotic cells have evolved elaborate mechanisms to buffer replication fork activity against stress. These mechanisms can be divided into two categories: those that enable bypass of damage, and those that allow stable pausing and on-demand resumption of replication once the obstacle is cleared or conditions are normalized. It is the latter group that we will focus on in this review. For additional discussion of the subject, we recommend these excellent recent reviews [10][11][12][13][14].
–
Stalled forks are remodeled
It is helpful to distinguish three processes that can occur at a stalled replication fork and involve DNA strand separation/annealing, degradation, and breakage. These are, respectively, fork reversal, resection, and collapse (for the mechanisms engaged at moving forks, i.e. translesion synthesis (TLS) and repriming, we recommend a recent review [15]). We will use the term fork remodeling to refer to these processes. Evidence suggests that reversal and resection are actively regulated, and not only are compatible with but in fact enable stabilization of forks and contribute to their ability to resist disruptions of DNA synthesis (see next section). Fork collapse may be viewed as an exhaustion or breakdown of fork remodeling over time. Likely however, it is an actively triggered pathway of last resort initiated by cleavage of one arm of a fork by the MUS81 endonuclease [16][17][18][19]. Collapsed forks may be resolved into unreplicated parental strands or repaired and reactivated [20]. Recent findings implicated Break-Induced Replication (BIR, [21]) in reactivation of collapsed replication forks in humans [22], and demonstrated MUS81-dependent BIR in S phase [19] as well as in mitosis [23][24]. In the interest of space, this review will predominantly focus on the mechanisms that preserve an active fork collapse-free. For an excellent overview on fork remodeling, see [25].
–
Reversal/restoration cycle helps to maintain forks’ ability to resume replication
Reversed forks (a.k.a. regressed forks, or “chicken feet”, Figure 1B, 2), in which two nascent strands pair with one another to form a forth branch have first been observed by electron microscopy (EM) in the preparations of genomic DNA decades ago [26]. More recently, their biological relevance came into focus with the studies from Lopes and Vindigni labs, which positively correlated presence and abundance of reversed forks in human genomic DNA with the ability of forks to resume replication in vivo after treatment with several different fork-interfering agents [27]. Lopes lab demonstrated that the RAD51 protein, a single-stranded (ss) DNA-binding RecA homolog of eukaryotes first known for its strand exchange activity in homologous recombination, is required for fork reversal [28] (for a recent review of RAD51, see [29]). RAD51 is also required for resumption of replication by stalled forks, which supports the idea that reversal preserves forks’ activity [30][31]. RAD51 probably does not execute reversal but rather, stabilizes the reversed fork [32]. However, several proteins can directly stimulate reversal of forks (Table 1). In vitro, the RAD54 DNA translocase [32][33], RECQ helicases WRN and BLM [34], and the FANCM helicase [35] showed this activity. The proteins FBH1, SMARCAL1, ZRANB3, and HLTF, as well as the MMS22L/TONSL complex were shown to reverse forks both in vitro and in vivo [19][36][37][38][39][40][41][42][43][44].
–
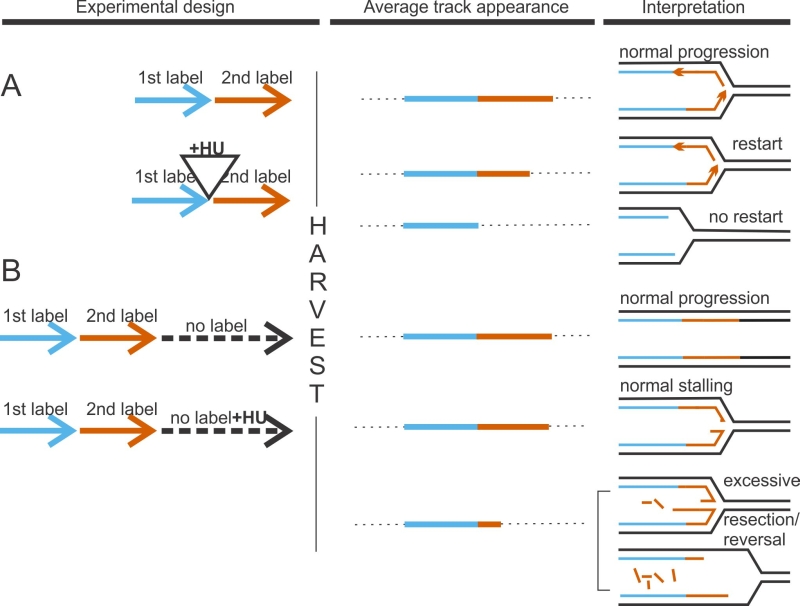 | FIGURE 1: DNA fiber analysis detects faulty fork activity. In a “classic” experiment to measure fork restart (A), cells are sequentially pulse-labeled for 15-30 min each with two nucleoside analogs, typically, chloro-deoxyuridine and iodo-deoxyuridine. In test samples, incubation with a fork-arresting dose of hydroxyurea (HU), a ribonucleotide reductase inhibitor, is introduced between the labels. Failure to restart after HU manifests as preponderance of tracks containing only 1st label. One way to measure degradation of nascent strands at stalled forks (B), is to follow sequential pulse-labeling with two labels by a label-free chase (usually 4-6hrs). In test samples, HU is introduced during this time. Extensive loss of nascent DNA manifests as shortening of 2nd label segments in tracks labeled prior to HU addition. |
–
FBH1 is a 3’-5’ DNA helicase capable of targeting its binding partners for ubiquitination [45]. SMARCAL1 and ZRANB3, two related members of the SNF2 family of ATP-dependent DNA helicase-like proteins, as well as their more distant relative HLTF, are required for stalled fork restart (see Figure 1A and BOX1 for technical background) [36][46][47][48]. SMARCAL1 is mutated in in a subset of Schimke’s Immunoosseous Dysplasia (SIOD) cases, and this mutation also abolishes SMARCAL1 fork reversal activity in vitro and triggers fork collapse and DNA damage in vivo, further supporting the notion that fork reversal is a protective mechanism [47][49]. SMARCAL1, ZRANB3, and HLTF show differential fork substrate preferences with regard to the presence and location of ssDNA gaps, and are hypothesized to have non-redundant roles in the cell. Also, only SMARCAL1 and ZRANB3 but not HTLF are able to perform the reaction opposite to fork reversal, i.e. fork restoration. For further detail on the roles of these proteins at forks, see Table 1 and [50][51]; also, Figure 3.
–
TABLE 1. Proteins capable of fork reversal and/or restoration. |
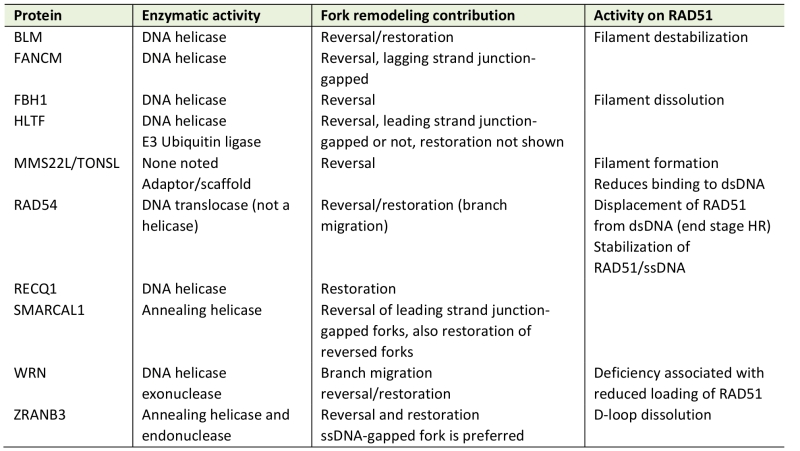 |
See main text for relevant references. |
–
While failure to reverse forks has deleterious consequences for fork activity and genome stability overall, studies find that excessive fork reversal, e.g. due to overexpression of SMARCAL1 or RAD51, can also result in DNA damage and threaten genome stability [52][53][54][55]. This suggests a notion that fork reversal is a “double-edged sword”, and needs to be balanced. Indeed, an additional control over fork reversal has been identified in poly(ADP-ribose)-polymerase 1 (PARP1) and the RECQ1 helicase. PARP1-dependent PARylation of DNA and proteins is involved in coordinating responses to DNA damage and replication disruptions [56][57]. Activated PARP1 was shown to stimulate accumulation of reversed forks in vivo [58]. The mechanism behind this phenotype was elucidated by Vindigni and colleagues, who showed that PARP1 does not directly promote fork reversal. Instead, it inhibits the RECQ helicase family member RECQ1, whose function is to preferentially restore reversed forks to their original three-armed configuration in vitro and in vivo [59]. In particular, under conditions of fork arrest/slowing, an increase in reversed forks was suppressed by inhibition of PARP1, but strikingly, not in the absence of RECQ1. Taken together, the data suggest that the duration and extent of fork reversal in vivo is orchestrated by counteracting activities and is regulated by a master-switch PARP1 that prevents premature restoration of a reversed fork.
–
Nascent strand resection at stalled forks
Schlacher and colleagues in the Jasin lab were the first to put degradation of nascent DNA at stalled forks in the spotlight [60][61]. Using DNA fiber assays, they observed loss of kilobases of newly-synthesized DNA from forks stalled by the nucleotide depleting drug hydroxyurea (HU), which manifested as pronounced shortening of replication tracts laid down immediately prior to fork arrest (Figure 1B and BOX1). This phenotype was present in cells defective for breast cancer susceptibility genes BRCA1 and BRCA2, or Fanconi Anemia genes FANCD2, or FANCA, but not in the wild type cells. The authors implicated the MRE11 nuclease known for its role in DNA end resection during double strand break (DSB) repair, in the loss of nascent DNA at forks. Moreover, they provided evidence that nucleolytic attack by MRE11 was upregulated without BRCA2 because no ssDNA/RAD51 filament was there to protect the fork. In particular, BRCA2 C-terminal domain involved in stabilizing RAD51 filament on ssDNA was critical for preventing degradation. Overexpression of BRC4 domain of BRCA2, which binds RAD51, or of the K133R mutant of RAD51, which forms hyper-stable filaments, respectively, enhanced and suppressed nascent strand degradation.
–
 |
| [27][44][62][63][64][65][66][67][68][69][70] |
–
The C-terminus mutant of BRCA2 that failed to protect forks from degradation was able to maintain homology-dependent DNA repair, suggesting, crucially, that this function of BRCA2 is at least somewhat separate and different from its DSB repair function. Schlacher et al. thereby referred to this distinct pathway as fork protection to highlight the protective functions of RAD51 and BRCA/FANC genes. Notably, BRCA2 absence did not inhibit fork restart after HU removal, though it increased DNA damage and genomic instability associated with HU treatment. Though BRCA2 C-terminal mutants were later found defective in fork restart [42][62], it has been observed time and again that resection does not necessarily result in a fork that is unable to restart replication, e.g. see [19][63].
–
Overall, stalled fork condition emerged as a counterbalance of protective activity of RAD51 and nucleolytic activity of MRE11, a kind of a battle between a hero and an anti-hero. However, several observations complicated the picture. For example, RAD51 depletion does not always result in upregulated nascent strand degradation at stalled forks (e.g. compare [28][64][65]). MRE11, on the other hand, has been shown to have a fork protective effect: without MRE11, fork stalling is associated with increased DNA damage, and forks are less capable of resuming replication [66]. MRE11 is recruited to stalled forks by PARP1, and this is a part of the fork-protective role of PARP1 [67].
–
In addition, Hashimoto et al. [68] observed both MRE11-dependent and independent ssDNA gap accumulation in replicating DNA upon loss of RAD51 in Xenopus egg extracts. Also, a publication by the Vindigni group demonstrated nascent strand degradation in U2OS cells upon depletion of RECQ1 and with the proficient BRCA1 and BRCA2 genes [64]. This degradation was executed by the DNA2 endonuclease in conjunction with the WRN helicase and surprisingly, did not involve MRE11. Moreover, depletion of RAD51 did not exacerbate this degradation but, on the contrary, prevented it. These observations suggested a more elaborate view of fork protection, as we will discuss below.
–
Fork remodeling as interlocked cycles of reversal and resection
To begin, resection and reversal can be viewed as interlocked, mutually constraining fork remodeling processes (Figure 2). The extent of resection or reversal as well as prevalence of one over the other may vary depending on relative abundances and activities of the involved proteins. In theory at least, reversal may precede and/or follow resection.
–
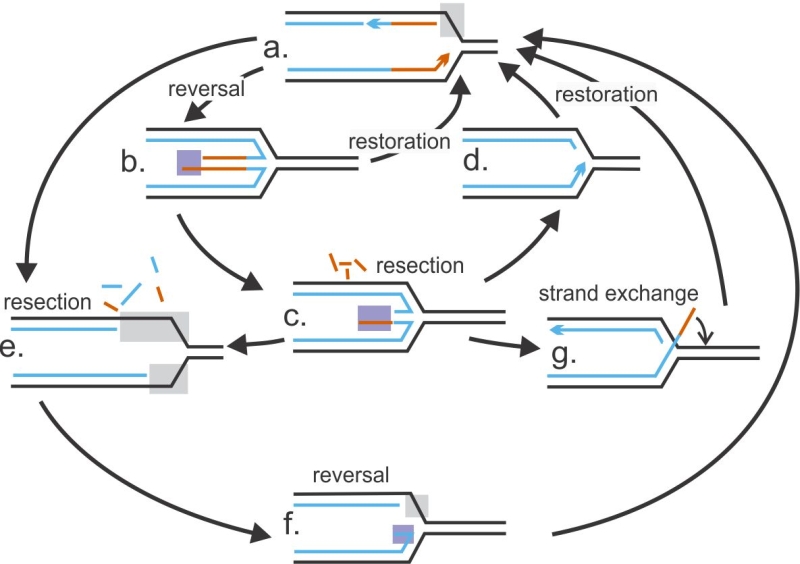 | FIGURE 2: Fork remodeling as interlocked cycles of reversal and resection. A framework to visualize remodeling of stalled forks as connected, dynamic states that are characterized by prevalence of reversal or restoration, and resection or protection. Gray rectangles mark exposure of parental ssDNA and purple rectangles – nascent ssDNA. Both can be used as markers of fork state. |
–
The timing of appearance and prevalence of single-stranded (ss)DNA at forks is a critical readout of fork reversal and resection. Parental ssDNA (gray boxes in Figure 2) can be initially exposed due to failure of DNA synthesis and excessive unwinding ahead of the fork by the replicative helicase, and then expand due to resection. Appearance of nascent ssDNA (purple boxes in Figure 2) strongly suggests reversal with subsequent resection. RAD51, MRE11, and other proteins may act either on the single-stranded portion of an extruded nascent strand duplex, i.e. the “forth” arm of a reversed fork (at structures b and c, Figure 2), or on the parental DNA over a single-stranded gap (structure e); the latter mode may be followed by reannealing of the parental duplex (structure f). Techniques such as Proximity Ligation Assay (see BOX1 for detail) can now begin to distinguish between these scenarios and will help delineate specific sequences of events in individual model systems. The order or prevalence of reversal versus resection may be the reason the phenotype of RAD51 deficiency can be either protective or sensitizing for resection. That is, RAD51 depletion will suppress resection if RAD51 acts only at reversal stage (i.e. at a →b path in Figure 2), and resection is strictly dependent on prior reversal (i.e. a →e path is not active) [19][69][64]. In the next sections we will use this framework as a guide to undertake an in-depth analysis of the rapidly growing body of data on the interplay between fork resection and reversal.
THE KEY PLAYERS AND PROCESSES
Figure 3 lists the proteins that contribute to remodeling of stalled forks. Some of these proteins were already mentioned in the previous sections. These and other proteins will be the subject of the discussion below.
–
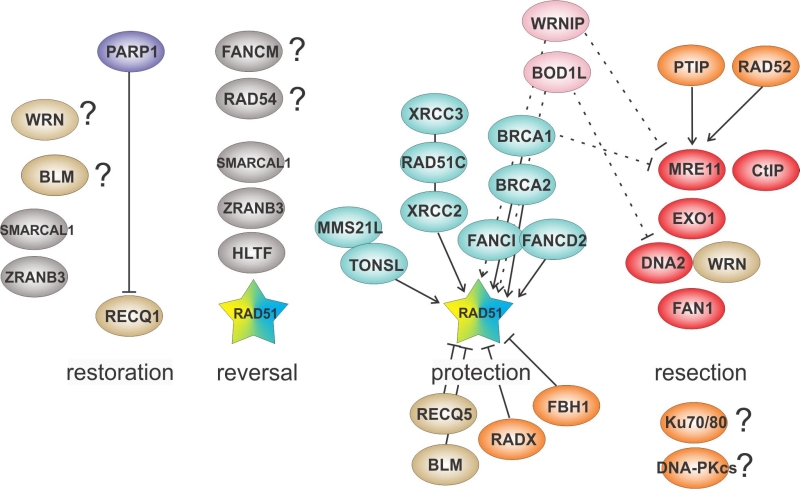 | FIGURE 3: An overview of proteins that mediate fork reversal/restoration and resection/protection. Functional relationships between proteins are indicated by solid lines, and dashed lines indicate likely inferences. |
–
MRE11 and other nucleases
Historically, many proteins that are active on stalled forks were first recognized for their roles in the repair of two-ended DSBs in DNA. MRE11 is no exception. Much of what is known about MRE11 at the molecular level comes from the studies of DSB end processing in the context of competition between homologous recombination (HR)-mediated repair and non-homologous end joining (NHEJ). MRE11’s ssDNA endonuclease activity initiates resection by introducing a nick internal to a DSB end. The nicked strand is then degraded in the 5’-3’ direction by the EXO1 exonuclease and in the 3’-5’ direction by MRE11’s exonuclease activity [70][71], which is the activity inhibited by the commonly used MRE11 inhibitor mirin. For long-range resection, MRE11 and EXO1 are joined by the DNA2 endonuclease assisted by the RECQ helicases BLM or WRN [72]. Neither EXO1 nor DNA2 complexes are strictly dependent on MRE11 for their recruitment to DSBs. Resection exposes a ssDNA 3’ overhang, onto which RAD51 is loaded for subsequent homology search and strand exchange. For further detail, see [73][74].
–
The extent of contribution of MRE11 exonuclease to resection appears to vary between experimental systems [75][76]. A recent report suggests that MRE11 exonuclease is a minor contributor to resection but, along with CtIP, is key to displacing Ku70/80 from DNA ends [77]. Without such displacement, RAD51 loading onto ssDNA is reduced [77], and processing of DNA ends by EXO1 is blocked [75]. In sum, MRE11 commits a DSB to resection, counteracts resection inhibitors such as Ku70/80, but also facilitates loading of RAD51 on ssDNA.
–
At replication forks, the forth arm that forms upon fork reversal bears resemblance to a one-ended DSB, and is expected to be a substrate for resection in a manner analogous to the above. Some evidence is also consistent with MRE11 targeting nascent DNA ends that flank ssDNA gaps within newly replicated parental/daughter DNA duplexes [42]. In contrast to a DSB however, a stalled fork may not require MRE11’s non-redundant function as an initiator of resection, because it already contains ssDNA nicks originating from the Okazaki fragments. In addition, unlike at DSBs, other resection-capable proteins, i.e. DNA2, BLM, and WRN are already present at forks, assisting in normal replication [16][31][78][79][80][81]. These functions may help explain why MRE11 may have a negligible input into resection at stalled forks at least in some cases [69][64]. On the other hand, unlike at DSBs, MRE11, which is not capable of performing long-range resection, may be able to degrade more DNA at forks during repeated cycles of resection – reversal – resection.
–
BRCA1 and BRCA2: many roles versus one?
At DSBs, BRCA1 both promotes and limits strand resection (for review, [82][83][84]. On the one hand, it assists in loading MRE11 and stimulating its endonuclease activity via CtIP [85]. BRCA1 also facilitates recruitment of EXO1 and suppresses DNA-PK activation [86]. One the other hand, one of BRCA1 complexes, BRCA1-A, also appears to inhibit resection once it has exceeded a certain distance from the original break site. Since MRE11’s role in initiating resection at forks may be dispensable, it is the restraining function of BRCA1 that may be prominent, explaining the increased resection and a higher level of MRE11 on nascent DNA in BRCA1-deficient cells. Lastly, via PALB2, BRCA1 also aids BRCA2 in loading RAD51 onto single stranded DNA [87]. The importance of BRCA1 at forks is underscored by a recent finding that BRCA1 is haploinsufficient for suppressing nascent strand degradation at forks [88].
–
Compared to many facets of BRCA1, the role of BRCA2 at forks seems straightforward enough: along with PALB2 and RAD51 paralogs it directs formation of the RAD51/ssDNA filament. BRCA2 also discourages binding of RAD51 to dsDNA [89][90], which may buffer RAD51 activity, since excess RAD51 and its resulting association with dsDNA may be inhibitory to strand exchange. Even so, some loading of RAD51 may occur independently of BRCA2, for example by MMS22L/TONSL [40]. Recent findings in the Xenopus egg extract system indicate that BRCA2 loss is associated with ssDNA gaps at and behind the forks even in the absence of exogenous fork-arresting agents [42]; and slowed fork progression was noted in a BRCA2-deficient human cell line [91]. It is possible that these defects in fact predispose forks in BRCA2-deficient cells to reversal upon stalling.
–
It should be noted that recent work also points at mechanistically important differences between BRCA1 and BRCA2 in the ways these proteins contribute to the fate of forks beyond the protection/resection cycle. BRCA2 deficiency has been associated with increased MUS81-mediated fork cleavage followed by BIR in S phase (during recovery from HU-mediated stalling) [19], or in mitosis (during an unperturbed S phase) [91]. On the other hand, BRCA1 deficiency was shown to suppress mitotic MUS81-coupled BIR after fork stalling [92]. These differences will have to be incorporated into the field’s developing view of BRCA-deficient cell survival and mutagenesis upon replication stress.
–
RAD51 versus the nucleases: reversal, protection, resection?
RAD51 is recruited to a stalled fork and is one of the factors that reverses it or stabilizes an already reversed fork [28]. Appearance of RAD51 at a stalled fork occurs at about the same time if not slightly before MRE11 [93]. MRE11 recruitment to a fork is promoted by PARP1 that also promotes fork reversal [59][67]. Thus, it is a reversed fork that can be a substrate for nucleolytic degradation by MRE11. In this scheme RAD51 is therefore upstream of MRE11 and provides a substrate for it. At the same time, RAD51 can limit the activity of MRE11 and other nucleases on this substrate, and if RAD51 is not there, the reversed fork is over-digested to the extent that a regressed arm disappears. Therefore, RAD51 can also act downstream of MRE11, and it is in this sense that MRE11 provides a substrate for RAD51, which agrees with the finding that inhibition of MRE11 exonuclease activity can reduce the amount of RAD51 loaded at HU-stalled forks [93]. Several new papers now provide strong evidence in support of this model. Using a Xenopus egg extract system [42] or several human cell lines [19][43][44] four labs have shown that upon depletion of BRCA2 or BRCA1, EXO1 and MRE11 non-redundantly degraded nascent DNA at forks, and strikingly, this degradation could be alleviated by reducing fork reversal. This was achieved when RAD51, or the SNF2 family members SMARCAL1, ZRANB3, or HLTF, were depleted or mutated. Thus, at least in the context of BRCA1/2 inactivation, reversed forks are a predominant substrate for MRE11 and EXO1 exonucleolytic degradation of nascent DNA and RAD51 is critical for fork reversal.
–
The dual role of RAD51 before/during and after fork reversal was supported by two observations. Mijic et al. found that the T131P mutation of RAD51 that prevents it from forming a stable nucleofilament [94] is a separation of function mutation: it retains RAD51’s ability to promote/stabilize fork reversal but fails to protect reversed forks from MRE11 [43]. Taglialatela et al. observed that B02 inhibitor of RAD51, which blocks its binding to DNA [95], elicited increased nascent strand degradation that was suppressed by depleting SMARCAL1 [44]. This suggested that B02 blocked fork protecting function of RAD51 downstream of fork reversal.
–
In sum, there is now a very strong case for a reversed fork as an entryway for nascent strand degradation by MRE11 and EXO1 (Figure 3). That said, the possibility that nascent strands can also be degraded in non-reversed forks by MRE11 or other nucleases is not ruled out, particularly for the case of BRCA1/2-proficient cells. If so, such nucleolytic activity will expose parental ssDNA that can serve as a substrate for RAD51, as it does during DSB repair. The final resolution of the question will require data on when parental and/or nascent ssDNA accumulate during fork remodeling, and which of these ssDNA are bound by RAD51, MRE11, and other proteins.
–
The nuclease crosstalk and the curious case of WRN
As we have seen, MRE11 and EXO1 are demonstrably active in fork resection in the BRCA1/2-deficient backgrounds. In contrast, Lemacon et a.l showed that DNA2 is not involved in BRCA1 or BRCA2-deficient cells [19]. Similarly, we found that in a BRCA1-deficient background of an ovarian cancer cell line, depleting WRN does not suppress upregulated resection, indicating that WRN is not involved in it (Sidorova, unpublished). Chaudhuri et al. made a similar observation in BRCA2-deficient cells, though also found that DNA2 was in fact involved (perhaps assisted by another RECQ helicase), and MRE11 and DNA2 were epistatic with respect to nascent strand degradation, suggesting a crosstalk between these nucleases [96].
–
On the other hand, DNA2/WRN and not MRE11 or EXO1 carried out resection in cases where nascent strand degradation was upregulated in BRCA-proficient cells [64]. Together, these findings suggest a possibility that BRCA1/2 status may influence involvement of the nucleases that degrade nascent DNA.
–
A study by Iannascoli et al. adds a helpful hint to this puzzle [69]. In particular, the WRN exonuclease-dead mutant E84A but not helicase or null mutants appears to trigger nascent strand degradation in BRCA-proficient cells and accumulates excess nascent ssDNA in SV40-transformed human fibroblasts (consistent with a reversed fork structure c in Figure 2). To our knowledge, this is the demonstration of restrained resection limited to the reversed arm. Moreover, both nascent ssDNA and nascent strand degradation phenotypes were suppressed by depletion of MRE11 (or EXO1) or by inhibition of MRE11 exonuclease activity. One possibility suggested by these data is that there is a regulatory link connecting DNA2/WRN to MRE11, EXO1 at stalled forks. Here it is worth mentioning that WRN has been associated with promoting RAD51 binding to stalled forks in two studies [63][97].
–
Based on the data reviewed in this and previous sections, we therefore hypothesize that there are two, potentially interconnected, pathways that are capable of resecting nascent DNA at reversed forks (Figure 4). Their relative prevalence may depend on the BRCA status and/or yet unknown genetic determinants that define the balance between fork reversal/restoration. Once DNA2/WRN are engaged, this inhibits MRE11 and EXO1. This model thus depicts a scenario very different from the HR-mediated DSB repair and will require the support of additional data for its validation.
–
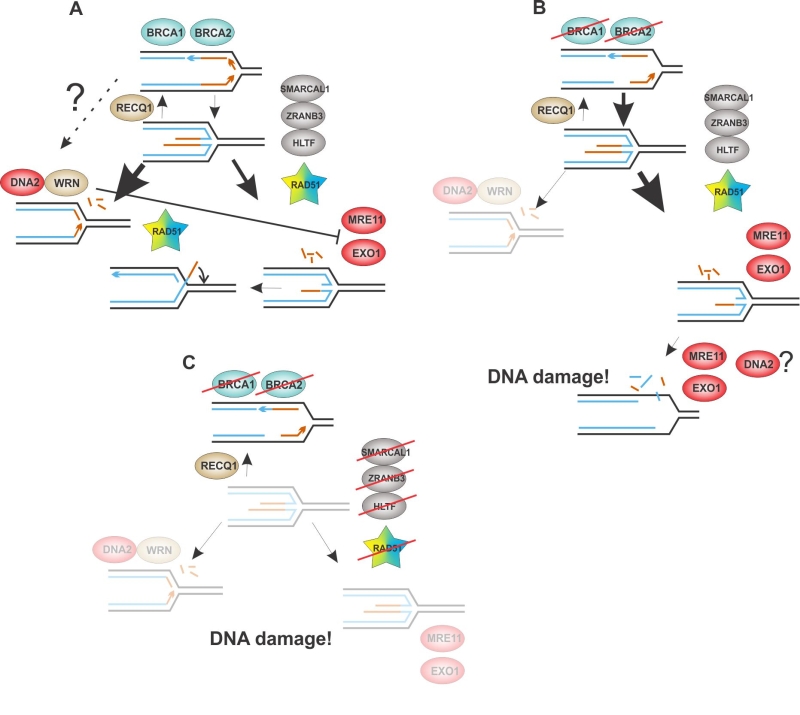 | FIGURE 4: A model for two interlinked branches of fork resection and their alterations upon mutational loss of the involved proteins. In a wild type cell the MRE11, EXO1 branch may be less prevalent than the DNA2/WRN branch, depending on the cell background. An inhibitory feedback from WRN to MRE11 may contribute to this arrangement. Fork resection may be limited by restorative branch migration of the fork junction, or reannealing of the 3’ overhang in a D-loop-like strand exchange (A). In BRCA1 or BRCA2-defective cells, the MRE11, EXO1 branch dominates and WRN is no longer a contributor. Fork resection proceeds beyond the four-way junction, either directly, or via cycles of reversal/resection, leading to DNA breakage either directly or via segregation of under-replicated DNA (B). Inhibition of fork reversal suppresses fork resection but also decreases stalled fork stability and ability to resume replication. DNA damage may result from fork collapse and segregation of under-replicated DNA and is exacerbated by BRCA1 or BRCA2 deficiency (C). Faded areas in the cartoon indicate pathway inactivity. See text of the sections “RAD51 versus the nucleases: reversal, protection, resection?” and “The nuclease crosstalk and the curious case of WRN” for a detailed discussion. |
FACILITATOR AND RESTRAINER PROTEINS ADD A LEVEL OF COMPLEXITY TO FORK REMODELING
In this section we will review the protein factors that facilitate or counteract RAD51 and MRE11 at stalled forks, and consider what these proteins can tell us about the dynamic interplay of reversal/restoration and protection/resection processes during fork remodeling, and their potential plasticity in different cells.
–
PTIP and RAD52 as facilitators of MRE11
In DSB repair, PTIP works as a facilitator of NHEJ counteracted by BRCA1 [98]. A recent report suggests that at DSBs PTIP recruits Artemis (DCLRE1C), an endonuclease that cleaves DNA at ss/dsDNA junctions and can trim 5’-3’ overhangs and well as attack ss gaps, generating DNA ends compatible with NHEJ [99]. Consistent with these findings, loss of PTIP in BRCA1-deficient cells renders them PARP inhibitor- and cisplatin-resistant and partially rescues end resection at DSBs [98]. Remarkably however, at stalled forks loss of PTIP suppresses nascent strand degradation in BRCA1 or BRCA2-deficient backgrounds, i.e. counteracts resection, and decreases fork-bound MRE11 [96]. Of note, loss of PTIP does not suppress fork reversal [43].
–
Understanding how PTIP performs its diverse functions at DSBs and stalled forks will require additional data. One possibility is that if PTIP facilitates recruitment of both MRE11 and Artemis at DSBs, it may thus restrain MRE11 at the stage of initiating resection. If so, this restraining function may be irrelevant at forks because, once again, the nick and gap-rich fork structure obviates the need for MRE11 as resection-initiator. On the other hand, Artemis may exacerbate nucleolysis at forks. In vitro, DNA-PKcs enables Artemis to cleave dsDNA [100], and it was shown to cleave stalled forks after prolonged arrest [101]. An alternative explanation for PTIP’s role at forks is that it may be more important as a recruiter of the histone methyltransferase MLL3/4, which generates histone H3 trimethylated on Lysine 4.
–
RAD52 is known for its involvement in the HR-mediated DSB repair, and becomes critical for cell survival in the absence of BRCA1/2 (for review, see [102]). In vitro, it can anneal ssDNA, facilitate strand exchange, and, under some conditions, stimulate RAD51 (ibid.) Remarkably, Mijic et al. showed that loss of RAD52 in BRCA2-deficient cells has a phenotype similar to PTIP loss: nascent strand resection at stalled forks is suppressed while fork reversal is unaffected. Moreover, without RAD52 less MRE11 is bound at stalled forks [43].
–
Restrainers of RAD51
The list of proteins whose role involves upregulating or downregulating the level of RAD51 at stalled forks in vivo and/or fork substrates in vitro continues to grow (Figure 3). Some of these proteins directly act on RAD51, while the role of others is more indirect – for example, they may compete with RAD51 for the same substrates.
–
RECQ helicases RECQ5 and BLM were perhaps the first to be demonstrated to disassemble RAD51/ssDNA filament in vitro [103][104][105][106], albeit RECQ5 inhibits RAD51 strand invasion (D-loop), while BLM promotes it. Interestingly, RECQ5 may be recruited by MRE11 [107]. However, RECQ5 and BLM are not known to affect nascent strand degradation, suggesting that these proteins may be employed at clearing RAD51 during fork restoration and restart.
–
FBH1, an F-box-containing 3’-5’ DNA helicase, was also shown to displace RAD51 from ssDNA in vitro [108]. Some in vivo activities of FBH1 are consistent with this role of a RAD51 restrainer: disruption of FBH1 has antirecombinogenic phenotype (ibid.), and also suppresses the phenotypes of two facilitators of fork protection, WRNIP and BOD1L (see below). However, FBH1 also reverses forks [38], thereby working in concert with RAD51, and promotes DSB formation at collapsed forks along with MUS81 [17], thereby terminating the processes associated with fork stabilization and recovery. A recent work showed that FBH1 participates in ubiquitination of RAD51, which leads to its degradation [109]. Assuming all these functions are distinct, further studies are needed to understand how they are selectively utilized in vivo.
–
PARI is another protein that can disrupt RAD51/DNA association in vitro and has anti-recombinogenic phenotype in vivo [110]. A novel ssDNA binding protein, RADX, was identified recently as another restrainer of RAD51, which likely competes with RAD51 for ssDNA [55]. Inactivation of RADX suppresses nascent strand degradation in BRCA2-deficient cells, suggesting that BRCA2/RADX double deficient cells may load enough RAD51 onto forks. RADX likely binds at reversed forks because suppression of reversal (by depletion of ZRANB3 or SMARCAL1) also suppresses phenotypes of RADX loss.
–
Facilitators of RAD51
Constraints on RAD51 activity are counterbalanced by several proteins that either load RAD51 independently of BRCA2, or protect loaded RAD51. In Chinese hamster cells, deficiency in RAD51 paralogs RAD51C, XRCC2, or XRCC3 upregulates nascent strand degradation and exposes excess of parental DNA in single stranded form at stalled forks [111]. Notably, deficiency in RAD51C and XRCC2 was additive with BRCA2 deficiency in nascent strand degradation, suggesting a BRCA2-independent contribution to RAD51 loading.
–
Fanconi Anemia proteins FANCD2 and FANCI likely enable RAD51 by protecting it on DNA. FANCD2 deficiency results in excessive nascent strand degradation at HU-arrested forks [61]. In vitro, FANCD2/FANCI can stabilize RAD51/ssDNA filament and protect the ds/ss DNA junction behind the 3’ ssDNA overhang from nucleolysis [112]. Work by Sobeck lab, using DNA polymerase inhibitor aphidicolin (APH) as means to arrest replication, suggests that the role of FANCD2 is also to control the activity of the structure-specific endonuclease/5’-3’ exonuclease FAN1 that is recruited to APH-arrested forks and can contribute to excessive resection of these forks in FANCD2-deficient cells [113]. Interestingly, FAN1 recruitment to forks was facilitated by MRE11 and FANCD2. Thereby, FANCD2 contributes to both bringing a nuclease to forks and limiting nucleolytic attack on forks.
–
Facilitators of fork protection
The following proteins contribute positively to fork protection, however, it is not yet clear, whether they facilitate RAD51 and/or restrain the nucleases.
–
In the absence of BOD1L, nascent strand degradation at forks stalled by HU is upregulated, as are the levels of parental ssDNA, albeit assayed using a different replication-stressing agent, mitomycin C [114][115]. Upregulated nascent strand degradation without BOD1L is epistatic with BRCA1 or BRCA2 absence, suggesting that these genes are acting in the same pathway and/or on the same substrate. Furthermore, upregulated strand degradation (and ssDNA formation) upon BODL1-deficiency is partially suppressed by depletion of FBH1 and BLM, suggesting that BOD1L counteracts anti-RAD51 activity of these two proteins. Surprisingly however, depletion of DNA2 but not MRE11 or EXO1, suppresses the nascent strand degradation phenotype of BOD1L loss. BOD1L does not have known enzymatic activities.
–
Depletion of the ATPase WRNIP1 known for its association with WRN, generates a phenotype very similar to that of BOD1L with respect to its interactions with BRCA2 and FBH1 [116]. The level of parental ssDNA is also elevated in WRNIP1-depleted cells, though less RAD51 is bound to it. Remarkably however, unlike BOD1L, WRNIP1’s upregulated nascent strand degradation is suppressed by inactivation of MRE11. These findings may suggest that BOD1L and WRNIP1 belong respectively to DNA2/WRN-dependent and MRE11-dependent branches of fork resection (Figures 3, 4). Addressing this will require further research.
–
The list of proteins that influence RAD51 binding and/or modulate resection will likely continue to grow. Among new additions are REV1 [117], which protects from nascent strand degradation and may be recruited by monoubiquitinated FANCD2; and the kinase NEK8 [118]. Another group of contributing proteins that should be mentioned are those that enable targeted degradation of the key participants, e.g. RAD51 [119], WRN [120], EXO1 [121], and others (reviewed in [122]).
–
How does it all come together? Toward an integrated view of fork remodeling
The overview of the proteins engaged in stalled fork remodeling suggests that many of them either contribute to both sides of the “standoff” between DNA protectors and DNA degraders, or are subject to counteracting activities of other proteins. In a system with so many moving parts, the exact balance between fork reversal, protection, and resection may depend on the sub-nuclear distribution, mutual ratios, and relative activity of the proteins involved, which may vary with the genetic and epigenetic background. The nature of the experimental perturbation may also play into this. For example, the level of RAD51 may influence how stringently fork reversal depends on it, while the degree of RAD51 depletion may affect its ultimate phenotype at forks. That is, only a complete loss of RAD51 may abolish fork reversal while partial depletion will only disrupt its function in fork protection.
–
It should be appreciated that most of the cell lines used in fork remodeling studies are transformed or cancer-derived, and fork remodeling in cancer cells may be already rewired or relaxed under the pressure of chronic replication stress. For example, lines HCT116 and U2OS exhibit baseline nascent strand degradation [64], unlike primary fibroblasts [63]. Genomic analysis of the U2OS line found potentially non-neutral SNVs in several genes [123] and in particular, in AND-1/WDHD1, which has roles in replication and homologous recombination, and was shown to interact with many proteins including BRCA1, CtIP, and MRE11, and regulate resection [124][125]. Genetic and epigenetic heterogeneity of our model systems should become not just an extra consideration as we face the necessity to organize the growing body of data, but also a resource for discovery as it becomes clear that the state of fork protection has translational significance as a determinant of cancer chemo-resistance, as we will discuss below.
SURVIVAL AND CHEMO-RESISTANCE: FORM REMODELING ENTERS THE TRANSLATIONAL SPOTLIGHT
Lethality of BRCA1 or BRCA2 deficiency when combined with PARP1 inhibition or cisplatin is perhaps the most famous of synthetic lethal interactions discovered [126] and translated into the clinic as treatment against breast and ovarian cancer [127][128]. Yet, resistant tumors arise frequently, which prompted efforts by many labs to identify the determinants of resistance [129]. Research focused on PARP1 inhibitor olaparib in the context of breast and ovarian cancer uncovered multiple pathways of partial or complete resistance that were utilized by cancer cells. In many cases resistance-causing mutations reversed HR defect of parental BRCA1 or BRCA2-deficient cells, but remarkably, in many other cases they restored stalled fork protection while leaving HR defective. In fact, in vivo and in vitro selection for cisplatin or olaparib resistance in BRCA2 or BRCA1-deficient backgrounds readily yields tumors or cell line clones in which the parental nascent strand degradation at forks is now suppressed (see refs. below). These important findings brought replication fork remodeling to the frontline of cancer chemotherapy. Collectively, the data point at the equilibria between fork reversal and restoration and between resection and protection as “breeding grounds” of chemo-resistance. However, intensifying research by many labs also poses additional questions about the nature of the lethality of PARP1 inhibition in BRCA-deficient cells, and about the role of PARP1 in nascent strand degradation.
–
The PARP1 paradox
Excessive nascent strand degradation correlates with increased DNA breakage [60][61]. Thus, it can be hypothesized that the reduced viability of BRCA-deficient cells subjected to PARP1 inhibitor (PARPi) can be at least in part mediated by the highly elevated nascent strand degradation. Indeed, some studies found that PARP1 inhibition by olaparib exacerbated MRE11-dependent nascent strand degradation at stalled forks in BRCA2-deficient cells [130] (Table 2). Also in agreement with this hypothesis is the observation that PARPi resistance in BRCA2-deficient cells can emerge due to secondary mutations that also suppress nascent strand degradation [96] (also, see next section). However, using different cell sources as well as combinations of gene knockouts, RNAi, or olaparib, several studies now have come to a different conclusion (Table 2): PARP1 deficiency or inhibition suppress nascent strand degradation and reduce MRE11 binding to forks in BRCA2-deficient cells [43][131][132]. Similarly, Chaudhuri et a.l observed that PARP1-/-, BRCA1-/- double knockout mouse B cells exhibited a suppressed nascent strand degradation compared to BRCA1-/- cells [96].
–
TABLE 2. Nascent strand degradation phenotypes of BRCA-deficient, PARP1-deficient cells. |
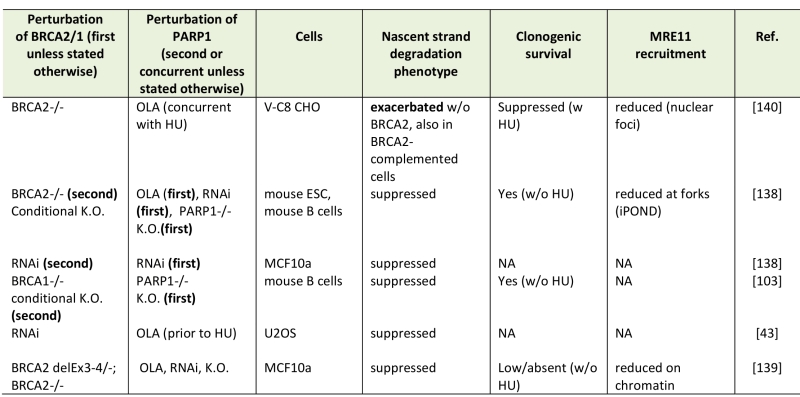 |
Abbreviations: OLA, olaparib, K.O., knockout, RNAi, siRNA or shRNA-mediated depletion, HU, hydroxyurea. [43][103][138][139][140] |
–
These findings are more in line with the consensus that PARP1 facilitates recruitment of MRE11 to stalled forks [67][133], and call for a more nuanced view of the relationship between PARP1 and BRCA proteins at replication forks. For example, PARP1 deficiency may block nascent strand degradation by blocking fork reversal, similar to the effect of the loss of SMARCAL1, ZRANB3, or HLTF [28][43][58]. If so, failure to reverse forks may in and of itself trigger DNA damage in the context of replication stress in BRCA-deficient cells, as described in previous sections. Indeed, Mijic et al. observed a mild increase in chromatid breaks and gaps after forks stalling in BRCA2-depleted cells treated with olaparib [43]. Combining ZRANB3 knockout with BRCA2 depletion also increased chromatid breaks compared to cells with only one of these two genes inactivated, and PARPi was epistatic with the ZRANB3 deficiency. However, Taglialatela et al. found that chromosomal aberra-tions and DNA damage were reduced upon depletion of SMARCAL1 or ZRANB3 in BRCA1 or BRCA2-depleted cells [44]. Overall, further research is required to elucidate the complex relationships between PARP1, BRCA proteins, fork remodeling processes, and DNA damage response and cell survival. Some potential areas of consideration are suggested below.
–
First, the unique nature of PARP1 loss may be in that it causes inappropriate restoration of reversed forks rather than blocking reversal in the first place, as the SNF2 family members or RAD51 do. Second, at least for some cases, trapping of PARP1 on DNA by olaparib may explain how a differential phenotype may emerge from PARP1 inhibition versus depletion in BRCA2-deficient cells [134]. Third, suppression of fork reversal upon PARP1 loss may well have a dual effect: at a high level of fork stalling (due to exogenous stressors like HU) failure to reverse forks is deleterious, while at a low level of fork stalling it may speed up S phase at the cost of decreased genomic stability. This logic may explain why PARP1 loss actually supports proliferation of BRCA2-deficeint non-cancer cells [96][131], given that these cells display constitutive fork progression problems [42][134] that trigger MUS81-mediated fork collapse and mitotic DNA synthesis [91][132]. Forth, the effect of PARP1 (like that of RAD51) may depend on whether it is a significant contributor to fork reversal in a given cell background, given that fork remodeling and DNA damage response may have undergone rewiring under selective pressure.
–
It should also be noted that the variability of PARP1 phenotypes may reflect its multiple, often counteracting inputs at the fork. For example, PARylation facilitates recruitment of BRCA1 to DSBs [135] and may do so at stalled forks. PARP1 also facilitates recruitment of NHEJ factors XRCC1 and DNA-PKcs to stalled and unresected forks [136]. Ku70/80 heterodimer binds to stalled forks, likely, at the end of a reversed arm that mimics a one-ended DSB, and protects it from the nucleases. Ku70/80 together with DNA-PKcs mediate timely fork restart [136][137][138][139]. These data are consistent with the expectation that PARP1 inhibition should upregulate resection. However, activated PARP1 also reduces Ku70/80 affinity for DNA ends (reviewed in [140], which would be consistent with PARP inhibition suppressing resection.
–
Alterations that confer chemo-resistance to BRCA-deficient cells correlate with suppressed nascent strand degradation
Perhaps the best testimony in favor of the association between fork protection and chemo-resistance is the accumulation of data that demonstrate this association in diverse, independently derived genetic backgrounds. For example, overexpression of FANCD2 was shown to suppress nascent strand degradation and confer PARPi resistance in a subset of BRCA1 or BRCA2-deficient cases [141]. Also, in BRCA1-deficient ovarian cancer cells, RAD51 paralogs XRCC2 and XRCC3 are capable of loading enough RAD51 onto stalled forks (in an ATR-dependent manner), thus providing proto-resistance to PARPi that can evolve to full resistance [142]. Increased loading of RAD51 at forks is also consistent with the olaparib-resistant phenotype of RADX loss [55]. PTIP, which affects MRE11 level at stalled forks, is another gene whose loss restores olaparib resistance and suppresses nascent strand degradation of BRCA1 and BRCA2-deficient cells [96]. This activity of PTIP depends on its ability to associate with MLL3/4, a histone methyltransferase that methylates histone H3 on Lysine 4 [143], and indeed Lysine 4 mono- and trimethylated H3 (H3K4me3) accumulates at resected forks in PTIP-proficient and not PTIP-deficient cells. Moreover, loss of MLL3/4 partially suppresses nascent strand degradation in BRCA2-deficient cells.
–
Discovery of the involvement of MLL3/4 implicates epigenetic regulation by histone modification in fork protection, adding another layer of complexity to the already elaborate system. Another prominent epigenetic regulator found to affect nascent strand degradation is CHD4, a member of the NuRD complex involved in gene regulation via nucleosome remodeling and histone deacetylation [144]. CHD4 depletion enhanced resistance to olaparib and cisplatin in BRCA2-deficient but not in BRCA1-deficient cells [145], and suppressed nascent strand degradation in BRCA2-deficient cells [96]. Lastly, a very recent study uncovered a similar role for the member of the Polycomb Repressive complex 2, EZH2, a histone H3 Lysine 27 methyltransferase, in BRCA2-deficient breast and ovarian cancer cells. Inhibition of EZH2 conferred PARPi and cisplatin resistance to these cells and suppressed nascent strand degradation at stalled forks [146] (reviewed in [147]). Intriguingly, the authors traced the effect of EZH2 to its ability to mediate recruitment of the endonuclease MUS81 rather than RAD51 or MRE11 to stalled forks, and showed that downregulation of EZH2 protects forks by suppressing recruitment of MUS81. Overall, our understanding of the epigenetic control of fork protection is far from complete, but it is safe to assume that it will reveal further heterogeneity and plasticity of fork protection mechanisms of cancer cells.
FORK REMODELING AND INNATE IMMUNITY
In this last section we would like to bring attention to a developing, exciting area of research that may well improve our understanding of the effects the deregulation of fork remodeling has on cancer and normal cells. This area is innate immune response.
–
Briefly, innate immune response is a universal cell-intrinsic response to bacteria and viruses. One of its jobs is to recognize and react to foreign DNA found in the cytoplasm, which involves multiple sensor molecules. Over the past decade, studies have begun to make connections between DNA damage response and the immune response to self-DNA [148][149] mediated by a cytosolic DNA sensor cGAS and a downstream adaptor STING (for review [150]). The crucial discovery that emerged is that endogenous short genomic DNA species produced in the course of checkpoint activation and DNA repair can trigger innate immunity if not properly degraded, and that this may contribute to the development of such autoimmune diseases as systemic lupus erythrematosus (SLE) and Aicardi Goutieres Syndrome (AGS) [151][152][153][154]. TREX1, a major single-stranded DNA exonuclease, has emerged as a key controller of the abundance of short DNA species, both single- and double-stranded, that are “shed” by the damaged and repairing genome. Without TREX1, both DNA damage checkpoint signaling and type I interferon (IFN) production (a hallmark of innate immune response) are upregulated [155][156][157]. Moreover, TREX1 controls abundance of short ssDNA species upon replication fork stalling by hydroxyurea [158].
–
Several studies have now made explicit associations between DNA resection during DNA repair or fork stalling and innate immune response to self-DNA. Erdal et al. detected a spike in short cytoplasmic ssDNA in cells treated with ionizing radiation (IR), mitomycin C or cisplatin, which activated innate immune response. Depletion of resection nucleases DNA2, EXO1, and BLM, but, interestingly, not MRE11 and CtIP, suppressed these responses, and TREX1 deficiency exacerbated them [159]. The authors also showed that inhibition of PARP1 boosted both basal and IR-induced levels of cytosolic ssDNA. Notably, TREX1 was found to associate with PARP1 in the nucleus upon DNA damage [160]. Non-involvement of MRE11 and CtIP may suggest that only long-range resection produces reactive ssDNA species. On the other hand, MRE11 [161] as well as Ku70/80 [162] were identified as dsDNA sensors in the cytoplasm that feed into the STING pathway. This role of MRE11 may, in theory, complicate interpretation of innate immune reaction to the depletion of the protein.
–
Pasero and colleagues identified SAMHD1 as a cofactor of MRE11 at stalled forks. SAMHD1 is a dNTPase with roles in cell-intrinsic antiviral response, and one of the genes mutated in AGS [163]. SAMHD1 was also shown to promote resection with MRE11 and CtIP at DSBs [164]. Pasero lab showed that deficiency in SAMHD1 negatively affects both fork restart and activation of the S phase checkpoint. Remarkably, it also upregulates release of resected DNA fragments into the cytoplasm where they trigger STING-dependent type I interferon response (P. Pasero, pers. comm.) Thus, SAMHD1 is one of the links between free DNA, checkpoint signaling, and inflammation.
–
In another study, RAD51 depletion was correlated with elevation of cytoplasmic ss and dsDNA, upregulation of innate immune response including STING, and enhanced nascent strand degradation at forks after IR [65]. All of these phenotypes were suppressed by inhibition of MRE11 exonuclease activity. An interesting addition to this line of evidence was generated by Wolf et al. [165], where the authors demonstrated that failure to clear free short ssDNA can take a toll on fork remodeling. The authors exploited a TREX1-deficient background to demonstrate that excess of short free ssDNA that accumulates in these cells is bound by RPA and RAD51 both in the cytosol and the nucleus. More remarkably, they also provided evidence suggesting that this condition can deplete the pools of RPA and RAD51 available for binding to genomic DNA at sites of damage and fork stalling, and thus can compromise DNA repair and fork protection. Overall, the data described above firmly establish innate immune response as part of a cellular stress response associated with deregulation of resection of stalled replication forks. Moreover, by affecting the availability of proteins like RAD51, RPA, and MRE11, innate immune response to replication stress-associated self-DNA may become another determinant of cancer susceptibility to chemotherapy.
CONCLUDING REMARKS
We hope to have shown that fork remodeling mechanisms emerge as balancing acts of many counteracting processes – a game of substrates that revolves around degradation versus protection of nascent DNA strands, and whose original main characters, MRE11 and RAD51, are surrounded by a whole cast of others. Moreover, fork remodeling moves into the spotlight as a major contributor to carcinogenesis, chemotherapy resistance, and inflammation.
–
We have seen that many HR and NHEJ proteins are involved in fork protection and curiously, often in a somewhat “off-label” way that is not exactly identical to their roles in DNA repair. We have also seen that close inspection of fork remodeling reveals cell line-dependent heterogeneity and malleability under selective pressures of carcinogenesis. While complete understanding of these complexities is still farther down the road, a few general considerations may guide us on the way.
–
First, it is worth remembering that fork remodeling phenotypes, as typically measured, are likely an average of heterogeneous responses to fork stalling in different genomic and epigenomic regions, next to actively transcribed genes or not, with different configurations of parental and daughter strands (ssDNA gaps, nicks, 5’ vs. 3’ overhangs), for different periods of time since the onset of fork-arresting treatment; and in early versus late S phase of the cell cycle. Aggregated together, these responses may create an impression of one too many proteins contributing semi-redundantly, while in reality cells may use “local” solutions to fork remodeling with only a subset of proteins involved in each case. To bring just one example, only a particular subset of stalled forks – those with leading strand gaps – may actually be reversed by SMARCAL1 [37]. Our ability to examine homogeneous subpopulations of cells and forks will be increasingly important in the future.
–
Second, it should be considered that the phenotypes often measured in perturbation studies of fork protection may not be a simple consequence of a protein’s absence, but a re-balancing act that the whole network undergoes because of it, i.e. an absence of function of one may actually cause a gain of function of several others.
–
And lastly – even if variability is widespread, the functional variables that define how fork remodeling is configured in cells are still few, and common. These are: 1) robustness and efficiency of loading of RAD51 at forks and the extent of its dependency on BRCA1/2; 2) robustness and dominance of fork reversal over fork restoration; 3) robustness and efficiency of nucleolytic activity at forks. The additional variables that are less studied at present but may come to the fore in the future are: efficiency of loading of NHEJ proteins as protective factors, the degree of nucleosome mobility and histone turnover at forks, and the efficiency of innate immune response. As the data show, while chemotherapeutic resistance in the fork protection-defective cancer cells arises frequently, it seems to work by adjusting the above-mentioned common variables. As such it is predictable – and thus can be preempted.
References
- S. Negrini, V.G. Gorgoulis, and T.D. Halazonetis, "Genomic instability — an evolving hallmark of cancer", Nature Reviews Molecular Cell Biology, vol. 11, pp. 220-228, 2010. http://dx.doi.org/10.1038/nrm2858
- D. Hanahan, and R. Weinberg, "Hallmarks of Cancer: The Next Generation", Cell, vol. 144, pp. 646-674, 2011. http://dx.doi.org/10.1016/j.cell.2011.02.013
- M.K. Zeman, and K.A. Cimprich, "Causes and consequences of replication stress", Nature Cell Biology, vol. 16, pp. 2-9, 2013. http://dx.doi.org/10.1038/ncb2897
- I. Magdalou, B.S. Lopez, P. Pasero, and S.A. Lambert, "The causes of replication stress and their consequences on genome stability and cell fate", Seminars in Cell & Developmental Biology, vol. 30, pp. 154-164, 2014. http://dx.doi.org/10.1016/j.semcdb.2014.04.035
- S. Hills, and J. Diffley, "DNA Replication and Oncogene-Induced Replicative Stress", Current Biology, vol. 24, pp. R435-R444, 2014. http://dx.doi.org/10.1016/j.cub.2014.04.012
- M. Macheret, and T.D. Halazonetis, "DNA Replication Stress as a Hallmark of Cancer", Annual Review of Pathology: Mechanisms of Disease, vol. 10, pp. 425-448, 2015. http://dx.doi.org/10.1146/annurev-pathol-012414-040424
- H. Técher, S. Koundrioukoff, A. Nicolas, and M. Debatisse, "The impact of replication stress on replication dynamics and DNA damage in vertebrate cells", Nature Reviews Genetics, vol. 18, pp. 535-550, 2017. http://dx.doi.org/10.1038/nrg.2017.46
- H. Gaillard, T. García-Muse, and A. Aguilera, "Replication stress and cancer", Nature Reviews Cancer, vol. 15, pp. 276-289, 2015. http://dx.doi.org/10.1038/nrc3916
- B. Blumenfeld, M. Ben-Zimra, and I. Simon, "Perturbations in the Replication Program Contribute to Genomic Instability in Cancer", International Journal of Molecular Sciences, vol. 18, pp. 1138, 2017. http://dx.doi.org/10.3390/ijms18061138
- K.J. Neelsen, and M. Lopes, "Replication fork reversal in eukaryotes: from dead end to dynamic response", Nature Reviews Molecular Cell Biology, vol. 16, pp. 207-220, 2015. http://dx.doi.org/10.1038/nrm3935
- M. Berti, and A. Vindigni, "Replication stress: getting back on track", Nature Structural & Molecular Biology, vol. 23, pp. 103-109, 2016. http://dx.doi.org/10.1038/nsmb.3163
-
A.M. Kolinjivadi, V. Sannino, A. de Antoni, H. Técher, G. Baldi, and V. Costanzo, "Moonlighting at replication forks – a new life for homologous recombination proteins
BRCA 1,BRCA 2 andRAD 51", FEBS Letters, vol. 591, pp. 1083-1100, 2017. http://dx.doi.org/10.1002/1873-3468.12556 - V. Bhatia, E. Herrera-Moyano, A. Aguilera, and B. Gómez-González, "The Role of Replication-Associated Repair Factors on R-Loops", Genes, vol. 8, pp. 171, 2017. http://dx.doi.org/10.3390/genes8070171
- E. Chang, and P. Stirling, "Replication Fork Protection Factors Controlling R-Loop Bypass and Suppression", Genes, vol. 8, pp. 33, 2017. http://dx.doi.org/10.3390/genes8010033
- E. Bournique, M. Dall’Osto, J. Hoffmann, and V. Bergoglio, "Role of specialized DNA polymerases in the limitation of replicative stress and DNA damage transmission", Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis, vol. 808, pp. 62-73, 2018. http://dx.doi.org/10.1016/j.mrfmmm.2017.08.002
- A. Franchitto, L.M. Pirzio, E. Prosperi, O. Sapora, M. Bignami, and P. Pichierri, "Replication fork stalling in WRN-deficient cells is overcome by prompt activation of a MUS81-dependent pathway", The Journal of Cell Biology, vol. 183, pp. 241-252, 2008. http://dx.doi.org/10.1083/jcb.200803173
- K. Fugger, W. Kit Chu, P. Haahr, A. Nedergaard Kousholt, H. Beck, M.J. Payne, K. Hanada, I.D. Hickson, and C. Storgaard Sørensen, "FBH1 co-operates with MUS81 in inducing DNA double-strand breaks and cell death following replication stress", Nature Communications, vol. 4, 2013. http://dx.doi.org/10.1038/ncomms2395
- A. Pepe, and S. West, "MUS81-EME2 Promotes Replication Fork Restart", Cell Reports, vol. 7, pp. 1048-1055, 2014. http://dx.doi.org/10.1016/j.celrep.2014.04.007
- D. Lemaçon, J. Jackson, A. Quinet, J.R. Brickner, S. Li, S. Yazinski, Z. You, G. Ira, L. Zou, N. Mosammaparast, and A. Vindigni, "MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells", Nature Communications, vol. 8, 2017. http://dx.doi.org/10.1038/s41467-017-01180-5
- K. Hanada, M. Budzowska, S.L. Davies, E. van Drunen, H. Onizawa, H.B. Beverloo, A. Maas, J. Essers, I.D. Hickson, and R. Kanaar, "The structure-specific endonuclease Mus81 contributes to replication restart by generating double-strand DNA breaks", Nature Structural & Molecular Biology, vol. 14, pp. 1096-1104, 2007. http://dx.doi.org/10.1038/nsmb1313
- R.P. Anand, S.T. Lovett, and J.E. Haber, "Break-Induced DNA Replication", Cold Spring Harbor Perspectives in Biology, vol. 5, pp. a010397-a010397, 2013. http://dx.doi.org/10.1101/cshperspect.a010397
- L. Costantino, S.K. Sotiriou, J.K. Rantala, S. Magin, E. Mladenov, T. Helleday, J.E. Haber, G. Iliakis, O.P. Kallioniemi, and T.D. Halazonetis, "Break-Induced Replication Repair of Damaged Forks Induces Genomic Duplications in Human Cells", Science, vol. 343, pp. 88-91, 2014. http://dx.doi.org/10.1126/science.1243211
- S.K. Sotiriou, I. Kamileri, N. Lugli, K. Evangelou, C. Da-Ré, F. Huber, L. Padayachy, S. Tardy, N.L. Nicati, S. Barriot, F. Ochs, C. Lukas, J. Lukas, V.G. Gorgoulis, L. Scapozza, and T.D. Halazonetis, "Mammalian RAD52 Functions in Break-Induced Replication Repair of Collapsed DNA Replication Forks", Molecular Cell, vol. 64, pp. 1127-1134, 2016. http://dx.doi.org/10.1016/j.molcel.2016.10.038
- R. Bhowmick, S. Minocherhomji, and I.D. Hickson, "RAD52 Facilitates Mitotic DNA Synthesis Following Replication Stress", Molecular Cell, vol. 64, pp. 1117-1126, 2016. http://dx.doi.org/10.1016/j.molcel.2016.10.037
- J.T.P. Yeeles, J. Poli, K.J. Marians, and P. Pasero, "Rescuing Stalled or Damaged Replication Forks", Cold Spring Harbor Perspectives in Biology, vol. 5, pp. a012815-a012815, 2013. http://dx.doi.org/10.1101/cshperspect.a012815
- N.P. Higgins, K. Kato, and B. Strauss, "A model for replication repair in mammalian cells.", Journal of molecular biology, 1976. http://www.ncbi.nlm.nih.gov/pubmed/1255724
- A. Vindigni, and M. Lopes, "Combining electron microscopy with single molecule DNA fiber approaches to study DNA replication dynamics", Biophysical Chemistry, vol. 225, pp. 3-9, 2017. http://dx.doi.org/10.1016/j.bpc.2016.11.014
- R. Zellweger, D. Dalcher, K. Mutreja, M. Berti, J.A. Schmid, R. Herrador, A. Vindigni, and M. Lopes, "Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells", Journal of Cell Biology, vol. 208, pp. 563-579, 2015. http://dx.doi.org/10.1083/jcb.201406099
- S.K. Godin, M.R. Sullivan, and K.A. Bernstein, "Novel insights into RAD51 activity and regulation during homologous recombination and DNA replication", Biochemistry and Cell Biology, vol. 94, pp. 407-418, 2016. http://dx.doi.org/10.1139/bcb-2016-0012
- E. Petermann, M.L. Orta, N. Issaeva, N. Schultz, and T. Helleday, "Hydroxyurea-Stalled Replication Forks Become Progressively Inactivated and Require Two Different RAD51-Mediated Pathways for Restart and Repair", Molecular Cell, vol. 37, pp. 492-502, 2010. http://dx.doi.org/10.1016/j.molcel.2010.01.021
- J.M. Sidorova, K. Kehrli, F. Mao, and R. Monnat, "Distinct functions of human RECQ helicases WRN and BLM in replication fork recovery and progression after hydroxyurea-induced stalling", DNA Repair, vol. 12, pp. 128-139, 2013. http://dx.doi.org/10.1016/j.dnarep.2012.11.005
- D.V. Bugreev, M.J. Rossi, and A.V. Mazin, "Cooperation of RAD51 and RAD54 in regression of a model replication fork", Nucleic Acids Research, vol. 39, pp. 2153-2164, 2010. http://dx.doi.org/10.1093/nar/gkq1139
- S.J. Ceballos, and W. Heyer, "Functions of the Snf2/Swi2 family Rad54 motor protein in homologous recombination", Biochimica et Biophysica Acta (BBA) - Gene Regulatory Mechanisms, vol. 1809, pp. 509-523, 2011. http://dx.doi.org/10.1016/j.bbagrm.2011.06.006
- A. Machwe, L. Xiao, J. Groden, and D.K. Orren, "The Werner and Bloom Syndrome Proteins Catalyze Regression of a Model Replication Fork", Biochemistry, vol. 45, pp. 13939-13946, 2006. http://dx.doi.org/10.1021/bi0615487
- K. Gari, C. Décaillet, M. Delannoy, L. Wu, and A. Constantinou, "Remodeling of DNA replication structures by the branch point translocase FANCM", Proceedings of the National Academy of Sciences, vol. 105, pp. 16107-16112, 2008. http://dx.doi.org/10.1073/pnas.0804777105
- A. Blastyák, I. Hajdú, I. Unk, and L. Haracska, "Role of Double-Stranded DNA Translocase Activity of Human HLTF in Replication of Damaged DNA", Molecular and Cellular Biology, vol. 30, pp. 684-693, 2010. http://dx.doi.org/10.1128/mcb.00863-09
- R. Bétous, F. Couch, A. Mason, B. Eichman, M. Manosas, and D. Cortez, "Substrate-Selective Repair and Restart of Replication Forks by DNA Translocases", Cell Reports, vol. 3, pp. 1958-1969, 2013. http://dx.doi.org/10.1016/j.celrep.2013.05.002
- K. Fugger, M. Mistrik, K. Neelsen, Q. Yao, R. Zellweger, A. Kousholt, P. Haahr, W. Chu, J. Bartek, M. Lopes, I. Hickson, and C. Sørensen, "FBH1 Catalyzes Regression of Stalled Replication Forks", Cell Reports, vol. 10, pp. 1749-1757, 2015. http://dx.doi.org/10.1016/j.celrep.2015.02.028
- A. Kile, D. Chavez, J. Bacal, S. Eldirany, D. Korzhnev, I. Bezsonova, B. Eichman, and K. Cimprich, "HLTF’s Ancient HIRAN Domain Binds 3′ DNA Ends to Drive Replication Fork Reversal", Molecular Cell, vol. 58, pp. 1090-1100, 2015. http://dx.doi.org/10.1016/j.molcel.2015.05.013
- W. Piwko, L.J. Mlejnkova, K. Mutreja, L. Ranjha, D. Stafa, A. Smirnov, M.M. Brodersen, R. Zellweger, A. Sturzenegger, P. Janscak, M. Lopes, M. Peter, and P. Cejka, "The MMS22L–TONSL heterodimer directly promotes RAD51‐dependent recombination upon replication stress", The EMBO Journal, vol. 35, pp. 2584-2601, 2016. http://dx.doi.org/10.15252/embj.201593132
- M. Vujanovic, J. Krietsch, M.C. Raso, N. Terraneo, R. Zellweger, J.A. Schmid, A. Taglialatela, J. Huang, C.L. Holland, K. Zwicky, R. Herrador, H. Jacobs, D. Cortez, A. Ciccia, L. Penengo, and M. Lopes, "Replication Fork Slowing and Reversal upon DNA Damage Require PCNA Polyubiquitination and ZRANB3 DNA Translocase Activity", Molecular Cell, vol. 67, pp. 882-890.e5, 2017. http://dx.doi.org/10.1016/j.molcel.2017.08.010
- A.M. Kolinjivadi, V. Sannino, A. De Antoni, K. Zadorozhny, M. Kilkenny, H. Técher, G. Baldi, R. Shen, A. Ciccia, L. Pellegrini, L. Krejci, and V. Costanzo, "Smarcal1-Mediated Fork Reversal Triggers Mre11-Dependent Degradation of Nascent DNA in the Absence of Brca2 and Stable Rad51 Nucleofilaments", Molecular Cell, vol. 67, pp. 867-881.e7, 2017. http://dx.doi.org/10.1016/j.molcel.2017.07.001
- S. Mijic, R. Zellweger, N. Chappidi, M. Berti, K. Jacobs, K. Mutreja, S. Ursich, A. Ray Chaudhuri, A. Nussenzweig, P. Janscak, and M. Lopes, "Replication fork reversal triggers fork degradation in BRCA2-defective cells", Nature Communications, vol. 8, 2017. http://dx.doi.org/10.1038/s41467-017-01164-5
- A. Taglialatela, S. Alvarez, G. Leuzzi, V. Sannino, L. Ranjha, J. Huang, C. Madubata, R. Anand, B. Levy, R. Rabadan, P. Cejka, V. Costanzo, and A. Ciccia, "Restoration of Replication Fork Stability in BRCA1- and BRCA2-Deficient Cells by Inactivation of SNF2-Family Fork Remodelers", Molecular Cell, vol. 68, pp. 414-430.e8, 2017. http://dx.doi.org/10.1016/j.molcel.2017.09.036
- J. Kim, J. Kim, S. Lee, D. Kim, H. Kang, S. Bae, Z. Pan, and Y. Seo, "The Novel Human DNA Helicase hFBH1 Is an F-box Protein", Journal of Biological Chemistry, vol. 277, pp. 24530-24537, 2002. http://dx.doi.org/10.1074/jbc.M201612200
- A. Ciccia, A.L. Bredemeyer, M.E. Sowa, M. Terret, P.V. Jallepalli, J.W. Harper, and S.J. Elledge, "The SIOD disorder protein SMARCAL1 is an RPA-interacting protein involved in replication fork restart", Genes & Development, vol. 23, pp. 2415-2425, 2009. http://dx.doi.org/10.1101/gad.1832309
- C.E. Bansbach, R. Bétous, C.A. Lovejoy, G.G. Glick, and D. Cortez, "The annealing helicase SMARCAL1 maintains genome integrity at stalled replication forks", Genes & Development, vol. 23, pp. 2405-2414, 2009. http://dx.doi.org/10.1101/gad.1839909
- A. Ciccia, A. Nimonkar, Y. Hu, I. Hajdu, Y. Achar, L. Izhar, S. Petit, B. Adamson, J. Yoon, S. Kowalczykowski, D. Livingston, L. Haracska, and S. Elledge, "Polyubiquitinated PCNA Recruits the ZRANB3 Translocase to Maintain Genomic Integrity after Replication Stress", Molecular Cell, vol. 47, pp. 396-409, 2012. http://dx.doi.org/10.1016/j.molcel.2012.05.024
- R. Bétous, A.C. Mason, R.P. Rambo, C.E. Bansbach, A. Badu-Nkansah, B.M. Sirbu, B.F. Eichman, and D. Cortez, "SMARCAL1 catalyzes fork regression and Holliday junction migration to maintain genome stability during DNA replication", Genes & Development, vol. 26, pp. 151-162, 2012. http://dx.doi.org/10.1101/gad.178459.111
- L.A. Poole, and D. Cortez, "Functions of SMARCAL1, ZRANB3, and HLTF in maintaining genome stability", Critical Reviews in Biochemistry and Molecular Biology, vol. 52, pp. 696-714, 2017. http://dx.doi.org/10.1080/10409238.2017.1380597
- N. Lugli, S.K. Sotiriou, and T.D. Halazonetis, "The role of SMARCAL1 in replication fork stability and telomere maintenance", DNA Repair, vol. 56, pp. 129-134, 2017. http://dx.doi.org/10.1016/j.dnarep.2017.06.015
- F.B. Couch, and D. Cortez, "Fork reversal, too much of a good thing", Cell Cycle, vol. 13, pp. 1049-1050, 2014. http://dx.doi.org/10.4161/cc.28212
- F.B. Couch, C.E. Bansbach, R. Driscoll, J.W. Luzwick, G.G. Glick, R. Bétous, C.M. Carroll, S.Y. Jung, J. Qin, K.A. Cimprich, and D. Cortez, "ATR phosphorylates SMARCAL1 to prevent replication fork collapse", Genes & Development, vol. 27, pp. 1610-1623, 2013. http://dx.doi.org/10.1101/gad.214080.113
- A.C. Parplys, J.I. Seelbach, S. Becker, M. Behr, A. Wrona, C. Jend, W.Y. Mansour, S.A. Joosse, H. Stuerzbecher, H. Pospiech, C. Petersen, E. Dikomey, and K. Borgmann, "High levels of RAD51 perturb DNA replication elongation and cause unscheduled origin firing due to impaired CHK1 activation", Cell Cycle, vol. 14, pp. 3190-3202, 2015. http://dx.doi.org/10.1080/15384101.2015.1055996
- H. Dungrawala, K.P. Bhat, R. Le Meur, W.J. Chazin, X. Ding, S.K. Sharan, S.R. Wessel, A.A. Sathe, R. Zhao, and D. Cortez, "RADX Promotes Genome Stability and Modulates Chemosensitivity by Regulating RAD51 at Replication Forks", Molecular Cell, vol. 67, pp. 374-386.e5, 2017. http://dx.doi.org/10.1016/j.molcel.2017.06.023
- A. Ray Chaudhuri, and A. Nussenzweig, "The multifaceted roles of PARP1 in DNA repair and chromatin remodelling", Nature Reviews Molecular Cell Biology, vol. 18, pp. 610-621, 2017. http://dx.doi.org/10.1038/nrm.2017.53
- F. Ciccarone, M. Zampieri, and P. Caiafa, "PARP1 orchestrates epigenetic events setting up chromatin domains", Seminars in Cell & Developmental Biology, vol. 63, pp. 123-134, 2017. http://dx.doi.org/10.1016/j.semcdb.2016.11.010
- A. Ray Chaudhuri, Y. Hashimoto, R. Herrador, K.J. Neelsen, D. Fachinetti, R. Bermejo, A. Cocito, V. Costanzo, and M. Lopes, "Topoisomerase I poisoning results in PARP-mediated replication fork reversal", Nature Structural & Molecular Biology, vol. 19, pp. 417-423, 2012. http://dx.doi.org/10.1038/nsmb.2258
- M. Berti, A. Ray Chaudhuri, S. Thangavel, S. Gomathinayagam, S. Kenig, M. Vujanovic, F. Odreman, T. Glatter, S. Graziano, R. Mendoza-Maldonado, F. Marino, B. Lucic, V. Biasin, M. Gstaiger, R. Aebersold, J.M. Sidorova, R.J. Monnat, M. Lopes, and A. Vindigni, "Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition", Nature Structural & Molecular Biology, vol. 20, pp. 347-354, 2013. http://dx.doi.org/10.1038/nsmb.2501
- K. Schlacher, N. Christ, N. Siaud, A. Egashira, H. Wu, and M. Jasin, "Double-Strand Break Repair-Independent Role for BRCA2 in Blocking Stalled Replication Fork Degradation by MRE11", Cell, vol. 145, pp. 529-542, 2011. http://dx.doi.org/10.1016/j.cell.2011.03.041
- K. Schlacher, H. Wu, and M. Jasin, "A Distinct Replication Fork Protection Pathway Connects Fanconi Anemia Tumor Suppressors to RAD51-BRCA1/2", Cancer Cell, vol. 22, pp. 106-116, 2012. http://dx.doi.org/10.1016/j.ccr.2012.05.015
- T.M. Kim, M.Y. Son, S. Dodds, L. Hu, and P. Hasty, "Deletion of BRCA2 exon 27 causes defects in response to both stalled and collapsed replication forks", Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis, vol. 766-767, pp. 66-72, 2014. http://dx.doi.org/10.1016/j.mrfmmm.2014.06.003
- K. Kehrli, M. Phelps, P. Lazarchuk, E. Chen, R. Monnat, and J.M. Sidorova, "Class I Histone Deacetylase HDAC1 and WRN RECQ Helicase Contribute Additively to Protect Replication Forks upon Hydroxyurea-induced Arrest", Journal of Biological Chemistry, vol. 291, pp. 24487-24503, 2016. http://dx.doi.org/10.1074/jbc.M115.708594
- S. Thangavel, M. Berti, M. Levikova, C. Pinto, S. Gomathinayagam, M. Vujanovic, R. Zellweger, H. Moore, E.H. Lee, E.A. Hendrickson, P. Cejka, S. Stewart, M. Lopes, and A. Vindigni, "DNA2 drives processing and restart of reversed replication forks in human cells", Journal of Cell Biology, vol. 208, pp. 545-562, 2015. http://dx.doi.org/10.1083/jcb.201406100
- S. Bhattacharya, K. Srinivasan, S. Abdisalaam, F. Su, P. Raj, I. Dozmorov, R. Mishra, E.K. Wakeland, S. Ghose, S. Mukherjee, and A. Asaithamby, "RAD51 interconnects between DNA replication, DNA repair and immunity", Nucleic Acids Research, vol. 45, pp. 4590-4605, 2017. http://dx.doi.org/10.1093/nar/gkx126
- K. Trenz, E. Smith, S. Smith, and V. Costanzo, "ATM and ATR promote Mre11 dependent restart of collapsed replication forks and prevent accumulation of DNA breaks", The EMBO Journal, vol. 25, pp. 1764-1774, 2006. http://dx.doi.org/10.1038/sj.emboj.7601045
- H.E. Bryant, E. Petermann, N. Schultz, A. Jemth, O. Loseva, N. Issaeva, F. Johansson, S. Fernandez, P. McGlynn, and T. Helleday, "PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination", The EMBO Journal, vol. 28, pp. 2601-2615, 2009. http://dx.doi.org/10.1038/emboj.2009.206
- Y. Hashimoto, A. Ray Chaudhuri, M. Lopes, and V. Costanzo, "Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis", Nature Structural & Molecular Biology, vol. 17, pp. 1305-1311, 2010. http://dx.doi.org/10.1038/nsmb.1927
- C. Iannascoli, V. Palermo, I. Murfuni, A. Franchitto, and P. Pichierri, "The WRN exonuclease domain protects nascent strands from pathological MRE11/EXO1-dependent degradation", Nucleic Acids Research, pp. gkv836, 2015. http://dx.doi.org/10.1093/nar/gkv836
- J. Chapman, M. Taylor, and S. Boulton, "Playing the End Game: DNA Double-Strand Break Repair Pathway Choice", Molecular Cell, vol. 47, pp. 497-510, 2012. http://dx.doi.org/10.1016/j.molcel.2012.07.029
- A. Shibata, D. Moiani, A. Arvai, J. Perry, S. Harding, M. Genois, R. Maity, S. van Rossum-Fikkert, A. Kertokalio, F. Romoli, A. Ismail, E. Ismalaj, E. Petricci, M. Neale, R. Bristow, J. Masson, C. Wyman, P. Jeggo, and J. Tainer, "DNA Double-Strand Break Repair Pathway Choice Is Directed by Distinct MRE11 Nuclease Activities", Molecular Cell, vol. 53, pp. 7-18, 2014. http://dx.doi.org/10.1016/j.molcel.2013.11.003
- A. Sturzenegger, K. Burdova, R. Kanagaraj, M. Levikova, C. Pinto, P. Cejka, and P. Janscak, "DNA2 Cooperates with the WRN and BLM RecQ Helicases to Mediate Long-range DNA End Resection in Human Cells", Journal of Biological Chemistry, vol. 289, pp. 27314-27326, 2014. http://dx.doi.org/10.1074/jbc.M114.578823
- J.M. Daley, H. Niu, A.S. Miller, and P. Sung, "Biochemical mechanism of DSB end resection and its regulation", DNA Repair, vol. 32, pp. 66-74, 2015. http://dx.doi.org/10.1016/j.dnarep.2015.04.015
- T. Liu, and J. Huang, "DNA End Resection: Facts and Mechanisms", Genomics, Proteomics & Bioinformatics, vol. 14, pp. 126-130, 2016. http://dx.doi.org/10.1016/j.gpb.2016.05.002
- N. Tomimatsu, B. Mukherjee, K. Deland, A. Kurimasa, E. Bolderson, K.K. Khanna, and S. Burma, "Exo1 plays a major role in DNA end resection in humans and influences double-strand break repair and damage signaling decisions", DNA Repair, vol. 11, pp. 441-448, 2012. http://dx.doi.org/10.1016/j.dnarep.2012.01.006
- N.N. Hoa, R. Akagawa, T. Yamasaki, K. Hirota, K. Sasa, T. Natsume, J. Kobayashi, T. Sakuma, T. Yamamoto, K. Komatsu, M.T. Kanemaki, Y. Pommier, S. Takeda, and H. Sasanuma, "Relative contribution of four nucleases, CtIP, Dna2, Exo1 and Mre11, to the initial step of DNA double‐strand break repair by homologous recombination in both the chicken DT40 and human TK6 cell lines", Genes to Cells, vol. 20, pp. 1059-1076, 2015. http://dx.doi.org/10.1111/gtc.12310
- P. Chanut, S. Britton, J. Coates, S.P. Jackson, and P. Calsou, "Coordinated nuclease activities counteract Ku at single-ended DNA double-strand breaks", Nature Communications, vol. 7, 2016. http://dx.doi.org/10.1038/ncomms12889
- S.L. Davies, P.S. North, and I.D. Hickson, "Role for BLM in replication-fork restart and suppression of origin firing after replicative stress", Nature Structural & Molecular Biology, vol. 14, pp. 677-679, 2007. http://dx.doi.org/10.1038/nsmb1267
- J.M. Sidorova, N. Li, A. Folch, and R.J. Monnat, Jr., "The RecQ helicase WRN is required for normal replication fork progression after DNA damage or replication fork arrest", Cell Cycle, vol. 7, pp. 796-807, 2008. http://dx.doi.org/10.4161/cc.7.6.5566
- F. Ammazzalorso, L.M. Pirzio, M. Bignami, A. Franchitto, and P. Pichierri, "ATR and ATM differently regulate WRN to prevent DSBs at stalled replication forks and promote replication fork recovery", The EMBO Journal, vol. 29, pp. 3156-3169, 2010. http://dx.doi.org/10.1038/emboj.2010.205
- J.P. Duxin, H.R. Moore, J. Sidorova, K. Karanja, Y. Honaker, B. Dao, H. Piwnica-Worms, J.L. Campbell, R.J. Monnat, and S.A. Stewart, "Okazaki Fragment Processing-independent Role for Human Dna2 Enzyme during DNA Replication", Journal of Biological Chemistry, vol. 287, pp. 21980-21991, 2012. http://dx.doi.org/10.1074/jbc.M112.359018
- A. Shibata, "Regulation of repair pathway choice at two-ended DNA double-strand breaks", Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis, vol. 803-805, pp. 51-55, 2017. http://dx.doi.org/10.1016/j.mrfmmm.2017.07.011
- J. Li, and X. Xu, "DNA double-strand break repair: a tale of pathway choices", Acta Biochimica et Biophysica Sinica, vol. 48, pp. 641-646, 2016. http://dx.doi.org/10.1093/abbs/gmw045
- R. Ceccaldi, B. Rondinelli, and A.D. D’Andrea, "Repair Pathway Choices and Consequences at the Double-Strand Break", Trends in Cell Biology, vol. 26, pp. 52-64, 2016. http://dx.doi.org/10.1016/j.tcb.2015.07.009
- R. Anand, L. Ranjha, E. Cannavo, and P. Cejka, "Phosphorylated CtIP Functions as a Co-factor of the MRE11-RAD50-NBS1 Endonuclease in DNA End Resection", Molecular Cell, vol. 64, pp. 940-950, 2016. http://dx.doi.org/10.1016/j.molcel.2016.10.017
- A.J. Davis, L. Chi, S. So, K. Lee, E. Mori, K. Fattah, J. Yang, and D.J. Chen, "BRCA1 modulates the autophosphorylation status of DNA-PKcs in S phase of the cell cycle", Nucleic Acids Research, vol. 42, pp. 11487-11501, 2014. http://dx.doi.org/10.1093/nar/gku824
- F. Zhang, J. Ma, J. Wu, L. Ye, H. Cai, B. Xia, and X. Yu, "PALB2 Links BRCA1 and BRCA2 in the DNA-Damage Response", Current Biology, vol. 19, pp. 524-529, 2009. http://dx.doi.org/10.1016/j.cub.2009.02.018
- S. Pathania, S. Bade, M. Le Guillou, K. Burke, R. Reed, C. Bowman-Colin, Y. Su, D.T. Ting, K. Polyak, A.L. Richardson, J. Feunteun, J.E. Garber, and D.M. Livingston, "BRCA1 haploinsufficiency for replication stress suppression in primary cells", Nature Communications, vol. 5, 2014. http://dx.doi.org/10.1038/ncomms6496
- R.B. Jensen, A. Carreira, and S.C. Kowalczykowski, "Purified human BRCA2 stimulates RAD51-mediated recombination", Nature, vol. 467, pp. 678-683, 2010. http://dx.doi.org/10.1038/nature09399
- T. Thorslund, M.J. McIlwraith, S.A. Compton, S. Lekomtsev, M. Petronczki, J.D. Griffith, and S.C. West, "The breast cancer tumor suppressor BRCA2 promotes the specific targeting of RAD51 to single-stranded DNA", Nature Structural & Molecular Biology, vol. 17, pp. 1263-1265, 2010. http://dx.doi.org/10.1038/nsmb.1905
- X. Lai, R. Broderick, V. Bergoglio, J. Zimmer, S. Badie, W. Niedzwiedz, J. Hoffmann, and M. Tarsounas, "MUS81 nuclease activity is essential for replication stress tolerance and chromosome segregation in BRCA2-deficient cells", Nature Communications, vol. 8, 2017. http://dx.doi.org/10.1038/ncomms15983
- Y. Xu, S. Ning, Z. Wei, R. Xu, X. Xu, M. Xing, R. Guo, and D. Xu, "53BP1 and BRCA1 control pathway choice for stalled replication restart", eLife, vol. 6, 2017. http://dx.doi.org/10.7554/eLife.30523
- B.M. Sirbu, F.B. Couch, J.T. Feigerle, S. Bhaskara, S.W. Hiebert, and D. Cortez, "Analysis of protein dynamics at active, stalled, and collapsed replication forks", Genes & Development, vol. 25, pp. 1320-1327, 2011. http://dx.doi.org/10.1101/gad.2053211
- A. Wang, T. Kim, J. Wagner, B. Conti, F. Lach, A. Huang, H. Molina, E. Sanborn, H. Zierhut, B. Cornes, A. Abhyankar, C. Sougnez, S. Gabriel, A. Auerbach, S. Kowalczykowski, and A. Smogorzewska, "A Dominant Mutation in Human RAD51 Reveals Its Function in DNA Interstrand Crosslink Repair Independent of Homologous Recombination", Molecular Cell, vol. 59, pp. 478-490, 2015. http://dx.doi.org/10.1016/j.molcel.2015.07.009
- F. Huang, O.M. Mazina, I.J. Zentner, S. Cocklin, and A.V. Mazin, "Inhibition of Homologous Recombination in Human Cells by Targeting RAD51 Recombinase", Journal of Medicinal Chemistry, vol. 55, pp. 3011-3020, 2012. http://dx.doi.org/10.1021/jm201173g
- A. Ray Chaudhuri, E. Callen, X. Ding, E. Gogola, A.A. Duarte, J. Lee, N. Wong, V. Lafarga, J.A. Calvo, N.J. Panzarino, S. John, A. Day, A.V. Crespo, B. Shen, L.M. Starnes, J.R.D. Ruiter, J.A. Daniel, P.A. Konstantinopoulos, D. Cortez, S.B. Cantor, O. Fernandez-Capetillo, K. Ge, J. Jonkers, S. Rottenberg, S.K. Sharan, and A. Nussenzweig, "Replication fork stability confers chemoresistance in BRCA-deficient cells", Nature, vol. 535, pp. 382-387, 2016. http://dx.doi.org/10.1038/nature18325
- F. Su, S. Mukherjee, Y. Yang, E. Mori, S. Bhattacharya, J. Kobayashi, S. Yannone, D. Chen, and A. Asaithamby, "Nonenzymatic Role for WRN in Preserving Nascent DNA Strands after Replication Stress", Cell Reports, vol. 9, pp. 1387-1401, 2014. http://dx.doi.org/10.1016/j.celrep.2014.10.025
- E. Callen, M. Di Virgilio, M. Kruhlak, M. Nieto-Soler, N. Wong, H. Chen, R. Faryabi, F. Polato, M. Santos, L. Starnes, D. Wesemann, J. Lee, A. Tubbs, B. Sleckman, J. Daniel, K. Ge, F. Alt, O. Fernandez-Capetillo, M. Nussenzweig, and A. Nussenzweig, "53BP1 Mediates Productive and Mutagenic DNA Repair through Distinct Phosphoprotein Interactions", Cell, vol. 153, pp. 1266-1280, 2013. http://dx.doi.org/10.1016/j.cell.2013.05.023
- J. Wang, A. Aroumougame, M. Lobrich, Y. Li, D. Chen, J. Chen, and Z. Gong, "PTIP associates with Artemis to dictate DNA repair pathway choice", Genes & Development, vol. 28, pp. 2693-2698, 2014. http://dx.doi.org/10.1101/gad.252478.114
- J. Gu, S. Li, X. Zhang, L. Wang, D. Niewolik, K. Schwarz, R.J. Legerski, E. Zandi, and M.R. Lieber, "DNA-PKcs regulates a single-stranded DNA endonuclease activity of Artemis", DNA Repair, vol. 9, pp. 429-437, 2010. http://dx.doi.org/10.1016/j.dnarep.2010.01.001
-
J. Unno, M. Takagi, J. Piao, M. Sugimoto, F. Honda, D. Maeda, M. Masutani, T. Kiyono, F. Watanabe, T. Morio, H. Teraoka, and S. Mizutani, "Artemis‐dependent
DNA double‐strand break formation at stalled replication forks", Cancer Science, vol. 104, pp. 703-710, 2013. http://dx.doi.org/10.1111/cas.12144 - K. Hanamshet, O. Mazina, and A. Mazin, "Reappearance from Obscurity: Mammalian Rad52 in Homologous Recombination", Genes, vol. 7, pp. 63, 2016. http://dx.doi.org/10.3390/genes7090063
- D.V. Bugreev, X. Yu, E.H. Egelman, and A.V. Mazin, "Novel pro- and anti-recombination activities of the Bloom’s syndrome helicase", Genes & Development, vol. 21, pp. 3085-3094, 2007. http://dx.doi.org/10.1101/gad.1609007
- D.V. Bugreev, O.M. Mazina, and A.V. Mazin, "Bloom Syndrome Helicase Stimulates RAD51 DNA Strand Exchange Activity through a Novel Mechanism", Journal of Biological Chemistry, vol. 284, pp. 26349-26359, 2009. http://dx.doi.org/10.1074/jbc.M109.029371
- Y. Hu, S. Raynard, M.G. Sehorn, X. Lu, W. Bussen, L. Zheng, J.M. Stark, E.L. Barnes, P. Chi, P. Janscak, M. Jasin, H. Vogel, P. Sung, and G. Luo, "RECQL5/Recql5 helicase regulates homologous recombination and suppresses tumor formation via disruption of Rad51 presynaptic filaments", Genes & Development, vol. 21, pp. 3073-3084, 2007. http://dx.doi.org/10.1101/gad.1609107
- S. Schwendener, S. Raynard, S. Paliwal, A. Cheng, R. Kanagaraj, I. Shevelev, J.M. Stark, P. Sung, and P. Janscak, "Physical Interaction of RECQ5 Helicase with RAD51 Facilitates Its Anti-recombinase Activity", Journal of Biological Chemistry, vol. 285, pp. 15739-15745, 2010. http://dx.doi.org/10.1074/jbc.M110.110478
- L. Zheng, R. Kanagaraj, B. Mihaljevic, S. Schwendener, A.A. Sartori, B. Gerrits, I. Shevelev, and P. Janscak, "MRE11 complex links RECQ5 helicase to sites of DNA damage", Nucleic Acids Research, vol. 37, pp. 2645-2657, 2009. http://dx.doi.org/10.1093/nar/gkp147
- J. Simandlova, J. Zagelbaum, M.J. Payne, W.K. Chu, I. Shevelev, K. Hanada, S. Chatterjee, D.A. Reid, Y. Liu, P. Janscak, E. Rothenberg, and I.D. Hickson, "FBH1 Helicase Disrupts RAD51 Filaments in Vitro and Modulates Homologous Recombination in Mammalian Cells", Journal of Biological Chemistry, vol. 288, pp. 34168-34180, 2013. http://dx.doi.org/10.1074/jbc.M113.484493
- W.K. Chu, M.J. Payne, P. Beli, K. Hanada, C. Choudhary, and I.D. Hickson, "FBH1 influences DNA replication fork stability and homologous recombination through ubiquitylation of RAD51", Nature Communications, vol. 6, 2015. http://dx.doi.org/10.1038/ncomms6931
- G. Moldovan, D. Dejsuphong, M. Petalcorin, K. Hofmann, S. Takeda, S. Boulton, and A. D'Andrea, "Inhibition of Homologous Recombination by the PCNA-Interacting Protein PARI", Molecular Cell, vol. 45, pp. 75-86, 2012. http://dx.doi.org/10.1016/j.molcel.2011.11.010
- K. Somyajit, S. Saxena, S. Babu, A. Mishra, and G. Nagaraju, "Mammalian RAD51 paralogs protect nascent DNA at stalled forks and mediate replication restart", Nucleic Acids Research, pp. gkv880, 2015. http://dx.doi.org/10.1093/nar/gkv880
- K. Sato, M. Shimomuki, Y. Katsuki, D. Takahashi, W. Kobayashi, M. Ishiai, H. Miyoshi, M. Takata, and H. Kurumizaka, "FANCI-FANCD2 stabilizes the RAD51-DNA complex by binding RAD51 and protects the 5′-DNA end", Nucleic Acids Research, vol. 44, pp. 10758-10771, 2016. http://dx.doi.org/10.1093/nar/gkw876
- I. Chaudhury, D.R. Stroik, and A. Sobeck, "FANCD2-Controlled Chromatin Access of the Fanconi-Associated Nuclease FAN1 Is Crucial for the Recovery of Stalled Replication Forks", Molecular and Cellular Biology, vol. 34, pp. 3939-3954, 2014. http://dx.doi.org/10.1128/mcb.00457-14
- M. Higgs, J. Reynolds, A. Winczura, A. Blackford, V. Borel, E. Miller, A. Zlatanou, J. Nieminuszczy, E. Ryan, N. Davies, T. Stankovic, S. Boulton, W. Niedzwiedz, and G. Stewart, "BOD1L Is Required to Suppress Deleterious Resection of Stressed Replication Forks", Molecular Cell, vol. 59, pp. 462-477, 2015. http://dx.doi.org/10.1016/j.molcel.2015.06.007
- M.R. Higgs, and G.S. Stewart, "Protection or resection: BOD1L as a novel replication fork protection factor", Nucleus, vol. 7, pp. 34-40, 2016. http://dx.doi.org/10.1080/19491034.2016.1143183
-
G. Leuzzi, V. Marabitti, P. Pichierri, and A. Franchitto, "
WRNIP 1 protects stalled forks from degradation and promotes fork restart after replication stress", The EMBO Journal, vol. 35, pp. 1437-1451, 2016. http://dx.doi.org/10.15252/embj.201593265 - Y. Yang, Z. Liu, F. Wang, P. Temviriyanukul, X. Ma, Y. Tu, L. Lv, Y. Lin, M. Huang, T. Zhang, H. Pei, B.P. Chen, J.G. Jansen, N. de Wind, P.L. Fischhaber, E.C. Friedberg, T. Tang, and C. Guo, "FANCD2 and REV1 cooperate in the protection of nascent DNA strands in response to replication stress", Nucleic Acids Research, vol. 43, pp. 8325-8339, 2015. http://dx.doi.org/10.1093/nar/gkv737
- A. Abeyta, M. Castella, C. Jacquemont, and T. Taniguchi, "NEK8 regulates DNA damage-induced RAD51 foci formation and replication fork protection", Cell Cycle, vol. 16, pp. 335-347, 2017. http://dx.doi.org/10.1080/15384101.2016.1259038
- S. Inano, K. Sato, Y. Katsuki, W. Kobayashi, H. Tanaka, K. Nakajima, S. Nakada, H. Miyoshi, K. Knies, A. Takaori-Kondo, D. Schindler, M. Ishiai, H. Kurumizaka, and M. Takata, "RFWD3-Mediated Ubiquitination Promotes Timely Removal of Both RPA and RAD51 from DNA Damage Sites to Facilitate Homologous Recombination", Molecular Cell, vol. 66, pp. 622-634.e8, 2017. http://dx.doi.org/10.1016/j.molcel.2017.04.022
- F. Su, S. Bhattacharya, S. Abdisalaam, S. Mukherjee, H. Yajima, Y. Yang, R. Mishra, K. Srinivasan, S. Ghose, D.J. Chen, S.M. Yannone, and A. Asaithamby, "Replication stress induced site-specific phosphorylation targets WRN to the ubiquitin-proteasome pathway", Oncotarget, vol. 7, pp. 46-65, 2015. http://dx.doi.org/10.18632/oncotarget.6659
- N. Tomimatsu, B. Mukherjee, J.L. Harris, F.L. Boffo, M.C. Hardebeck, P.R. Potts, K.K. Khanna, and S. Burma, "DNA-damage-induced degradation of EXO1 exonuclease limits DNA end resection to ensure accurate DNA repair", Journal of Biological Chemistry, vol. 292, pp. 10779-10790, 2017. http://dx.doi.org/10.1074/jbc.M116.772475
- J. Sommers, A. Suhasini, and R. Brosh, "Protein Degradation Pathways Regulate the Functions of Helicases in the DNA Damage Response and Maintenance of Genomic Stability", Biomolecules, vol. 5, pp. 590-616, 2015. http://dx.doi.org/10.3390/biom5020590
- P. Akan, A. Alexeyenko, P. Costea, L. Hedberg, B. Solnestam, S. Lundin, J. Hällman, E. Lundberg, M. Uhlén, and J. Lundeberg, "Comprehensive analysis of the genome transcriptome and proteome landscapes of three tumor cell lines", Genome Medicine, vol. 4, pp. 86, 2012. http://dx.doi.org/10.1186/gm387
- Y. Chen, H. Liu, H. Zhang, C. Sun, Z. Hu, Q. Tian, C. Peng, P. Jiang, H. Hua, X. Li, and H. Pei, "And-1 coordinates with CtIP for efficient homologous recombination and DNA damage checkpoint maintenance", Nucleic Acids Research, vol. 45, pp. 2516-2530, 2016. http://dx.doi.org/10.1093/nar/gkw1212
- Y. Li, Z. Li, R. Wu, Z. Han, and W. Zhu, "And-1 is required for homologous recombination repair by regulating DNA end resection", Nucleic Acids Research, vol. 45, pp. 2531-2545, 2016. http://dx.doi.org/10.1093/nar/gkw1241
- H.E. Bryant, N. Schultz, H.D. Thomas, K.M. Parker, D. Flower, E. Lopez, S. Kyle, M. Meuth, N.J. Curtin, and T. Helleday, "Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase", Nature, vol. 434, pp. 913-917, 2005. http://dx.doi.org/10.1038/nature03443
- C.J. Lord, A.N. Tutt, and A. Ashworth, "Synthetic Lethality and Cancer Therapy: Lessons Learned from the Development of PARP Inhibitors", Annual Review of Medicine, vol. 66, pp. 455-470, 2015. http://dx.doi.org/10.1146/annurev-med-050913-022545
- K.K. Dhillon, I. Bajrami, T. Taniguchi, and C.J. Lord, "Synthetic lethality: the road to novel therapies for breast cancer", Endocrine-Related Cancer, vol. 23, pp. T39-T55, 2016. http://dx.doi.org/10.1530/erc-16-0228
- C.J. Lord, and A. Ashworth, "Mechanisms of resistance to therapies targeting BRCA-mutant cancers", Nature Medicine, vol. 19, pp. 1381-1388, 2013. http://dx.doi.org/10.1038/nm.3369
- S. Ying, F.C. Hamdy, and T. Helleday, "Mre11-Dependent Degradation of Stalled DNA Replication Forks Is Prevented by BRCA2 and PARP1", Cancer Research, vol. 72, pp. 2814-2821, 2012. http://dx.doi.org/10.1158/0008-5472.can-11-3417
- X. Ding, A.R. Chaudhuri, E. Callen, Y. Pang, K. Biswas, K.D. Klarmann, B.K. Martin, S. Burkett, L. Cleveland, S. Stauffer, T. Sullivan, A. Dewan, H. Marks, A.T. Tubbs, N. Wong, E. Buehler, K. Akagi, S.E. Martin, J.R. Keller, A. Nussenzweig, and S.K. Sharan, "Synthetic viability by BRCA2 and PARP1/ARTD1 deficiencies", Nature Communications, vol. 7, 2016. http://dx.doi.org/10.1038/ncomms12425
- W. Feng, and M. Jasin, "BRCA2 suppresses replication stress-induced mitotic and G1 abnormalities through homologous recombination", Nature Communications, vol. 8, 2017. http://dx.doi.org/10.1038/s41467-017-00634-0
- J. Haince, D. McDonald, A. Rodrigue, U. Déry, J. Masson, M.J. Hendzel, and G.G. Poirier, "PARP1-dependent Kinetics of Recruitment of MRE11 and NBS1 Proteins to Multiple DNA Damage Sites", Journal of Biological Chemistry, vol. 283, pp. 1197-1208, 2008. http://dx.doi.org/10.1074/jbc.M706734200
- P.M. Schoonen, F. Talens, C. Stok, E. Gogola, A.M. Heijink, P. Bouwman, F. Foijer, M. Tarsounas, S. Blatter, J. Jonkers, S. Rottenberg, and M.A.T.M. van Vugt, "Progression through mitosis promotes PARP inhibitor-induced cytotoxicity in homologous recombination-deficient cancer cells", Nature Communications, vol. 8, 2017. http://dx.doi.org/10.1038/ncomms15981
- M. Li, and X. Yu, "Function of BRCA1 in the DNA Damage Response Is Mediated by ADP-Ribosylation", Cancer Cell, vol. 23, pp. 693-704, 2013. http://dx.doi.org/10.1016/j.ccr.2013.03.025
- S. Ying, Z. Chen, A.L. Medhurst, J.A. Neal, Z. Bao, O. Mortusewicz, J. McGouran, X. Song, H. Shen, F.C. Hamdy, B.M. Kessler, K. Meek, and T. Helleday, "DNA-PKcs and PARP1 Bind to Unresected Stalled DNA Replication Forks Where They Recruit XRCC1 to Mediate Repair", Cancer Research, vol. 76, pp. 1078-1088, 2016. http://dx.doi.org/10.1158/0008-5472.can-15-0608
- S. Park, S.L. Ciccone, B. Freie, A. Kurimasa, D.J. Chen, G.C. Li, D.W. Clapp, and S. Lee, "A Positive Role for the Ku Complex in DNA Replication Following Strand Break Damage in Mammals", Journal of Biological Chemistry, vol. 279, pp. 6046-6055, 2004. http://dx.doi.org/10.1074/jbc.M311054200
- A.K. Ashley, M. Shrivastav, J. Nie, C. Amerin, K. Troksa, J.G. Glanzer, S. Liu, S.O. Opiyo, D.D. Dimitrova, P. Le, B. Sishc, S.M. Bailey, G.G. Oakley, and J.A. Nickoloff, "DNA-PK phosphorylation of RPA32 Ser4/Ser8 regulates replication stress checkpoint activation, fork restart, homologous recombination and mitotic catastrophe", DNA Repair, vol. 21, pp. 131-139, 2014. http://dx.doi.org/10.1016/j.dnarep.2014.04.008
- S. Liu, S.O. Opiyo, K. Manthey, J.G. Glanzer, A.K. Ashley, C. Amerin, K. Troksa, M. Shrivastav, J.A. Nickoloff, and G.G. Oakley, "Distinct roles for DNA-PK, ATM and ATR in RPA phosphorylation and checkpoint activation in response to replication stress", Nucleic Acids Research, vol. 40, pp. 10780-10794, 2012. http://dx.doi.org/10.1093/nar/gks849
- G.J. Grundy, H.A. Moulding, K.W. Caldecott, and S.L. Rulten, "One ring to bring them all—The role of Ku in mammalian non-homologous end joining", DNA Repair, vol. 17, pp. 30-38, 2014. http://dx.doi.org/10.1016/j.dnarep.2014.02.019
- Z. Kais, B. Rondinelli, A. Holmes, C. O’Leary, D. Kozono, A. D’Andrea, and R. Ceccaldi, "FANCD2 Maintains Fork Stability in BRCA1/2-Deficient Tumors and Promotes Alternative End-Joining DNA Repair", Cell Reports, vol. 15, pp. 2488-2499, 2016. http://dx.doi.org/10.1016/j.celrep.2016.05.031
- S.A. Yazinski, V. Comaills, R. Buisson, M. Genois, H.D. Nguyen, C.K. Ho, T. Todorova Kwan, R. Morris, S. Lauffer, A. Nussenzweig, S. Ramaswamy, C.H. Benes, D.A. Haber, S. Maheswaran, M.J. Birrer, and L. Zou, "ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells", Genes & Development, vol. 31, pp. 318-332, 2017. http://dx.doi.org/10.1101/gad.290957.116
- S.R. Patel, D. Kim, I. Levitan, and G.R. Dressler, "The BRCT-Domain Containing Protein PTIP Links PAX2 to a Histone H3, Lysine 4 Methyltransferase Complex", Developmental Cell, vol. 13, pp. 580-592, 2007. http://dx.doi.org/10.1016/j.devcel.2007.09.004
- A.Y. Lai, and P.A. Wade, "Cancer biology and NuRD: a multifaceted chromatin remodelling complex", Nature Reviews Cancer, vol. 11, pp. 588-596, 2011. http://dx.doi.org/10.1038/nrc3091
- S. Guillemette, R.W. Serra, M. Peng, J.A. Hayes, P.A. Konstantinopoulos, M.R. Green, and S.B. Cantor, "Resistance to therapy in BRCA2 mutant cells due to loss of the nucleosome remodeling factor CHD4", Genes & Development, vol. 29, pp. 489-494, 2015. http://dx.doi.org/10.1101/gad.256214.114
- B. Rondinelli, E. Gogola, H. Yücel, A.A. Duarte, M. van de Ven, R. van der Sluijs, P.A. Konstantinopoulos, J. Jonkers, R. Ceccaldi, S. Rottenberg, and A.D. D’Andrea, "EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation", Nature Cell Biology, vol. 19, pp. 1371-1378, 2017. http://dx.doi.org/10.1038/ncb3626
- K. Schlacher, "PARPi focus the spotlight on replication fork protection in cancer", Nature Cell Biology, vol. 19, pp. 1309-1310, 2017. http://dx.doi.org/10.1038/ncb3638
- S. Gasser, S. Orsulic, E.J. Brown, and D.H. Raulet, "The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor", Nature, vol. 436, pp. 1186-1190, 2005. http://dx.doi.org/10.1038/nature03884
- S. Gasser, and D.H. Raulet, "The DNA Damage Response Arouses the Immune System", Cancer Research, vol. 66, pp. 3959-3962, 2006. http://dx.doi.org/10.1158/0008-5472.can-05-4603
- Q. Chen, L. Sun, and Z.J. Chen, "Regulation and function of the cGAS–STING pathway of cytosolic DNA sensing", Nature Immunology, vol. 17, pp. 1142-1149, 2016. http://dx.doi.org/10.1038/ni.3558
- D. Kavanagh, D. Spitzer, P. Kothari, A. Shaikh, M.K. Liszewski, A. Richards, and J.P. Atkinson, "New roles for the major human 3′-5′ exonuclease TREX1 in human disease", Cell Cycle, vol. 7, pp. 1718-1725, 2008. http://dx.doi.org/10.4161/cc.7.12.6162
- Y.J. Crow, and J. Rehwinkel, "Aicardi-Goutieres syndrome and related phenotypes: linking nucleic acid metabolism with autoimmunity", Human Molecular Genetics, vol. 18, pp. R130-R136, 2009. http://dx.doi.org/10.1093/hmg/ddp293
- G. Chatzinikolaou, I. Karakasilioti, and G.A. Garinis, "DNA damage and innate immunity: links and trade-offs", Trends in Immunology, vol. 35, pp. 429-435, 2014. http://dx.doi.org/10.1016/j.it.2014.06.003
- I.S. Pateras, S. Havaki, X. Nikitopoulou, K. Vougas, P.A. Townsend, M.I. Panayiotidis, A.G. Georgakilas, and V.G. Gorgoulis, "The DNA damage response and immune signaling alliance: Is it good or bad? Nature decides when and where", Pharmacology & Therapeutics, vol. 154, pp. 36-56, 2015. http://dx.doi.org/10.1016/j.pharmthera.2015.06.011
- D.A. Lehtinen, S. Harvey, M.J. Mulcahy, T. Hollis, and F.W. Perrino, "The TREX1 Double-stranded DNA Degradation Activity Is Defective in Dominant Mutations Associated with Autoimmune Disease", Journal of Biological Chemistry, vol. 283, pp. 31649-31656, 2008. http://dx.doi.org/10.1074/jbc.M806155200
- C. Vanpouille-Box, A. Alard, M.J. Aryankalayil, Y. Sarfraz, J.M. Diamond, R.J. Schneider, G. Inghirami, C.N. Coleman, S.C. Formenti, and S. Demaria, "DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity", Nature Communications, vol. 8, 2017. http://dx.doi.org/10.1038/ncomms15618
- N. Yan, "Immune Diseases Associated with TREX1 and STING Dysfunction", Journal of Interferon & Cytokine Research, vol. 37, pp. 198-206, 2017. http://dx.doi.org/10.1089/jir.2016.0086
- Y. Yang, T. Lindahl, and D.E. Barnes, "Trex1 Exonuclease Degrades ssDNA to Prevent Chronic Checkpoint Activation and Autoimmune Disease", Cell, vol. 131, pp. 873-886, 2007. http://dx.doi.org/10.1016/j.cell.2007.10.017
- E. Erdal, S. Haider, J. Rehwinkel, A.L. Harris, and P.J. McHugh, "A prosurvival DNA damage-induced cytoplasmic interferon response is mediated by end resection factors and is limited by Trex1", Genes & Development, vol. 31, pp. 353-369, 2017. http://dx.doi.org/10.1101/gad.289769.116
- T. Miyazaki, Y. Kim, J. Yoon, H. Wang, T. Suzuki, and H.C. Morse, "The 3′–5′ DNA Exonuclease TREX1 Directly Interacts with Poly(ADP-ribose) Polymerase-1 (PARP1) during the DNA Damage Response", Journal of Biological Chemistry, vol. 289, pp. 32548-32558, 2014. http://dx.doi.org/10.1074/jbc.M114.547331
- T. Kondo, J. Kobayashi, T. Saitoh, K. Maruyama, K.J. Ishii, G.N. Barber, K. Komatsu, S. Akira, and T. Kawai, "DNA damage sensor MRE11 recognizes cytosolic double-stranded DNA and induces type I interferon by regulating STING trafficking", Proceedings of the National Academy of Sciences, vol. 110, pp. 2969-2974, 2013. http://dx.doi.org/10.1073/pnas.1222694110
- H. Sui, M. Zhou, H. Imamichi, X. Jiao, B.T. Sherman, H.C. Lane, and T. Imamichi, "STING is an essential mediator of the Ku70-mediated production of IFN-λ1 in response to exogenous DNA", Science Signaling, vol. 10, 2017. http://dx.doi.org/10.1126/scisignal.aah5054
- G.I. Rice, J. Bond, A. Asipu, R.L. Brunette, I.W. Manfield, I.M. Carr, J.C. Fuller, R.M. Jackson, T. Lamb, T.A. Briggs, M. Ali, H. Gornall, L.R. Couthard, A. Aeby, S.P. Attard-Montalto, E. Bertini, C. Bodemer, K. Brockmann, L.A. Brueton, P.C. Corry, I. Desguerre, E. Fazzi, A.G. Cazorla, B. Gener, B.C.J. Hamel, A. Heiberg, M. Hunter, M.S. van der Knaap, R. Kumar, L. Lagae, P.G. Landrieu, C.M. Lourenco, D. Marom, M.F. McDermott, W. van der Merwe, S. Orcesi, J.S. Prendiville, M. Rasmussen, S.A. Shalev, D.M. Soler, M. Shinawi, R. Spiegel, T.Y. Tan, A. Vanderver, E.L. Wakeling, E. Wassmer, E. Whittaker, P. Lebon, D.B. Stetson, D.T. Bonthron, and Y.J. Crow, "Mutations involved in Aicardi-Goutières syndrome implicate SAMHD1 as regulator of the innate immune response", Nature Genetics, vol. 41, pp. 829-832, 2009. http://dx.doi.org/10.1038/ng.373
- W. Daddacha, A.E. Koyen, A.J. Bastien, P.E. Head, V.R. Dhere, G.N. Nabeta, E.C. Connolly, E. Werner, M.Z. Madden, M.B. Daly, E.V. Minten, D.R. Whelan, A.J. Schlafstein, H. Zhang, R. Anand, C. Doronio, A.E. Withers, C. Shepard, R.K. Sundaram, X. Deng, W.S. Dynan, Y. Wang, R.S. Bindra, P. Cejka, E. Rothenberg, P.W. Doetsch, B. Kim, and D.S. Yu, "SAMHD1 Promotes DNA End Resection to Facilitate DNA Repair by Homologous Recombination", Cell Reports, vol. 20, pp. 1921-1935, 2017. http://dx.doi.org/10.1016/j.celrep.2017.08.008
- C. Wolf, A. Rapp, N. Berndt, W. Staroske, M. Schuster, M. Dobrick-Mattheuer, S. Kretschmer, N. König, T. Kurth, D. Wieczorek, K. Kast, M.C. Cardoso, C. Günther, and M.A. Lee-Kirsch, "RPA and Rad51 constitute a cell intrinsic mechanism to protect the cytosol from self DNA", Nature Communications, vol. 7, 2016. http://dx.doi.org/10.1038/ncomms11752
ACKNOWLEDGMENTS
I am grateful to Drs. Lopes, Vindigni, and Schlacher for discussions, and to Dr. Pasero for com-municating results prior to publication. I hope to have captured the most recent developments in this rapidly moving field, and I apologize to the researchers whose work was not included in the interests of space and specific focus, or not discussed in a greater detail. The work in the author’s lab is supported by NIH grants GM115482, CA193808, and CA215647.
COPYRIGHT
© 2017

A game of substrates: replication fork remodeling and its roles in genome stability and chemo-resistance by Sidorova is licensed under a Creative Commons Attribution 4.0 International License.


