Reviews:
Cell Stress, Vol. 4, No. 1, pp. 9 - 23; doi: 10.15698/cst2020.01.209
Fatty acids – from energy substrates to key regulators of cell survival, proliferation and effector function

1 Barts Cancer Institute, Queen Mary University of London, Charterhouse Square, London EC1M 6BQ, UK.
2 Laboratory for Lipidomics and Lipid Biology, Division of Pharmacy and Optometry, Faculty of Biology, Medicine and Health, School of Health sciences, The University of Manchester, Manchester Academic Health Science Centre, Oxford Road, Manchester M13 9PT, UK.
3 Institute of Inflammation and Ageing, College of Medical and Dental Sciences, University of Birmingham, Mindelsohn Way, Birmingham B15 2WB, UK.
4 Lydia Becker Institute of Immunology and Inflammation, Faculty of Biology, Medicine and Health, The University of Manchester, Manchester Academic Health Science Centre, Oxford Road, Manchester M13 9PT, UK.
5 Institute of Cardiovascular Sciences, College of Medical and Dental Sciences, University of Birmingham, Mindelsohn Way, Birmingham B15 2WB, UK.
6 Institute of Metabolism and Systems Research, College of Medical and Dental Sciences, University of Birmingham, Mindelsohn Way, Birmingham B15 2WB, UK.
Keywords: fatty acids; immune cells; T cells; cancer cells; metastasis; cancer immunology.
Abbreviatons:
ACC – acetyl-CoA carboxylase,
ACLY – ATP citrate lyase,
CPT – carnitine palmitoyl transferases,
CRPC – castration-resistant prostate cancer,
DHA – docosahexaenoic acid,
EAE – experimental autoimmune encephalomyelitis,
EMT – epithelial to mesenchymal transition, EPA – eicosapentaenoic acid,
FABP – Fatty acid binding protein,
FADS – fatty acyl-CoA desaturase,
FAS – fatty acid synthase,
GA – gastric adenocarcinoma,
HCC – hepatocellular carcinoma,
HDAC – histone deacetylase,
HFD – high fat diet,
ILC – innate lymphoid cell,
LAL – lysosomal acid lipase,
LMW-E – low molecular weight isoform of cyclin E,
LPS – lipopolysaccharide,
mCRPC – metastatic CRCP,
NK – natural killer,
PI(4,5)P2 – phosphatidylinositol (4,5) bisphosphate,
PLD – phospholipase D,
PPAR – peroxisome proliferator-activated receptor,
PUFA – polyunsaturated fatty acid,
SCD – stearoyl-CoA desaturase,
SREBP – sterol regulatory element binding protein,
Tconv – conventional T cell,
TCR – T cell receptor,
Teff – T effector cell,
Th – T helper cell,
Tm – T memory cell, TNBC – triple-negative breast cancer,
Treg – regulatory T cell,
wt – wild type.
Received originally: 07/10/2019 Received in revised form: 02/12/2019
Accepted: 04/12/2019
Published: 10/12/2019
Correspondence:
Anna Nicolaou, Stopford Building, Oxford Road, Manchester M13 9PT, UK; anna.nicolaou@manchester.ac.uk
Claudio Mauro, Queen Elizabeth Hospital, Mindelsohn Way, Birmingham B15 2WB, UK; c.mauro@bham.ac.uk
Conflict of interest statement: The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Please cite this article as: Danilo Cucchi, Dolores Camacho-Muñoz, Michelangelo Certo, Valentina Pucino, Anna Nicolaou and Claudio Mauro (2019). Fatty acids - from energy substrates to key regu-lators of cell survival, proliferation and effector function. Cell Stress 4(1): 9-23. doi: 10.15698/cst2020.01.209
Abstract
Recent advances in immunology and cancer research show that fatty acids, their metabolism and their sensing have a crucial role in the biology of many different cell types. Indeed, they are able to affect cellular behaviour with great implications for pathophysiology. Both the catabolic and anabolic pathways of fatty acids present us with a number of enzymes, receptors and agonists/antagonists that are potential therapeutic targets, some of which have already been successfully pursued. Fatty acids can affect the differentiation of immune cells, particularly T cells, as well as their activation and function, with important consequences for the balance between anti- and pro-inflammatory signals in immune diseases, such as rheumatoid arthritis, psoriasis, diabetes, obesity and cardiovascular conditions. In the context of cancer biology, fatty acids mainly provide substrates for energy production, which is of crucial importance to meet the energy demands of these highly proliferating cells. Fatty acids can also be involved in a broader transcriptional programme as they trigger signals necessary for tumorigenesis and can confer to cancer cells the ability to migrate and generate distant metastasis. For these reasons, the study of fatty acids represents a new research direction that can generate detailed insight and provide novel tools for the understanding of immune and cancer cell biology, and, more importantly, support the development of novel, efficient and fine-tuned clinical interventions. Here, we review the recent literature focusing on the involvement of fatty acids in the biology of immune cells, with emphasis on T cells, and cancer cells, from sensing and binding, to metabolism and downstream effects in cell signalling.
INTRODUCTION
Lipids are important biomolecules involved in a plethora of biological processes, spanning from the production and storage of energy [1] and assembly and function of the cellular membranes [2] to activation of genes [3] and modulation of signalling pathways [4]. This diverse group of compounds comprises fatty acids, glycerolipids, glycerophospholipids, sphingolipids, sterol lipids, prenol lipids, saccharolipids and polyketides [5][6][7]; typically, the cellular lipidome comprises more than 2,000 species. This structural diversity endows lipids with varied properties that enable and support a plethora of structural and functional roles.
–
As lipids are not encoded, their levels are regulated both by nutritional intake and biosynthetic pathways found in almost all cell types. Fatty acids are simple lipids biosynthesised by the complex enzyme fatty acid synthase (FAS) through the sequential elongation of acetyl-CoA [8]. The resulting 16-carbon (C) acyl chain palmitic acid can be further elongated and desaturated. However, humans and other animals require nutritional intake of two essential fatty acids -linoleic acid and alpha-linolenic acid – precursors to long and very long polyunsaturated fatty acids (PUFA). As well as contributing to the structural diversity of membrane glycerophospholipids and sphingolipids, PUFA are metabolised to potent autacoids, hormone-like mediators known for their involvement in inflammation and immunity [9][10][11], while the catabolism of fatty acids via β-oxidation provides the cells with an efficient way for energy production. Membrane glycerophospholipids and sphingolipids, as well as acylglycerols (e.g. triacylglycerols) act as cellular pools of esterified fatty acids that can be liberated and further metabolised to meet the energy and other biosynthetic needs of different cell types.
–
In this review, we are discussing the involvement of fatty acids in immunity, particularly in T cells, and cancer, with focus on their roles as energy substrates, regulators of cell survival, proliferation and effector function. Increasing evidence supports the notion that fatty acids can influence the biological behaviour of immune and other cell types when involved in pathophysiological conditions, such as metabolic disorders, autoimmune diseases and cancer; therefore, exploring the metabolism and properties of fatty acids can provide new potential pharmacological targets in the treatment of different clinical conditions.
–
FATTY ACIDS IN IMMUNITY
Growing evidence points to the importance of lipid metabolism and signalling in the function of different types of immune cells, both in a homeostatic state and during the immune response. Fatty acids and their derivatives are of particular importance and recent discoveries have highlighted their role in the regulation and function of immune cells, in particular T lymphocytes (Figure 1).
–
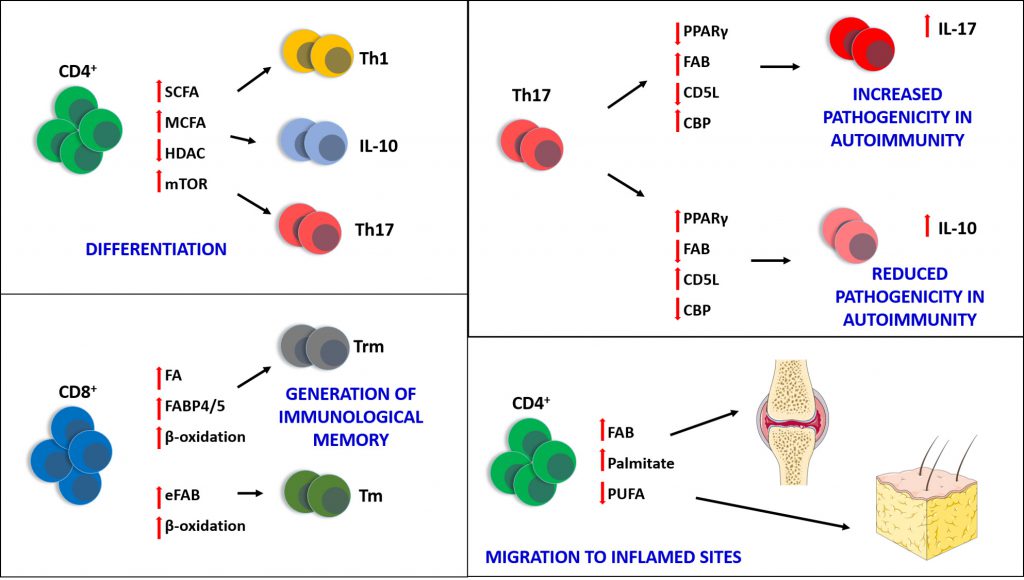 |
FIGURE 1: Fatty acids are crucial to many biological processes occurring in immune cells, particularly T lymphocytes. Short (C2-4)- and medium (C6-14) chain fatty acids (SCFA and MCFA) are able to influence the differentiation of CD4+ in Th1, Th17 and IL-10+ T cells, by downregulating histone deacetylase (HDAC) activity and activating mammalian target of rapamycin (mTOR). The generation of CD8+ tissue resident memory T cells (Trm) also relies upon the availability of fatty acids (FA) available for β-oxidation and the presence of the fatty acid binding proteins FABP4 and FABP5. The generation of central memory CD8+ T cells (Tm) instead relies upon endogenous fatty acid biosynthesis (eFAB), which also provides substrates for β-oxidation, crucial for the long-term survival of these cells. The pathogenicity of Th17 can be also regulated by fatty acids: by increasing the activity of fatty acid biosynthesis (FAB) and cholesterol biosynthesis (CBP), and downregulating the expression of PPARγ and CD5L, Th17 cells produce IL17 and become more pathogenic, especially in auto-immune diseases such as multiple sclerosis and rheumatoid arthritis. Conversely, upregulation of PPARγ and CD5L, and blocking of FAB and CBP, can reduce the pathogenicity of Th17 and increase production of IL10. Finally, saturated fatty acids (e.g. palmitate) can enable the migration of T cells towards non-lymphoid inflamed sites (e.g. joints in arthritis and fat tissue in obesity) where they sustain chronic inflammation; polyunsaturated fatty acids (PUFA) can have the opposite effect and inhibit T cell migration, exerting anti-inflammatory properties. |
–
Sensing and binding
Fatty acids bind to several surface receptor members of the family of G protein coupled receptors (GPCR) [12][13]. Of particular interest is GPR84, which has affinity for fatty acyl chain length of 6-14 C (medium chain fatty acids) and is expressed by a variety of immune cells, mainly neutrophils and monocytes/macrophages, and to a lesser extent, by CD4+ and CD8+ T cells. Expression of GPR84 both in human and mouse monocytes is upregulated upon lipopolysaccharide (LPS) stimulation, suggesting that signals triggered by medium chain fatty acids may have a role in monocyte/macrophage activation [14]. Moreover, medium chain saturated fatty acids such as caproic acid (C6), undecanoic acid (C11) and lauric acid (C12), are able to stimulate the secretion of interleukin 12 p40 subunit (IL12 p40) in mouse LPS-stimulated RAW264.7 cells, showing that they can have a role in regulating the activity of different immune cells during inflammation [14].
–
It has been recently reported that the short chain fatty acids acetate (C2), propionate (C3) and butyrate (C4) are able to orchestrate the differentiation of CD4+ T cells in effector or regulatory cells modulating the activity of histone deacetylases (HDAC) [15]. Short chain fatty acids can also inhibit HDAC in T cells leading to the amplification of the mTOR pathway, through acetylation of p70 S6 kinase and consequent phosphorylation of rS6, all events required for the differentiation in T helper 17 (Th17), T helper 1 (Th1) and IL-10+ T cells [15]. As short chain fatty acids can be readily uptaken through the plasma membrane [16], their effect was found to be independent of the surface receptors GPR41 and GPR43, which can sense acetate, butyrate and propionate [17], and have been shown to be important in regulating intestinal inflammatory responses [18]. These findings identify short chain fatty acids as crucial gut metabolites affecting the balance of effector and regulatory T cells and orchestrating the immune response in the gut.
–
Long chain fatty acids (14-22 C), such as the saturated palmitic acid (C16:0) and monounsaturated oleic acid (C18:1), can also be sensed by CD36, a fatty acid translocase required for the uptake of fatty acids in the gut [19], liver [20], myocardium, skeletal muscle and adipose tissue [21][22]. CD36 expression in endothelial cells (EC) is instrumental for the optimal translocation of long chain fatty acids from circulation to cardiomyocytes, skeletal muscle and adipose tissue, with important consequences for fatty acid metabolism, glucose utilization, glucose tolerance and insulin sensitivity [23]. CD36 also binds to a range of lipidic ligands, such as oxidized phospholipids [24], oxidized LDL [25] and native lipoproteins [26]. CD36 was found to be particularly important for the activity of phagocytes during inflammation, by functioning as a scavenger receptor [27] and by cooperating with several Toll-like receptors [28][29][30]. CD36 mutations in humans have been associated with CD36 deficiency in platelets and monocytes, with potential consequences for their functions [31]. CD36 loss has been reported to reduce macrophage infiltration in adipose tissue [32], while pharmacological inhibition of CD36 in vitro reduces saturated fatty acid uptake (e.g. palmitic acid (16:0) and stearic acid (18:0)) in macrophages and ameliorates insulin signalling in adipocytes. More importantly, genetic ablation of CD36 in the hematopoietic compartment led to a reduced infiltration of macrophages and improved insulin signalling in the adipose tissue of mice fed a high fat diet (HFD) [32], although it did not reduce the accumulation of long chain fatty acids [32][33], suggesting that some of the CD36-mediated functions in macrophages do not depend on its fatty acid translocase activity. All these findings highlight the importance of CD36 as a target for the treatment of metabolic disorders with an inflammatory component, such as obesity and diabetes. T cells also express CD36 on their surface, with T memory (Tm) cells showing lower levels than T effector (Teff) cells [34].
–
Fatty acid binding proteins (FABP) are a family of intracellular and extracellular proteins that bind saturated and unsaturated fatty acids [35]. It is now clear that these proteins not only buffer and transport fatty acids, but are also deeply involved in the regulation of their metabolism with consequences for cell signalling, particularly during inflammation [36][37]. Recently, tissue-resident memory Trm cells have been shown to be dependent on the activity of FABP4 and FABP5 for long-term survival. Pan et al. [38] demonstrated that the deficiency of FABP4/5 impairs the uptake of fatty acids such as palmitate, by skin CD8+ Trm cells, thus reducing their long-term survival in vivo. CD8+ Trm cells lacking FABP4/5 fail to increase mitochondrial oxidative metabolism in the presence of fatty acids in vitro, and their persistence in vivo was significantly reduced due to inhibition of β-oxidation. Finally, FABP4 and FABP5 were also found upregulated in human CD8+ Trm cells isolated from normal and psoriatic skin, confirming the importance of fatty acids in the maintenance and longevity of this tissue-resident protective immune population [38].
–
Cellular fatty acids and their metabolites activate different signals via binding peroxisome proliferator-activated receptors (PPAR), nuclear receptors involved in the regulation of transcription of genes linked to lipid metabolism [39]. PPARα and β/δ are particularly important in cardiac muscle, brown adipose tissue and liver, whilst PPARγ is more ubiquitously expressed [40][41][42]. These receptors have been proven to be important in the differentiation of a number of T cell subsets [43], particularly in informing the decision of CD4+ T cells toward differentiating to Th17 or T regulatory (Treg) cells [44]. Consistently, Klotz et al. [45] have shown that PPARγ regulates the differentiation of Th17 T cells, by negatively controlling the activity of RORγt. The same report shows that loss of PPARγ increases the severity of experimental autoimmune encephalomyelitis (EAE) and multiple sclerosis in mouse models, leading to a greater infiltration of Th17 cells into the central nervous system [45]. Overall, these findings indicate that activation of PPARγ with selective agonists can inhibit the differentiation of Th17 cells in autoimmune conditions with a strong Th17 component, such as multiple sclerosis, but also rheumatoid arthritis and psoriasis, making PPAR receptors a very promising pharmacological target in autoimmunity.
–
PPARγ was also found to be crucially important for the activity of adipose tissue associated- Treg cells, which express PPARγ at higher level than Treg originating from lymphoid organs [46]. Expression of PPARγ was associated with a cluster of mRNAs involved mainly in leukocytes migration and extravasation (Ccr1, Ccr3, Cxcr6, Cxcl2 and Cxcl3), lipid metabolism (Pcyt1a, Dgat1) and Il10 transcript. Expression of Foxp3 and PPARγ in naïve CD4+ T cells was sufficient to induce the same cluster of mRNAs while additional treatment with PPARγ agonist pioglitazone and rosiglitazone further enhanced the enrichment of transcripts involved in fatty acid transport (Cd36, Slc27a2), biosynthesis (Lipe, Scd1) and oxidation (Cpt1a). Genetic deletion of PPARγ in vivo led to a contraction of the Treg population in adipose tissue with a relative increase in the number of pro-inflammatory macrophages. Furthermore, in obese mice, treatment with pioglitazone enhanced the accumulation of Treg in epididymal adipose tissue and their expression of CD36. This phenotype was abrogated in obese PPAR deficient mice, which also showed a less marked reduction of pro-inflammatory macrophages upon treatment with pioglitazone, as compared to their wild type (wt) counterpart. PPARγ deficient mice did not show an improvement in metabolic parameters (insulin resistance, glucose and insulin tolerance) when treated with pioglitazone [46]. These findings clearly demonstrate that PPARγ is a crucial regulator of the properties of adipose tissue associated- Treg cells and its expression in Treg is necessary for the insulin-sensitizing activity of pioglitazone, with important consequences for the management of obesity-induced insulin resistance.
–
Activation and differentiation
T lymphocytes undergo a clonal expansion upon antigen recognition, which is necessary to mount an appropriate immune response. This process demands not just energy, but also the activation of specific signals required for the proper differentiation and activation of the cell. It has been shown that activation of the T cell receptor (TCR) is accompanied by upregulation of genes involved in the biosynthesis of cholesterol and fatty acids [47]. This anabolic programme is orchestrated by sterol regulatory element binding protein (SREBP) [47]. Interestingly, lack of SREBP by genetic inactivation is particularly detrimental to T cells undergoing clonal expansion after activation, as it does not allow for biosynthesis of cholesterol and fatty acids required to sustain their energy demands. Moreover, SREBP is required for the biosynthesis of cellular membranes, as shown by experiments where addition of cholesterol was able to rescue the growth of T cells [47]. Also, in vivo, the lack of SREBP was responsible for a poor anti-viral response against the lymphocytic choriomeningitis virus, again demonstrating that the ability of activating an anabolic lipid programme for the biosynthesis of cholesterol and fatty acids is a requirement for a proper immune response [47].
–
The generation of Tm cells has also been shown to rely upon mitochondrial fatty acid β-oxidation [34]. It has been shown that Tm cells do not take up extracellular palmitate, unlike Teff, highlighting how different T cell subtypes express different preferences for fatty acid substrates. Survival of Tm cells was reduced in the presence of the FAS inhibitor C75 [48], confirming that these cells rely upon de novo fatty acid biosynthesis for survival. Moreover, Tm cells utilize glucose to newly synthesise fatty acids to fuel oxidative phosphorylation, thus being independent from extracellular fatty acid uptake. The lipolysis occurring in these cells is crucial to their function, and it has been demonstrated that in Tm cells, lipids associate with lysosomes, where lysosomal acid lipase (LAL) produces fatty acids necessary to support β-oxidation. Furthermore, loss of LAL activity reduced the survival of Tm cells, without affecting Teff cells, and also inhibited the development of Tm cells upon infection in vivo [34].
–
Michalek et al. [49] have reported that Teff cells and Treg cells exhibit different metabolic requirements for differentiation and function, with the former being more glycolytic and the latter relying more on lipid oxidation [49], while Berod et al. [50] showed how de novo fatty acid biosynthesis plays a crucial role in determining the differentiation of CD4+ T cells to Th17 or Treg Foxp3+ cells. These authors showed that inhibition of acetyl-CoA carboxylase (ACC) in vitro, using the specific inhibitor Sorafen A, leads to an impaired differentiation of Th17, favouring instead the differentiation of Foxp3+ Treg cells. CD4+ naïve T cells, cultured in Th17 polarizing conditions in the presence of Sorafen A, failed to increase the production of IL-17 and upregulate genes associated with this lineage, such as Hif1alpha and Stat3. To further evaluate the role of ACC as a potential therapeutic target, the phenotype of mice lacking ACC1 (cytosolic isoform of the carboxylase required for the de novo synthesis of fatty acids [51]) in T cells was analysed. When compared to wt mice, and after induction of EAE, mice lacking ACC1 were found to be protected and did not develop any clinical signs of the disease. Similarly, administration of the ACC1 inhibitor Sor-S1036 in wt mice affected by EAE, resulted in a significant delay in disease onset and severity [50].
–
The possibility of exploiting ACC1 as a promising pharmacological target has also been shown in the context of chronic malaria infection [52]. Injection of CD4+ T cells lacking ACC1 in mice infected with Plasmodium chabaudi, resulted in reduced cell survival and was accompanied by reduced generation of Tm cells. A similar result was obtained using TOFA, an inhibitor of ACC1, during T cell priming that increased the number of Teff cells and reduced parasitemia. Inhibition of fatty acid synthesis was shown to be particularly detrimental for Tm cells as they rely more on newly synthesised fatty acids as compared to Teff cells, suggesting that targeting fatty acid biosynthesis in Tm cells may be important in the development of new strategies for fighting chronic infections like malaria [52]. These findings highlight the importance of fatty acid metabolism for the generation of immunological memory and suggest that regulation of this pathway is an interesting prospect for the development of vaccines.
–
The importance of fatty acids in balancing the protective effects of T cell responses and their pathogenicity was addressed by Wang et al., with particular regard to Th17 cells, which can lead to autoimmunity when they are aberrantly activated [53]. In this study, CD5L, a member of the scavenger receptor cysteine-rich superfamily [54], was identified in a single cell RNA-seq screening as a regulator of pathogenicity in Th17 cells and was found expressed only in non-pathogenic Th17 cells, both in vitro and in vivo. CD5L-/- T cells injected in mice affected by EAE led to a more severe phenotype, whilst mice that received wt cells did not show any clinical signs of disease. As CD5L can bind to FAS [55], the lipidome of wt and CD5L-/- Th17 cells was analysed and showed two completely distinct fatty acid profiles: the presence of CD5L increased the levels of PUFA versus saturated fatty acids and mono-unsaturated fatty acids, and reduced cholesterol content. In detail, CD5L is able to suppress the cholesterol synthesis pathway. These changes affect the activity of RORγt by modulating the availability of its ligands and affecting its ability to bind to target genes, such as IL-17a and IL-23r, whilst increasing binding to IL-10, making of CD5L a repressor of pathogenicity of Th17 cells [53]. Overall, these findings show how potent the effect of fatty acid metabolism is on the biology of immune cells, not only as source of energy, but also by sustaining the molecular signals required for differentiation and function.
–
Direct exposure to palmitate was found to be responsible for the biased differentiation of CD4+ T cells towards the expansion of a detrimental, pro-inflammatory effector memory population in obesity [56]. These aberrant T lymphocytes eagerly migrate to non-lymphoid, inflammatory sites (such as the fat tissue during obesity), where they sustain a low-grade chronic inflammation which is a hallmark of many metabolic disorders, such as obesity, type II diabetes and atherosclerosis [56].
–
In obesity, there are many different immune cells that play a role in sustaining the inflammatory response within the adipose tissue. The stress response triggered by hypertrophy in both adipocytes and stromal cells [57][58] is responsible for the recruitment and activation of CD8+ T cells, Th1 cells, and natural killer (NK) cells, leading to the accumulation of pro-inflammatory M1 macrophages [59][60][61]. In 2016, another population of immune cells was demonstrated to play an important role in the maintenance of obesity, the group 1 innate lymphoid cells (ILC1) [62]. The authors showed that residing ILC1 proliferate and accumulate locally, in the adipose depots, during HFD-induced obesity and are the main producer of IFN-γ. The sustained production of IFN-γ is responsible for the polarization of macrophages towards a pro-inflammatory M1 phenotype which then contributes to the development of obesity-associated insulin resistance. The axis IL12-STAT4 was found to be the molecular switch for the production of IFN-γ and the consequent M1 polarization of macrophages [62]. This is another example of how fat accumulation can deeply change the biological behaviour of immune cells, interfering with their homeostatic function, with great consequences for the establishment of pathological conditions.
–
B lymphocytes have also been shown to have an important role in the development of age-induced fat accumulation and insulin resistance [63]. Carter and co-workers [63] found that aging mice (between 6 and 12 months old) have higher number of follicular B2 cells in circulation and epididymal white adipose tissue. The expansion of this B cell population correlated with a higher plasma level of IgG (in particular IgG2c) and glucose intolerance. Moreover, these cells displayed a greater expression of OcaB (B-cell-specific nuclear cofactor Oct coactivator – crucial for B cell maturation [64] and IgG production [65]). Genetic ablation of OcaB resulted in the abrogation of this phenotype, with improvement of glucose intolerance and insulin sensitivity, and reduced fat accumulation during aging due to increased energy expenditure. OcaB-/- mice showed a higher core body temperature and increased metabolic activity in their adipose tissue depots, as shown by enhanced uptake of bromopalmitate and glucose. Replenishment of OcaB-/- mice with OcaB+/+ B cells, via bone marrow transfer, was sufficient to re-induce body weight gain, glucose intolerance and insulin insensitivity [63].
–
Recently, omega-3 (n-3) PUFA have been shown to alter the trafficking of activated CD4+ T cells to fat tissue [66]. Eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) were both able to reduce the number of effector memory CD4+ T cells and change the array of bioactive lipids produced in lymphoid organs and adipose tissues of animals following nutritional supplementation. EPA and DHA were also able to prevent the polarisation of T cells and activation of the small Rho GTPases Rhoα and Rac1, both events required for migration to target/inflamed tissue [66]. EPA and DHA have also been shown to increase the infiltration of CD4+ and CD8+ T cells in UV irradiated human skin, potentially through changes in the network of cutaneous bioactive lipids [67].
–
N-3 PUFA have also been shown to boost B cell antibody production [68] and B cell response [69], but the differential contribution of EPA and DHA to this phenotype was only recently addressed by Teague et al. [70]. The authors showed that a diet enriched with either EPA or DHA resulted in accumulation of EPA and DHA ethyl esters in B cells at the expense of n-6 PUFA. EPA and DHA were also able to increase the frequency of specific subsets of B cells (in particular IgM+ IgD– CD21low CD23– B cells) and while both of them increased the production of IgM, only EPA boosted the production of IgA. Moreover, after ten weeks supplementation, EPA and DHA increased B cell TNFα and IL6 production, but only DHA induced the production of IL10 [70]. The differential activity of n-3 PUFAs on B cells bears important consequences for application in clinical settings, being now clear that EPA and DHA are not biologically equivalent.
–
The availability of fatty acids can impact the function of many proteins through post translational modifications including S-acylation [71]. The most common protein acylation is palmitoylation although use of different acyl groups has been reported [72][73]. Palmitoylation is catalysed by aspartate–histidine–histidine–cysteine (DHHC) acyl transferases and can deeply affect the trafficking, localisation and activity of proteins [74]. Recently, Chopard et al. have highlighted how palmitoylation is important for HIV-1 infection [75]. The viral protein Tat can be secreted by infected cells and subsequently be endocytosed by many different cell types altering the expression of genes related to HIV-associated cancers [76]. Tat can also inhibit the cellular processes that rely upon phosphatidylinositol (4,5) bisphosphate (PI(4,5)P2) for phagocytosis and neurosecretion [77]. Accumulation of Tat in uninfected cells (e.g. T cells, macrophages and neurosecretory cells) is attributed to its palmitoylation by S-acyl transferase DHHC-20 on Cys31, which stabilises the interaction with PI(4,5)P2 thus preventing Tat secretion. The persistence of Tat on plasma membranes and its stable interaction with PI(4,5)P2 interferes with PI(4,5)P2-dependent membrane trafficking, with important consequences for Tat-mediated toxicity in HIV-1 infected individuals [75].
–
FATTY ACIDS IN CANCER
It is widely accepted that cancer cells undergo a profound metabolic reprogramming, which is now considered a hallmark of the disease [78]. The so-called Warburg effect is one of the most remarkable metabolic phenotypes of cancer cells, consisting of prompt uptake of glucose and upregulation of glycolysis even in the presence of oxygen, with consequent production of lactate and its release into the tumour microenvironment [79][80].
–
Growing evidence demonstrates the importance of lipid metabolism and lipid signalling in cancer cell biology, highlighting the great potential of lipid metabolising enzymes, transporters and receptors as therapeutic targets. Cancer cells show changes in fatty acid and cholesterol metabolism, and this impacts upon their ability to grow and proliferate [81][82][83] (Figure 2).
–
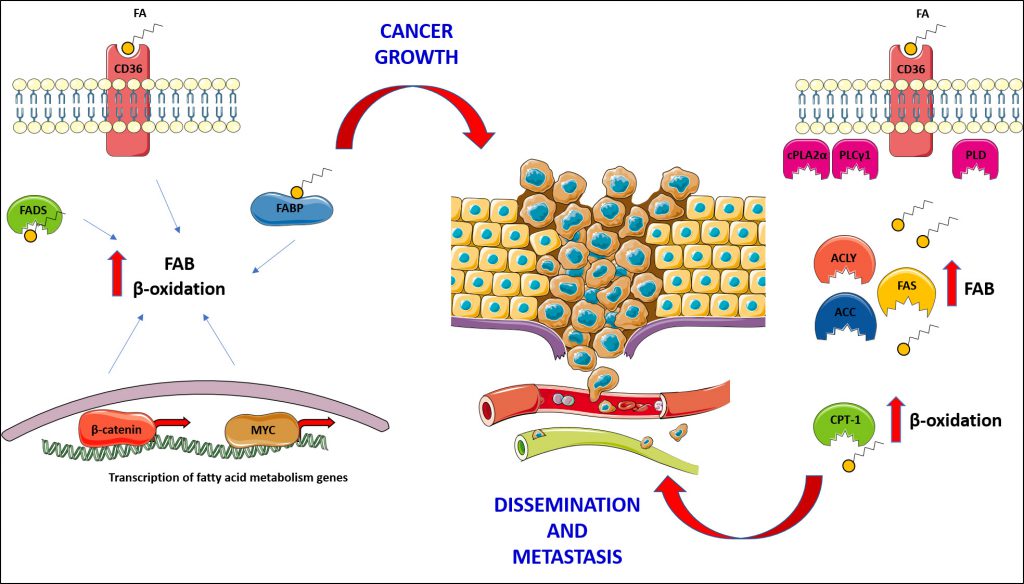 |
FIGURE 2: Cancer cells rely upon fatty acids (FA) for proliferation, survival and metastasis, and utilise them for the production of energy and membrane maintenance; consequently, both fatty acid biosynthesis (FAB) and β-oxidation are found increased in many cancers. The expression of CD36 and fatty acid binding proteins (FABP) has been associated with cancer growth. The activity of fatty acyl-CoA desaturases (FADS) sustains FAB, promoting cancer growth and survival. As shown on the left side of the figure, some cancers put in place a transcriptional programme, governed by master regulators such as MYC and β-catenin, in order to boost the activity of FAB and β-oxidation and, generally, utilisation of fatty acids, supporting cancer growth. Finally, on the right side of the figure how cancer cells utilise fatty acids to support their migration and growth in secondary sites is shown; in this context, the expression of CD36 is central for the dissemination of cancer cells from the primary tumour and the establishment and growth of metastasis. The activity of enzymes involved in phospholipid hydrolysis (Phospholipase D – PLD; cytosolic phospholipase A2α – cPLA2α; phospholipase Cγ1 – PLCγ1), in fatty acid biosynthesis (ATP citrate lyase – ACLY; acetyl-CoA carboxylase – ACC; fatty acid synthase – FAS) and in their catabolism (Carnitine palmitoyl transferase 1 – CPT-1), has been proven fundamental for dissemination and metastasis establishment. |
–
Sensing and binding
Fatty acid sensing is crucial for cancer cells, and different studies have shown the importance of lipoprotein receptors and FABP for cancer cell survival [84][85][86][87]. Saturated and unsaturated long chain fatty acids differentially regulate the transcriptional activity of the retinoic acid receptor RAR and PPARβ/δ, through FABP5. Although saturated fatty acids block FABP5 and inhibit PPARβ/δ, unsaturated fatty acids activate PPARβ/δ. The result is that saturated fatty acids, by activating RAR and inhibiting PPARβ/δ, are able to suppress the growth of cancer cells [88]. Recently, a role for FABP5 in promoting cancer growth through the estrogen-related receptor α (ERRα)-dependent upregulation of metabolic genes has been reported in prostate cancer [89].
–
CD36 represents another promising pharmacological target for interfering with the fatty acid uptake by cancer cells, and very recently, Watt and co-workers showed how targeting CD36 could be an effective strategy against prostate cancer [90]. In this study, the authors used patients' samples and xenograft models to demonstrate that increase in uptake and utilization of fatty acids in prostate cancer is due, at least in part, to expression of CD36, and that the presence of this transporter correlates with aggressive disease. Deleting CD36 or using a blocking monoclonal antibody slowed cancer progression and disease severity [90].
–
Biosynthesis and metabolism
Fatty acid biosynthesis plays a pivotal role in cancer biology and it is not surprising that inhibition of this process represents an appealing pharmacological avenue [91]. SREBPs have been shown to be important for cancer survival and progression [92][93]. More recently, Li et al. [94] have pointed to their importance in hepatocellular carcinoma (HCC). By blocking SREBP, either genetically or pharmacologically, the progression of HCC was remarkably inhibited together with downregulation of tumour-promoting inflammatory cytokines, such as IL-6, TNFα and IL-1β [94].
–
Inhibition of FAS, leading to impaired de novo biosynthesis has been explored as a therapeutic strategy for metastatic castration-resistant prostate cancer (mCRPC) [95], as demonstrated by the development of inhibitors that reduced the growth of human CRPC xenograft and human mCRPC-derived organoids [95]. The importance of fatty acid biosynthesis was further addressed in a recent study showing how cancer cells can utilize different fatty acyl-CoA desaturases (FADS) in order to sustain proliferation [96]. Several cell lines, including mouse HCC and primary human liver and lung cancer cell lines, were found to be independent from the activity of stearoyl-CoA desaturase (SCD), instead they were able to desaturate palmitate to the unusual sapienic acid (rather than to palmitoleic acid) through the activity of FADS2 [97]. Inhibition of both desaturases, SCD and FADS2, resulted in reduced cancer growth in vivo, suggesting that targeting of multiple pathways may be needed in order to effectively impair cancer metabolic plasticity [96].
–
Utilising fatty acids for energy production, in particular via β-oxidation, is important for cancer cell growth and has been considered a target for the development of new therapies in various cancers including B cell lymphoma [98], leukemia [99], prostate cancer [100], ovarian cancer [101] and pancreatic cancer [102]. Of particular interest in this context is the understanding of the transcriptional regulation of β-oxidation in cancer, as this has been associated with the over-expression of certain oncogenic transcription factors, such as MYC and β-catenin.
–
The oncogene MYC is responsible for alteration of metabolism during tumorigenesis [103][104]. Recently, Camarda et al. [105] showed that MYC-overexpressing triple-negative breast cancer (TNBC) is highly dependent on β-oxidation for survival. To assess the role of MYC in TNBC, the authors analysed the metabolites produced by breast tumours and normal mammary gland from a conditional transgenic model of MYC-overexpressing tumours and reported that MYC-TNBC tumours are enriched in acylcarnitines, metabolites of the β-oxidation pathway [105]. In human samples, TNBC tumours also appear to upregulate genes responsible for the activation of β-oxidation and downregulate genes of fatty acid biosynthesis, both at mRNA and protein level. Finally, inhibiting β-oxidation, either using the inhibitor etomoxir or knocking-down carnitine palmitoyl transferase 1B, proved to be detrimental for the energy production and survival of cancer cells in vitro and also in vivo [105].
–
Of particular interest is also the finding of β-catenin (CTNNB1)-mutated HCC dependence on fatty acids [106]. It was reported that these tumours do not comply with the Warburg effect, they are not glycolytic, and activate a strong fatty acid catabolic programme by increasing the flux of β-oxidation. This phenotype was observed in mice recapitulating β-catenin mutated HCC (by inactivation of Apc – [107]) and further confirmed in human samples, where the same metabolic signature was found. Loss of PPARα, a downstream target of β-catenin, was sufficient to blunt the upregulation of β-oxidation and reduce the formation and progression of tumours. Pharmacological inhibition of β-oxidation with etomoxir exerted the same effect on tumour growth [106].
–
Cancer immunology
The importance of fatty acid metabolism in regulating the functions of immune cells was recently shown to influence the ability of the immune system to clear tumours. The interaction between immune and cancer cells takes place in the tumour microenvironment, which is able to influence the proliferation and activation of immune cells. Pacella et al. have shown how the tumour microenvironment favours the expansion of a Treg cell population over a conventional T cell (Tconv) population, which would be able to fight the tumour [108]. Treg are able to survive better than Tconv in the tumour microenvironment because of their ability to utilize glucose for fatty acid biosynthesis. Treg indeed rely on fatty acid biosynthesis more than their conventional counterparts and the authors confirmed that activated Treg isolated from human liver cancers express a unique gene signature supporting glycolysis and lipid biosynthesis [108].
–
The relationship between lipid metabolism, cancer and the immune system has become the focus of growing research activity in recent years, following epidemiological studies revealing the association of cancer and obesity, and showing that obesity is a potent risk factor for developing cancer [109][110]. Recently, a new mechanism explaining how fatty acid accumulation blunts the immune response in obesity has been described, suggesting that obesity can significantly impair the cytotoxic activity of NK cells [111]. Feeding mice a HFD altered in NK cells the expression of genes involved in lipid metabolism and trafficking, and upregulated PPARβ/δ target genes involved in lipid-droplet formation (Lipe and Plin2) and lipid uptake (Cd36, Lpl and Lrp4). Human NK cells from obese patients were found to accumulate more lipid droplets and were less able to kill cancer cells, an effect mediated via lipid driven mTORC1 inhibition. Blocking the translocation of fatty acids to mitochondria using etomoxir was sufficient to restore the cytotoxic activity of NK cells [111].
–
However, obesity is also well known to induce a state of low-grade chronic inflammation, with prolonged and exacerbated innate and adaptive immune responses [56][112], which may promote obesity-driven diseases, including cancer. How these aspects of obesity control of innate and adaptive response are reconciled, and whether the effect on NK is specific to this cell type, are points of interest and remain unanswered.
–
Palmitoylation was found to be important in the context of cancer immunotherapy. Yao and colleagues demonstrated that programmed death ligand 1 (PD-L1), one of the main targets in cancer immunotherapy [113], can be palmitoylated in colorectal cancer cells by DHHC3 and that this modification increases its stability by preventing lysosomal degradation [114]. Inhibition of PD-L1 palmitoylation, either pharmacologically or by genetic ablation of DHHC3, was able to increase the T cell cytotoxicity against cancer cells in vitro, and to reduce tumour growth also in vivo. The authors developed a synthetic peptide able to specifically target DHHC3 and block PD-L1 palmitoylation, demonstrating that targeting this post-translational modification can be a feasible way of suppressing PD-L1-dependent immune evasion of tumour cells [114].
–
Metastatic disease
Of particular relevance, especially from a clinical and therapeutic standpoint, is the association between dysregulated fatty acid metabolism and cancer metastasis. Several studies have highlighted the importance of fatty acids in the growth of secondary tumours, showing how enzymes involved in fatty acid biosynthesis and catabolism are crucial in this process (Figure 2). In the context of fatty acid synthesis, the enzyme ATP citrate lyase (ACLY – responsible for the conversion of citrate to oxaloacetate and cytosolic acetyl-CoA) has been associated with metastatic disease in gastric adenocarcinoma (GA) [115]. It has been proposed that ACLY could be used as biomarker for the prediction of progression and prognosis of GA, as it was found overexpressed in patients' cancer tissue as compared to normal tissues, and its expression correlated negatively with patient survival [115]. In another study, ACLY was shown to be a potential therapeutic target. Indeed, its inhibition by microRNA-22 was able to suppress growth and invasion in different types of cancer cells [116]. In clinical samples, the authors observed that upregulation of ACLY correlates with the downregulation of miR-22. Moreover, in animal models of osteosarcoma and prostate cancer, treatment with miR-22 reduced tumour growth and formation of distant metastasis, and prolonged survival [116]. ACLY was also found to be a novel interactor of the low molecular weight isoform of cyclin E (LMW-E). This interaction allows breast cancer cells to store lipids necessary for growth, migration and invasion both in vitro and in vivo, while in clinical samples a strong correlation between worse prognosis, expression of LMW-E and accumulation of lipids droplets (a consequence of the increased activity of ACLY) was observed [117].
–
Another enzyme that could prove to be a good diagnostic marker as well as a potential therapeutic target is the ACC (responsible for the carboxylation of acetyl-CoA to malonyl-CoA). Expression of phospho-ACC was found to correlate with a worse overall survival in squamous cell carcinoma of the head and neck, in patients with node metastasis [118]. Recently, ACC was associated with aggressiveness in HCC and also with poor survival and disease recurrence, making it a potential prognostic marker and therapeutic target as well [119].
–
FAS is upregulated and associated with malignant progression in cancer: inhibition of FAS reduced invasion and migration of HCC [120], and was found responsible of promoting peritoneal metastasis in ovarian cancer through induction of epithelial to mesenchymal transition (EMT) [121]. Similarly, breast cancer cells undergo EMT in a FAS -dependent manner [122]. FAS was also associated with Wnt signalling and metastatic progression in colorectal cancer [123]. In the context of prostate cancer, Ahmad and co-workers found that PPARγ-mediated expression of FAS sustains the growth of prostate cancer cells and confers poor prognosis for metastatic prostate cancer [124]. Furthermore, Sounni et al. have shown how inhibition of FAS can confer advantage in overcoming tumour adaptation to anti-angiogenic treatment [125]. In this study, the authors found that interrupting the administration of anti-angiogenic drugs, such as sunitinib and sorafenib, led to a shift in the tumour metabolism responsible for an increase in fatty acid biosynthesis associated with tumour re-growth and dissemination, which often occurs in cancer patients after anti-angiogenic treatment withdrawal [125]. Pharmacological or genetic inhibition of FAS was able to suppress tumour growth and dissemination in different cancer models, highlighting a potential new strategy for treating cancer after withdrawal of anti-angiogenic drugs.
–
Phospholipid hydrolysis is important for metastatic progression, thus providing different potential targets for therapeutic intervention. Phospholipase D (PLD), through the release of phosphatidic acid and cooperation with Grb2 and Rac2 (events that affect the plasma-membrane plasticity and actin polymerization), was found to play a role in metastasis in several cancers. Henkels et al. [126] have demonstrated that PLD drives tumour growth and metastasis formation in a human breast cancer xenograft model. Deletion of PLD2 from metastatic cells suppressed the formation of metastasis and, consistently, its overexpression in non-metastatic cells led to the acquisition of a malignant metastatic phenotype. Furthermore, pharmacological inhibition of PLD2 was sufficient to suppress tumour growth and dissemination [126] while PLD2 has also been shown to regulate the expression of HIF1α in renal cancer cells [127]. Other phospholipases have been found to be important in metastasis formation; pharmacological inhibition of cytosolic phospholipase A2α (cPLA2α) with the inhibitor CIX, was able to reduce the migratory capabilities of a murine metastatic breast cancer cell line by interfering with Toll-like receptor- and type I interferon-mediated signals [128]. cPLA2α was found overexpressed also in human breast cancer samples and in invasive breast cancer cell lines [129]. Its inhibition significantly reduced the migration of metastatic cell lines in vitro, whilst its stable knockdown inhibited the EMT necessary for the acquisition of the migratory phenotype. Indeed, cPLA2α was found to be necessary for the TGFβ-induced EMT by activating the PI3K/Akt/GSK3β pathway, both in vitro and in vivo [129]. Phospholipase Cγ1 (PLCγ1) was also associated to metastatic risk in breast cancer patients [130]. Higher expression of PLCγ1 and its activated forms in tumour samples, was associated with a higher frequency of distant metastasis, highlighting how PLCγ1 not only could be a potential therapeutic target, but also a prognostic marker for metastatic risk in breast cancer patients [130].
–
Inhibition of β-oxidation also represents a potential therapeutic for interventions preventing cancer metastasis. Carnitine palmitoyl transferases (CPT) are responsible for the transport of fatty acids to the mitochondria for β-oxidation. In TNBC cells, mitochondrial β-oxidation is upregulated in order to produce sufficient amounts of ATP necessary for survival and proliferation, leading to activation of Src. Inhibition of CPT-1 and -2, by specific inhibitors or genetic knock-down, was able to prevent Src activation, tumour growth and also metastasis formation [131].
–
The importance of lipids in metastatic spreading was further demonstrated in a recent report exploring lesions generated in the oral cavity of mice after implantation of cells isolated from human oral carcinomas [132]. They found that a subset of CD44+ cells was characterized by the expression of genes involved in proliferation, lymphatic metastasis and neoplasm metastasis. Furthermore, these cells also upregulated genes involved in the metabolism and translocation of fatty acids, as well as receptors involved in fatty acid uptake, such as CD36. CD36 was found to be crucial for metastasis formation, enabling cells with low metastatic potential to significantly increase their capacity to colonize lymph nodes upon overexpression. Consistently, knock down of CD36 greatly reduced metastasis formation. Importantly, this effect was only observed in secondary tumours, with little or no effect on the growth of primary lesions, pointing to the specific role of CD36 in metastasis. When mice were fed a HFD (enriched in fatty acids) or cells were treated with saturated fatty acids such as palmitic acid, the number of CD36+ cells and metastasis was significantly increased. While both CD36+ and CD36– cells were able to form primary tumours, only the CD36+ population produced metastasis; administration of two different neutralizing antibodies against CD36 completely inhibited metastasis formation or their size and number when metastasis where already established. Finally, as upregulation of CD36 correlates with poor prognosis and survival rate in patients with different cancers, and that its amplification correlates with metastasis in many human cancers, CD36 appears to be an interesting target for the management of metastatic disease [132]. Several other reports have demonstrated a strong correlation between aberrant lipid metabolism, EMT and metastasis formation [133][134][135][136], also highlighting the importance of the research in this field to exploit lipid metabolism as a therapeutic target.
–
CONCLUSIONS AND FUTURE PERSPECTIVES
In the context of immunology, two main conclusions can be drawn regarding the importance of fatty acids for immune cells: the generation of immunological memory and the differentiation in specific T cell subsets (Figure 1).
–
With regard to the generation of Tm cells, the majority of reports to date suggest the dependence of these cells on fatty acid β-oxidation and fatty acid biosynthesis mostly as energy source for survival. This finding is of great importance for research questions trying to better understand and manipulate the generation of memory in different pathological contexts. As inhibition of fatty acid biosynthesis is highly detrimental to Tm cells, improving or protecting the activity of this pathway through the development of agonists or via administering substrates to boost it and help building an efficient memory compartment, can provide opportunities for the development of therapeutics to combat chronic disease.
–
Equally important is the variety of fatty acid substrates and their role in dictating the differentiation of T cells, which can be exploited for the generation of clinical interventions aimed at controlling the pathogenicity of aberrant T cells in inflammatory conditions. This may be of particular relevance to auto-immune diseases, where the balance between Th17 and Treg cells is crucial. Activation of PPARγ and inhibition of ACC1 could become a good strategy to block the generation of Th17 cells [45][50]. This approach can also improve the generation of protective Treg cells [50]. The receptor CD5L has also been found to be a repressor of the pathogenicity of Th17 cells by regulating fatty acid and cholesterol metabolism [53].
–
Other immune cell types, such as ILC1 and B cells, are also important in the establishment of metabolic disorders, particularly obesity and insulin resistance [62][63]. Understanding the mechanisms by which these cell types utilize fatty acids and appreciating their exact involvement in maintaining adipose tissue inflammation, typical of obesity, will provide useful insights for studies aiming to combat this condition. Further research in the field of immunology and fatty acid metabolism will provide valuable new information and, more importantly, identify new targets to manipulate the fatty acid metabolic pathways in order to fine balance the generation and function of diverse immunological populations.
–
Cancer cells also rely upon fatty acids for the production of ATP, which they require in great amounts to meet the energy demand necessary for a high proliferation rate, in the relatively nutrient-poor tumour microenvironment. Inhibiting uptake, biosynthesis and/or utilisation of fatty acids, via targeting the relevant enzymes, receptors and FABP, represents possible strategies to fight cancer (Figure 2).
–
The inhibition of receptors like CD36 and enzymes involved in fatty acid biosynthesis has shown encouraging results in certain cancers (e.g. prostate cancer- [90]) where fatty acids appear to be important sources of energy to sustain proliferation and survival [91]. The consumption of fatty acids via β-oxidation is also a very important metabolic pathway in cancer cells and that is why some cancers display an entire transcriptional programme aimed at utilizing fatty acids for proliferation [105][106]. This is the case of the two transcription factors MYC and β-catenin in breast cancer [105] and HCC [106], respectively, both strongly promoting β-oxidation.
–
Despite their dependence on certain metabolites, such as glucose and diverse types of fatty acids, cancer cells can easily adapt to the fluctuation of nutrients in the tumour microenvironment. The work from Vriens et al. [96] shows that cancer cells can use alternative pathways and substrates and that only inhibiting both canonical and alternative signals can be a successful strategy to overcome the metabolic plasticity of cancer cells. Thus, it is evident that along trying to discover new ways of inhibiting canonical pathways, we also need to explore new non-canonical pathways that tumours may exploit for growth and metastasis.
–
S-acylation is a modification important for the regulation of protein trafficking and function, providing new pharmacological targets that can be exploited in the context of a wide range of pathological conditions, spanning from infection to cancer [75][114]. Inhibition of this process by limiting the availability of fatty acids or direct pharmacological inhibition of acyltransferases, could prove a viable approach applicable to different clinical scenarios.
–
Metastatic disease represents one of the greatest challenges of our time, and it is exciting to see an increased number of reports showing that fatty acid uptake and metabolism are actively involved in this process. Inhibition of several enzymes, both of the fatty acid biosynthesis and β-oxidation pathways, can reduce metastasis in many different cancers, while targeting the receptor CD36 represents an efficient strategy for blocking their generation and growth [132].
–
Future research needs to focus on the most promising targets in order to develop efficient pharmacological tools that can be used in a number of pathological contexts. We also need to further dissect the molecular mechanisms regulating the biosynthesis and catabolism of fatty acids, in order to discover new targets and improve the strategies that would allow us to modulate these processes when they become aberrant.
–
REFERENCES
- Fielding BA, Frayn KN (2003). Lipid metabolism. Curr Opin Lipidol 14(4): 389-391. 10.1097/01.mol.0000083770.66245.76
- van Meer G, Voelker DR, Feigenson GW (2008). Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol 9(2): 112-124. 10.1038/nrm2330
- Calder PC (2013). Long chain fatty acids and gene expression in inflammation and immunity. Curr Opin Clin Nutr Metab Care 16(4): 425-433. 10.1097/MCO.0b013e3283620616
- Kendall AC, Pilkington SM, Massey KA, Sassano G, Rhodes LE, Nicolaou A (2015). Distribution of bioactive lipid mediators in human skin. J Invest Dermatol 135(6): 1510-1520. 10.1038/jid.2015.41
- Fahy E, Subramaniam S, Brown HA, Glass CK, Merrill AH, Jr., Murphy RC, Raetz CR, Russell DW, Seyama Y, Shaw W, Shimizu T, Spener F, van Meer G, VanNieuwenhze MS, White SH, Witztum JL, Dennis EA (2005). A comprehensive classification system for lipids. J Lipid Res 46(5): 839-861. 10.1194/jlr.E400004-JLR200
- Fahy E, Subramaniam S, Murphy RC, Nishijima M, Raetz CR, Shimizu T, Spener F, van Meer G, Wakelam MJ, Dennis EA (2009). Update of the LIPID MAPS comprehensive classification system for lipids. J Lipid Res 50 (Suppl): S9-14. 10.1194/jlr.R800095-JLR200
- Liebisch G, Vizcaino JA, Kofeler H, Trotzmuller M, Griffiths WJ, Schmitz G, Spener F, Wakelam MJ (2013). Shorthand notation for lipid structures derived from mass spectrometry. J Lipid Res 54(6): 1523-1530. 10.1194/jlr.M033506
- Lodhi IJ, Wei X, Semenkovich CF (2011). Lipoexpediency: de novo lipogenesis as a metabolic signal transmitter. Trends Endocrinol Metab 22(1): 1-8. 10.1016/j.tem.2010.09.002
- Calder PC (2008). The relationship between the fatty acid composition of immune cells and their function. Prostaglandins Leukot Essent Fatty Acids 79(3-5): 101-108. 10.1016/j.plefa.2008.09.016
- Nicolaou A, Mauro C, Urquhart P, Marelli-Berg F (2014). Polyunsaturated Fatty Acid-derived lipid mediators and T cell function. Front Immunol 5: 75. 10.3389/fimmu.2014.00075
- Serhan CN, Chiang N, Van Dyke TE (2008). Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol 8(5): 349-361. 10.1038/nri2294
- Itoh Y, Kawamata Y, Harada M, Kobayashi M, Fujii R, Fukusumi S, Ogi K, Hosoya M, Tanaka Y, Uejima H, Tanaka H, Maruyama M, Satoh R, Okubo S, Kizawa H, Komatsu H, Matsumura F, Noguchi Y, Shinohara T, Hinuma S, Fujisawa Y, Fujino M (2003). Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature 422(6928): 173-176. 10.1038/nature01478
- Le Poul E, Loison C, Struyf S, Springael JY, Lannoy V, Decobecq ME, Brezillon S, Dupriez V, Vassart G, Van Damme J, Parmentier M, Detheux M (2003). Functional characterization of human receptors for short chain fatty acids and their role in polymorphonuclear cell activation. J Biol Chem 278(28): 25481-25489. 10.1074/jbc.M301403200
- Wang J, Wu X, Simonavicius N, Tian H, Ling L (2006). Medium-chain fatty acids as ligands for orphan G protein-coupled receptor GPR84. J Biol Chem 281(45): 34457-34464. 10.1074/jbc.M608019200
- Park J, Kim M, Kang SG, Jannasch AH, Cooper B, Patterson J, Kim CH (2015). Short-chain fatty acids induce both effector and regulatory T cells by suppression of histone deacetylases and regulation of the mTOR-S6K pathway. Mucosal Immunol 8(1): 80-93. 10.1038/mi.2014.44
- Thwaites DT, Anderson CM (2007). H+-coupled nutrient, micronutrient and drug transporters in the mammalian small intestine. Exp Physiol 92(4): 603-619. 10.1113/expphysiol.2005.029959
- Brown AJ, Goldsworthy SM, Barnes AA, Eilert MM, Tcheang L, Daniels D, Muir AI, Wigglesworth MJ, Kinghorn I, Fraser NJ, Pike NB, Strum JC, Steplewski KM, Murdock PR, Holder JC, Marshall FH, Szekeres PG, Wilson S, Ignar DM, Foord SM, Wise A, Dowell SJ (2003). The Orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J Biol Chem 278(13): 11312-11319. 10.1074/jbc.M211609200
- Kim MH, Kang SG, Park JH, Yanagisawa M, Kim CH (2013). Short-chain fatty acids activate GPR41 and GPR43 on intestinal epithelial cells to promote inflammatory responses in mice. Gastroenterology 145(2): 396-406 e391-310. 10.1053/j.gastro.2013.04.056
- Drover VA, Nguyen DV, Bastie CC, Darlington YF, Abumrad NA, Pessin JE, London E, Sahoo D, Phillips MC (2008). CD36 mediates both cellular uptake of very long chain fatty acids and their intestinal absorption in mice. J Biol Chem 283(19): 13108-13115. 10.1074/jbc.M708086200
- Zhou J, Febbraio M, Wada T, Zhai Y, Kuruba R, He J, Lee JH, Khadem S, Ren S, Li S, Silverstein RL, Xie W (2008). Hepatic fatty acid transporter Cd36 is a common target of LXR, PXR, and PPARgamma in promoting steatosis. Gastroenterology 134(2): 556-567. 10.1053/j.gastro.2007.11.037
- Koonen DP, Glatz JF, Bonen A, Luiken JJ (2005). Long-chain fatty acid uptake and FAT/CD36 translocation in heart and skeletal muscle. Biochim Biophys Acta 1736(3): 163-180. 10.1016/j.bbalip.2005.08.018
- Coburn CT, Knapp FF, Jr., Febbraio M, Beets AL, Silverstein RL, Abumrad NA (2000). Defective uptake and utilization of long chain fatty acids in muscle and adipose tissues of CD36 knockout mice. J Biol Chem 275(42): 32523-32529. 10.1074/jbc.M003826200
- Son NH, Basu D, Samovski D, Pietka TA, Peche VS, Willecke F, Fang X, Yu SQ, Scerbo D, Chang HR, Sun F, Bagdasarov S, Drosatos K, Yeh ST, Mullick AE, Shoghi KI, Gumaste N, Kim K, Huggins LA, Lhakhang T, Abumrad NA, Goldberg IJ (2018). Endothelial cell CD36 optimizes tissue fatty acid uptake. J Clin Invest 128(10): 4329-4342. 10.1172/JCI99315
- Podrez EA, Poliakov E, Shen Z, Zhang R, Deng Y, Sun M, Finton PJ, Shan L, Gugiu B, Fox PL, Hoff HF, Salomon RG, Hazen SL (2002). Identification of a novel family of oxidized phospholipids that serve as ligands for the macrophage scavenger receptor CD36. J Biol Chem 277(41): 38503-38516. 10.1074/jbc.M203318200
- Endemann G, Stanton LW, Madden KS, Bryant CM, White RT, Protter AA (1993). CD36 is a receptor for oxidized low density lipoprotein. J Biol Chem 268(16): 11811-11816. 7685021
- Calvo D, Gomez-Coronado D, Suarez Y, Lasuncion MA, Vega MA (1998). Human CD36 is a high affinity receptor for the native lipoproteins HDL, LDL, and VLDL. J Lipid Res 39(4): 777-788. 9555943
- Silverstein RL, Febbraio M (2009). CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci Signal 2(72): re3. 10.1126/scisignal.272re3
- Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, Rayner KJ, Boyer L, Zhong R, Frazier WA, Lacy-Hulbert A, El Khoury J, Golenbock DT, Moore KJ (2010). CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol 11(2): 155-161. 10.1038/ni.1836
- Hoebe K, Georgel P, Rutschmann S, Du X, Mudd S, Crozat K, Sovath S, Shamel L, Hartung T, Zahringer U, Beutler B (2005). CD36 is a sensor of diacylglycerides. Nature 433(7025): 523-527. 10.1038/nature03253
- Stuart LM, Deng J, Silver JM, Takahashi K, Tseng AA, Hennessy EJ, Ezekowitz RA, Moore KJ (2005). Response to Staphylococcus aureus requires CD36-mediated phagocytosis triggered by the COOH-terminal cytoplasmic domain. J Cell Biol 170(3): 477-485. 10.1083/jcb.200501113
- Kashiwagi H, Tomiyama Y, Honda S, Kosugi S, Shiraga M, Nagao N, Sekiguchi S, Kanayama Y, Kurata Y, Matsuzawa Y (1995). Molecular basis of CD36 deficiency. Evidence that a 478C—>T substitution (proline90—>serine) in CD36 cDNA accounts for CD36 deficiency. J Clin Invest 95(3): 1040-1046. 10.1172/JCI117749
- Nicholls HT, Kowalski G, Kennedy DJ, Risis S, Zaffino LA, Watson N, Kanellakis P, Watt MJ, Bobik A, Bonen A, Febbraio M, Lancaster GI, Febbraio MA (2011). Hematopoietic cell-restricted deletion of CD36 reduces high-fat diet-induced macrophage infiltration and improves insulin signaling in adipose tissue. Diabetes 60(4): 1100-1110. 10.2337/db10-1353
- Huang SC, Everts B, Ivanova Y, O'Sullivan D, Nascimento M, Smith AM, Beatty W, Love-Gregory L, Lam WY, O'Neill CM, Yan C, Du H, Abumrad NA, Urban JF, Jr., Artyomov MN, Pearce EL, Pearce EJ (2014). Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol 15(9): 846-855. 10.1038/ni.2956
- O'Sullivan D, van der Windt GJ, Huang SC, Curtis JD, Chang CH, Buck MD, Qiu J, Smith AM, Lam WY, DiPlato LM, Hsu FF, Birnbaum MJ, Pearce EJ, Pearce EL (2014). Memory CD8(+) T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity 41(1): 75-88. 10.1016/j.immuni.2014.06.005
- Storch J, Thumser AE (2010). Tissue-specific functions in the fatty acid-binding protein family. J Biol Chem 285(43): 32679-32683. 10.1074/jbc.R110.135210
- Furuhashi M, Hotamisligil GS (2008). Fatty acid-binding proteins: role in metabolic diseases and potential as drug targets. Nat Rev Drug Discov 7(6): 489-503. 10.1038/nrd2589
- Hotamisligil GS, Bernlohr DA (2015). Metabolic functions of FABPs—mechanisms and therapeutic implications. Nat Rev Endocrinol 11(10): 592-605. 10.1038/nrendo.2015.122
- Pan Y, Tian T, Park CO, Lofftus SY, Mei S, Liu X, Luo C, O'Malley JT, Gehad A, Teague JE, Divito SJ, Fuhlbrigge R, Puigserver P, Krueger JG, Hotamisligil GS, Clark RA, Kupper TS (2017). Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature 543(7644): 252-256. 10.1038/nature21379
- Berger J, Moller DE (2002). The mechanisms of action of PPARs. Annu Rev Med 53: 409-435. 10.1146/annurev.med.53.082901.104018
- Sonoda J, Pei L, Evans RM (2008). Nuclear receptors: decoding metabolic disease. FEBS Lett 582(1): 2-9. 10.1016/j.febslet.2007.11.016
- Zieleniak A, Wojcik M, Wozniak LA (2008). Structure and physiological functions of the human peroxisome proliferator-activated receptor gamma. Arch Immunol Ther Exp 56(5): 331-345. 10.1007/s00005-008-0037-y
- Wang YX (2010). PPARs: diverse regulators in energy metabolism and metabolic diseases. Cell Res 20(2): 124-137. 10.1038/cr.2010.13
- Choi JM, Bothwell AL (2012). The nuclear receptor PPARs as important regulators of T-cell functions and autoimmune diseases. Mol Cells 33(3): 217-222. 10.1007/s10059-012-2297-y
- Wohlfert EA, Nichols FC, Nevius E, Clark RB (2007). Peroxisome proliferator-activated receptor gamma (PPARgamma) and immunoregulation: enhancement of regulatory T cells through PPARgamma-dependent and -independent mechanisms. J Immunol 178(7): 4129-4135. 10.4049/jimmunol.178.7.4129
- Klotz L, Burgdorf S, Dani I, Saijo K, Flossdorf J, Hucke S, Alferink J, Nowak N, Beyer M, Mayer G, Langhans B, Klockgether T, Waisman A, Eberl G, Schultze J, Famulok M, Kolanus W, Glass C, Kurts C, Knolle PA (2009). The nuclear receptor PPAR gamma selectively inhibits Th17 differentiation in a T cell-intrinsic fashion and suppresses CNS autoimmunity. J Exp Med 206(10): 2079-2089. 10.1084/jem.20082771
- Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, Shoelson SE, Benoist C, Mathis D (2012). PPAR-gamma is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature 486(7404): 549-553. 10.1038/nature11132
- Kidani Y, Elsaesser H, Hock MB, Vergnes L, Williams KJ, Argus JP, Marbois BN, Komisopoulou E, Wilson EB, Osborne TF, Graeber TG, Reue K, Brooks DG, Bensinger SJ (2013). Sterol regulatory element-binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity. Nat Immunol 14(5): 489-499. 10.1038/ni.2570
- Hansen M, Flatt T, Aguilaniu H (2013). Reproduction, fat metabolism, and life span: what is the connection? Cell Metab 17(1): 10-19. 10.1016/j.cmet.2012.12.003
- Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, Sullivan SA, Nichols AG, Rathmell JC (2011). Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol 186(6): 3299-3303. 10.4049/jimmunol.1003613
- Berod L, Friedrich C, Nandan A, Freitag J, Hagemann S, Harmrolfs K, Sandouk A, Hesse C, Castro CN, Bahre H, Tschirner SK, Gorinski N, Gohmert M, Mayer CT, Huehn J, Ponimaskin E, Abraham WR, Muller R, Lochner M, Sparwasser T (2014). De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med 20(11): 1327-1333. 10.1038/nm.3704
- Wakil SJ, Abu-Elheiga LA (2009). Fatty acid metabolism: target for metabolic syndrome. J Lipid Res 50 Suppl(S138-143. 10.1194/jlr.R800079-JLR200
- Ibitokou SA, Dillon BE, Sinha M, Szczesny B, Delgadillo A, Reda Abdelrahman D, Szabo C, Abu-Elheiga L, Porter C, Tuvdendorj D, Stephens R (2018). Early Inhibition of Fatty Acid Synthesis Reduces Generation of Memory Precursor Effector T Cells in Chronic Infection. J Immunol 200(2): 643-656. 10.4049/jimmunol.1602110
- Wang C, Yosef N, Gaublomme J, Wu C, Lee Y, Clish CB, Kaminski J, Xiao S, Meyer Zu Horste G, Pawlak M, Kishi Y, Joller N, Karwacz K, Zhu C, Ordovas-Montanes M, Madi A, Wortman I, Miyazaki T, Sobel RA, Park H, Regev A, Kuchroo VK (2015). CD5L/AIM Regulates Lipid Biosynthesis and Restrains Th17 Cell Pathogenicity. Cell 163(6): 1413-1427. 10.1016/j.cell.2015.10.068
- Miyazaki T, Hirokami Y, Matsuhashi N, Takatsuka H, Naito M (1999). Increased susceptibility of thymocytes to apoptosis in mice lacking AIM, a novel murine macrophage-derived soluble factor belonging to the scavenger receptor cysteine-rich domain superfamily. J Exp Med 189(2): 413-422. 10.1084/jem.189.2.413
- Kurokawa J, Arai S, Nakashima K, Nagano H, Nishijima A, Miyata K, Ose R, Mori M, Kubota N, Kadowaki T, Oike Y, Koga H, Febbraio M, Iwanaga T, Miyazaki T (2010). Macrophage-derived AIM is endocytosed into adipocytes and decreases lipid droplets via inhibition of fatty acid synthase activity. Cell Metab 11(6): 479-492. 10.1016/j.cmet.2010.04.013
- Mauro C, Smith J, Cucchi D, Coe D, Fu H, Bonacina F, Baragetti A, Cermenati G, Caruso D, Mitro N, Catapano AL, Ammirati E, Longhi MP, Okkenhaug K, Norata GD, Marelli-Berg FM (2017). Obesity-Induced Metabolic Stress Leads to Biased Effector Memory CD4(+) T Cell Differentiation via PI3K p110delta-Akt-Mediated Signals. Cell Metab 25(3): 593-609. 10.1016/j.cmet.2017.01.008
- Hotamisligil GS (2006). Inflammation and metabolic disorders. Nature 444(7121): 860-867. 10.1038/nature05485
- Khan T, Muise ES, Iyengar P, Wang ZV, Chandalia M, Abate N, Zhang BB, Bonaldo P, Chua S, Scherer PE (2009). Metabolic dysregulation and adipose tissue fibrosis: role of collagen VI. Mol Cell Biol 29(6): 1575-1591. 10.1128/MCB.01300-08
- Lee BC, Kim MS, Pae M, Yamamoto Y, Eberle D, Shimada T, Kamei N, Park HS, Sasorith S, Woo JR, You J, Mosher W, Brady HJ, Shoelson SE, Lee J (2016). Adipose Natural Killer Cells Regulate Adipose Tissue Macrophages to Promote Insulin Resistance in Obesity. Cell Metab 23(4): 685-698. 10.1016/j.cmet.2016.03.002
- Cho KW, Morris DL, DelProposto JL, Geletka L, Zamarron B, Martinez-Santibanez G, Meyer KA, Singer K, O'Rourke RW, Lumeng CN (2014). An MHC II-dependent activation loop between adipose tissue macrophages and CD4+ T cells controls obesity-induced inflammation. Cell Rep 9(2): 605-617. 10.1016/j.celrep.2014.09.004
- Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, Otsu M, Hara K, Ueki K, Sugiura S, Yoshimura K, Kadowaki T, Nagai R (2009). CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med 15(8): 914-920. 10.1038/nm.1964
- O'Sullivan TE, Rapp M, Fan X, Weizman OE, Bhardwaj P, Adams NM, Walzer T, Dannenberg AJ, Sun JC (2016). Adipose-Resident Group 1 Innate Lymphoid Cells Promote Obesity-Associated Insulin Resistance. Immunity 45(2): 428-441. 10.1016/j.immuni.2016.06.016
- Carter S, Miard S, Caron A, Salle-Lefort S, St-Pierre P, Anhe FF, Lavoie-Charland E, Blais-Lecours P, Drolet MC, Lefebvre JS, Lacombe J, Deshaies Y, Couet J, Laplante M, Ferron M, Bosse Y, Marette A, Richard D, Marsolais D, Picard F (2018). Loss of OcaB Prevents Age-Induced Fat Accretion and Insulin Resistance by Altering B-Lymphocyte Transition and Promoting Energy Expenditure. Diabetes 67(7): 1285-1296. 10.2337/db17-0558
- Jankovic M, Nussenzweig MC (2003). OcaB regulates transitional B cell selection. Int Immunol 15(9): 1099-1104. 10.1093/intimm/dxg109
- Kim U, Qin XF, Gong S, Stevens S, Luo Y, Nussenzweig M, Roeder RG (1996). The B-cell-specific transcription coactivator OCA-B/OBF-1/Bob-1 is essential for normal production of immunoglobulin isotypes. Nature 383(6600): 542-547. 10.1038/383542a0
- Cucchi D, Camacho-Munoz D, Certo M, Niven J, Smith J, Nicolaou A, Mauro C (2019). Omega-3 polyunsaturated fatty acids impinge on CD4+ T cell motility and adipose tissue distribution via direct and lipid mediator-dependent effects. Cardiovasc Res. 10.1093/cvr/cvz208.
- Kendall AC, Pilkington SM, Murphy SA, Del Carratore F, Sunarwidhi AL, Kiezel-Tsugunova M, Urquhart P, Watson REB, Breitling R, Rhodes LE, Nicolaou A (2019). Dynamics of the human skin mediator lipidome in response to dietary omega-3 fatty acid supplementation. FASEB J: fj201901501R. 10.1096/fj.201901501R
- Gurzell EA, Teague H, Harris M, Clinthorne J, Shaikh SR, Fenton JI (2013). DHA-enriched fish oil targets B cell lipid microdomains and enhances ex vivo and in vivo B cell function. J Leukoc Biol 93(4): 463-470. 10.1189/jlb.0812394
- Tomasdottir V, Thorleifsdottir S, Vikingsson A, Hardardottir I, Freysdottir J (2014). Dietary omega-3 fatty acids enhance the B1 but not the B2 cell immune response in mice with antigen-induced peritonitis. J Nutr Biochem 25(2): 111-117. 10.1016/j.jnutbio.2013.09.010
- Teague H, Harris M, Fenton J, Lallemand P, Shewchuk BM, Shaikh SR (2014). Eicosapentaenoic and docosahexaenoic acid ethyl esters differentially enhance B-cell activity in murine obesity. J Lipid Res 55(7): 1420-1433. 10.1194/jlr.M049809
- Resh MD (2006). Trafficking and signaling by fatty-acylated and prenylated proteins. Nat Chem Biol 2(11): 584-590. 10.1038/nchembio834
- Liang X, Nazarian A, Erdjument-Bromage H, Bornmann W, Tempst P, Resh MD (2001). Heterogeneous fatty acylation of Src family kinases with polyunsaturated fatty acids regulates raft localization and signal transduction. J Biol Chem 276(33): 30987-30994. 10.1074/jbc.M104018200
- Kordyukova LV, Serebryakova MV, Baratova LA, Veit M (2008). S acylation of the hemagglutinin of influenza viruses: mass spectrometry reveals site-specific attachment of stearic acid to a transmembrane cysteine. J Virol 82(18): 9288-9292. 10.1128/JVI.00704-08
- Greaves J, Chamberlain LH (2011). DHHC palmitoyl transferases: substrate interactions and (patho)physiology. Trends Biochem Sci 36(5): 245-253. 10.1016/j.tibs.2011.01.003
- Chopard C, Tong PBV, Toth P, Schatz M, Yezid H, Debaisieux S, Mettling C, Gross A, Pugniere M, Tu A, Strub JM, Mesnard JM, Vitale N, Beaumelle B (2018). Cyclophilin A enables specific HIV-1 Tat palmitoylation and accumulation in uninfected cells. Nat Commun 9(1): 2251. 10.1038/s41467-018-04674-y
- Gopal S, Achenbach CJ, Yanik EL, Dittmer DP, Eron JJ, Engels EA (2014). Moving forward in HIV-associated cancer. J Clin Oncol 32(9): 876-880. 10.1200/JCO.2013.53.1376
- Beaumelle B, Toth P, Malak OA, Chopard C, Loussouarn G, Vitale N (2017). Phosphatidylinositol (4,5)-bisphosphate-mediated pathophysiological effect of HIV-1 Tat protein. Biochimie 141: 80-85. 10.1016/j.biochi.2017.05.014
- Hanahan D, Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144(5): 646-674. 10.1016/j.cell.2011.02.013
- Warburg O (1956). On the origin of cancer cells. Science 123(3191): 309-314. doi.
- Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, Yan H, Wang W, Chen S, Viale A, Zheng H, Paik JH, Lim C, Guimaraes AR, Martin ES, Chang J, Hezel AF, Perry SR, Hu J, Gan B, Xiao Y, Asara JM, Weissleder R, Wang YA, Chin L, Cantley LC, et al. (2012). Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 149(3): 656-670. 10.1016/j.cell.2012.01.058
- Baenke F, Peck B, Miess H, Schulze A (2013). Hooked on fat: the role of lipid synthesis in cancer metabolism and tumour development. Dis Model Mech 6(6): 1353-1363. 10.1242/dmm.011338
- Cruz PM, Mo H, McConathy WJ, Sabnis N, Lacko AG (2013). The role of cholesterol metabolism and cholesterol transport in carcinogenesis: a review of scientific findings, relevant to future cancer therapeutics. Front Pharmacol 4: 119. 10.3389/fphar.2013.00119
- Ackerman D, Simon MC (2014). Hypoxia, lipids, and cancer: surviving the harsh tumor microenvironment. Trends Cell Biol 24(8): 472-478. 10.1016/j.tcb.2014.06.001
- Sundelin JP, Stahlman M, Lundqvist A, Levin M, Parini P, Johansson ME, Boren J (2012). Increased expression of the very low-density lipoprotein receptor mediates lipid accumulation in clear-cell renal cell carcinoma. PLoS One 7(11): e48694. 10.1371/journal.pone.0048694
- Montel V, Gaultier A, Lester RD, Campana WM, Gonias SL (2007). The low-density lipoprotein receptor-related protein regulates cancer cell survival and metastasis development. Cancer Res 67(20): 9817-9824. 10.1158/0008-5472.CAN-07-0683
- Bensaad K, Favaro E, Lewis CA, Peck B, Lord S, Collins JM, Pinnick KE, Wigfield S, Buffa FM, Li JL, Zhang Q, Wakelam MJO, Karpe F, Schulze A, Harris AL (2014). Fatty acid uptake and lipid storage induced by HIF-1alpha contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep 9(1): 349-365. 10.1016/j.celrep.2014.08.056
- Adamson J, Morgan EA, Beesley C, Mei Y, Foster CS, Fujii H, Rudland PS, Smith PH, Ke Y (2003). High-level expression of cutaneous fatty acid-binding protein in prostatic carcinomas and its effect on tumorigenicity. Oncogene 22(18): 2739-2749. 10.1038/sj.onc.1206341
- Levi L, Wang Z, Doud MK, Hazen SL, Noy N (2015). Saturated fatty acids regulate retinoic acid signalling and suppress tumorigenesis by targeting fatty acid-binding protein 5. Nat Commun 6: 8794. 10.1038/ncomms9794
- Senga S, Kawaguchi K, Kobayashi N, Ando A, Fujii H (2018). A novel fatty acid-binding protein 5-estrogen-related receptor alpha signaling pathway promotes cell growth and energy metabolism in prostate cancer cells. Oncotarget 9(60): 31753-31770. 10.18632/oncotarget.25878
- Watt MJ, Clark AK, Selth LA, Haynes VR, Lister N, Rebello R, Porter LH, Niranjan B, Whitby ST, Lo J, Huang C, Schittenhelm RB, Anderson KE, Furic L, Wijayaratne PR, Matzaris M, Montgomery MK, Papargiris M, Norden S, Febbraio M, Risbridger GP, Frydenberg M, Nomura DK, Taylor RA (2019). Suppressing fatty acid uptake has therapeutic effects in preclinical models of prostate cancer. Sci Transl Med 11(478). 10.1126/scitranslmed.aau5758
- Mashima T, Seimiya H, Tsuruo T (2009). De novo fatty-acid synthesis and related pathways as molecular targets for cancer therapy. Br J Cancer 100(9): 1369-1372. 10.1038/sj.bjc.6605007.
- Sun Y, He W, Luo M, Zhou Y, Chang G, Ren W, Wu K, Li X, Shen J, Zhao X, Hu Y (2015). SREBP1 regulates tumorigenesis and prognosis of pancreatic cancer through targeting lipid metabolism. Tumour Biol 36(6): 4133-4141. 10.1007/s13277-015-3047-5
- Li X, Wu JB, Chung LW, Huang WC (2015). Anti-cancer efficacy of SREBP inhibitor, alone or in combination with docetaxel, in prostate cancer harboring p53 mutations. Oncotarget 6(38): 41018-41032. 10.18632/oncotarget.5879
- Li N, Zhou ZS, Shen Y, Xu J, Miao HH, Xiong Y, Xu F, Li BL, Luo J, Song BL (2017). Inhibition of the sterol regulatory element-binding protein pathway suppresses hepatocellular carcinoma by repressing inflammation in mice. Hepatology 65(6): 1936-1947. 10.1002/hep.29018
- Zadra G, Ribeiro CF, Chetta P, Ho Y, Cacciatore S, Gao X, Syamala S, Bango C, Photopoulos C, Huang Y, Tyekucheva S, Bastos DC, Tchaicha J, Lawney B, Uo T, D'Anello L, Csibi A, Kalekar R, Larimer B, Ellis L, Butler LM, Morrissey C, McGovern K, Palombella VJ, Kutok JL, Mahmood U, Bosari S, Adams J, Peluso S, Dehm SM, et al. (2019). Inhibition of de novo lipogenesis targets androgen receptor signaling in castration-resistant prostate cancer. Proc Natl Acad Sci U S A 116(2): 631-640. 10.1073/pnas.1808834116
- Vriens K, Christen S, Parik S, Broekaert D, Yoshinaga K, Talebi A, Dehairs J, Escalona-Noguero C, Schmieder R, Cornfield T, Charlton C, Romero-Perez L, Rossi M, Rinaldi G, Orth MF, Boon R, Kerstens A, Kwan SY, Faubert B, Mendez-Lucas A, Kopitz CC, Chen T, Fernandez-Garcia J, Duarte JAG, Schmitz AA, Steigemann P, Najimi M, Hagebarth A, Van Ginderachter JA, Sokal E, et al. (2019). Evidence for an alternative fatty acid desaturation pathway increasing cancer plasticity. Nature 566(7744): 403-406. 10.1038/s41586-019-0904-1
- Ge L, Gordon JS, Hsuan C, Stenn K, Prouty SM (2003). Identification of the delta-6 desaturase of human sebaceous glands: expression and enzyme activity. J Invest Dermatol 120(5): 707-714. 10.1046/j.1523-1747.2003.12123.x
- Caro P, Kishan AU, Norberg E, Stanley IA, Chapuy B, Ficarro SB, Polak K, Tondera D, Gounarides J, Yin H, Zhou F, Green MR, Chen L, Monti S, Marto JA, Shipp MA, Danial NN (2012). Metabolic signatures uncover distinct targets in molecular subsets of diffuse large B cell lymphoma. Cancer Cell 22(4): 547-560. 10.1016/j.ccr.2012.08.014
- Samudio I, Harmancey R, Fiegl M, Kantarjian H, Konopleva M, Korchin B, Kaluarachchi K, Bornmann W, Duvvuri S, Taegtmeyer H, Andreeff M (2010). Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J Clin Invest 120(1): 142-156. 10.1172/JCI38942
- Wu X, Daniels G, Lee P, Monaco ME (2014). Lipid metabolism in prostate cancer. Am J Clin Exp Urol 2(2): 111-120. 25374912
- Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, Romero IL, Carey MS, Mills GB, Hotamisligil GS, Yamada SD, Peter ME, Gwin K, Lengyel E (2011). Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med 17(11): 1498-1503. 10.1038/nm.2492
- Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sanchez N, Marchesini M, Carugo A, Green T, Seth S, Giuliani V, Kost-Alimova M, Muller F, Colla S, Nezi L, Genovese G, Deem AK, Kapoor A, Yao W, Brunetto E, Kang Y, Yuan M, Asara JM, Wang YA, Heffernan TP, Kimmelman AC, Wang H, Fleming JB, Cantley LC, DePinho RA, Draetta GF (2014). Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 514(7524): 628-632. 10.1038/nature13611
- Hu S, Balakrishnan A, Bok RA, Anderton B, Larson PE, Nelson SJ, Kurhanewicz J, Vigneron DB, Goga A (2011). 13C-pyruvate imaging reveals alterations in glycolysis that precede c-Myc-induced tumor formation and regression. Cell Metab 14(1): 131-142. 10.1016/j.cmet.2011.04.012
- Wahlstrom T, Henriksson MA (2015). Impact of MYC in regulation of tumor cell metabolism. Biochim Biophys Acta 1849(5): 563-569. 10.1016/j.bbagrm.2014.07.004
- Camarda R, Zhou AY, Kohnz RA, Balakrishnan S, Mahieu C, Anderton B, Eyob H, Kajimura S, Tward A, Krings G, Nomura DK, Goga A (2016). Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer. Nat Med 22(4): 427-432. 10.1038/nm.4055
- Senni N, Savall M, Cabrerizo Granados D, Alves-Guerra MC, Sartor C, Lagoutte I, Gougelet A, Terris B, Gilgenkrantz H, Perret C, Colnot S, Bossard P (2019). beta-catenin-activated hepatocellular carcinomas are addicted to fatty acids. Gut 68(2): 322-334. 10.1136/gutjnl-2017-315448
- Colnot S, Decaens T, Niwa-Kawakita M, Godard C, Hamard G, Kahn A, Giovannini M, Perret C (2004). Liver-targeted disruption of Apc in mice activates beta-catenin signaling and leads to hepatocellular carcinomas. Proc Natl Acad Sci U S A 101(49): 17216-17221. 10.1073/pnas.0404761101
- Pacella I, Procaccini C, Focaccetti C, Miacci S, Timperi E, Faicchia D, Severa M, Rizzo F, Coccia EM, Bonacina F, Mitro N, Norata GD, Rossetti G, Ranzani V, Pagani M, Giorda E, Wei Y, Matarese G, Barnaba V, Piconese S (2018). Fatty acid metabolism complements glycolysis in the selective regulatory T cell expansion during tumor growth. Proc Natl Acad Sci U S A 115(28): E6546-E6555. 10.1073/pnas.1720113115
- Calle EE, Kaaks R (2004). Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat Rev Cancer 4(8): 579-591. 10.1038/nrc1408
- Renehan AG, Tyson M, Egger M, Heller RF, Zwahlen M (2008). Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet 371(9612): 569-578. 10.1016/S0140-6736(08)60269-X
- Michelet X, Dyck L, Hogan A, Loftus RM, Duquette D, Wei K, Beyaz S, Tavakkoli A, Foley C, Donnelly R, O'Farrelly C, Raverdeau M, Vernon A, Pettee W, O'Shea D, Nikolajczyk BS, Mills KHG, Brenner MB, Finlay D, Lynch L (2018). Metabolic reprogramming of natural killer cells in obesity limits antitumor responses. Nat Immunol 19(12): 1330-1340. 10.1038/s41590-018-0251-7
- Christ A, Gunther P, Lauterbach MAR, Duewell P, Biswas D, Pelka K, Scholz CJ, Oosting M, Haendler K, Bassler K, Klee K, Schulte-Schrepping J, Ulas T, Moorlag S, Kumar V, Park MH, Joosten LAB, Groh LA, Riksen NP, Espevik T, Schlitzer A, Li Y, Fitzgerald ML, Netea MG, Schultze JL, Latz E (2018). Western Diet Triggers NLRP3-Dependent Innate Immune Reprogramming. Cell 172(1-2): 162-175 e114. 10.1016/j.cell.2017.12.013
- Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, Pitot HC, Hamid O, Bhatia S, Martins R, Eaton K, Chen S, Salay TM, Alaparthy S, Grosso JF, Korman AJ, Parker SM, Agrawal S, Goldberg SM, Pardoll DM, Gupta A, Wigginton JM (2012). Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 366(26): 2455-2465. 10.1056/NEJMoa1200694
- Yao H, Lan J, Li C, Shi H, Brosseau JP, Wang H, Lu H, Fang C, Zhang Y, Liang L, Zhou X, Wang C, Xue Y, Cui Y, Xu J (2019). Inhibiting PD-L1 palmitoylation enhances T-cell immune responses against tumours. Nat Biomed Eng 3(4): 306-317. 10.1038/s41551-019-0375-6
- Qian X, Hu J, Zhao J, Chen H (2015). ATP citrate lyase expression is associated with advanced stage and prognosis in gastric adenocarcinoma. Int J Clin Exp Med 8(5): 7855-7860. 26221340
- Xin M, Qiao Z, Li J, Liu J, Song S, Zhao X, Miao P, Tang T, Wang L, Liu W, Yang X, Dai K, Huang G (2016). miR-22 inhibits tumor growth and metastasis by targeting ATP citrate lyase: evidence in osteosarcoma, prostate cancer, cervical cancer and lung cancer. Oncotarget 7(28): 44252-44265. 10.18632/oncotarget.10020
- Lucenay KS, Doostan I, Karakas C, Bui T, Ding Z, Mills GB, Hunt KK, Keyomarsi K (2016). Cyclin E Associates with the Lipogenic Enzyme ATP-Citrate Lyase to Enable Malignant Growth of Breast Cancer Cells. Cancer Res 76(8): 2406-2418. 10.1158/0008-5472.CAN-15-1646
- Su YW, Lin YH, Pai MH, Lo AC, Lee YC, Fang IC, Lin J, Hsieh RK, Chang YF, Chen CL (2014). Association between phosphorylated AMP-activated protein kinase and acetyl-CoA carboxylase expression and outcome in patients with squamous cell carcinoma of the head and neck. PLoS One 9(4): e96183. 10.1371/journal.pone.0096183
- Wang MD, Wu H, Fu GB, Zhang HL, Zhou X, Tang L, Dong LW, Qin CJ, Huang S, Zhao LH, Zeng M, Wu MC, Yan HX, Wang HY (2016). Acetyl-coenzyme A carboxylase alpha promotion of glucose-mediated fatty acid synthesis enhances survival of hepatocellular carcinoma in mice and patients. Hepatology 63(4): 1272-1286. 10.1002/hep.28415
- Hao Q, Li T, Zhang X, Gao P, Qiao P, Li S, Geng Z (2014). Expression and roles of fatty acid synthase in hepatocellular carcinoma. Oncol Rep 32(6): 2471-2476. 10.3892/or.2014.3484
- Jiang L, Wang H, Li J, Fang X, Pan H, Yuan X, Zhang P (2014). Up-regulated FASN expression promotes transcoelomic metastasis of ovarian cancer cell through epithelial-mesenchymal transition. Int J Mol Sci 15(7): 11539-11554. 10.3390/ijms150711539
- Li J, Dong L, Wei D, Wang X, Zhang S, Li H (2014). Fatty acid synthase mediates the epithelial-mesenchymal transition of breast cancer cells. Int J Biol Sci 10(2): 171-180. 10.7150/ijbs.7357
- Wang H, Xi Q, Wu G (2016). Fatty acid synthase regulates invasion and metastasis of colorectal cancer via Wnt signaling pathway. Cancer Med 5(7): 1599-1606. 10.1002/cam4.711
- Ahmad I, Mui E, Galbraith L, Patel R, Tan EH, Salji M, Rust AG, Repiscak P, Hedley A, Markert E, Loveridge C, van der Weyden L, Edwards J, Sansom OJ, Adams DJ, Leung HY (2016). Sleeping Beauty screen reveals Pparg activation in metastatic prostate cancer. Proc Natl Acad Sci U S A 113(29): 8290-8295. 10.1073/pnas.1601571113
- Sounni NE, Cimino J, Blacher S, Primac I, Truong A, Mazzucchelli G, Paye A, Calligaris D, Debois D, De Tullio P, Mari B, De Pauw E, Noel A (2014). Blocking lipid synthesis overcomes tumor regrowth and metastasis after antiangiogenic therapy withdrawal. Cell Metab 20(2): 280-294. 10.1016/j.cmet.2014.05.022
- Henkels KM, Boivin GP, Dudley ES, Berberich SJ, Gomez-Cambronero J (2013). Phospholipase D (PLD) drives cell invasion, tumor growth and metastasis in a human breast cancer xenograph model. Oncogene 32(49): 5551-5562. 10.1038/onc.2013.207
- Toschi A, Edelstein J, Rockwell P, Ohh M, Foster DA (2008). HIF alpha expression in VHL-deficient renal cancer cells is dependent on phospholipase D. Oncogene 27(19): 2746-2753. 10.1038/sj.onc.1210927
- Tunset HM, Feuerherm AJ, Selvik LM, Johansen B, Moestue SA (2019). Cytosolic Phospholipase A2 Alpha Regulates TLR Signaling and Migration in Metastatic 4T1 Cells. Int J Mol Sci 20(19): E4800. 10.3390/ijms20194800
- Chen L, Fu H, Luo Y, Chen L, Cheng R, Zhang N, Guo H (2017). cPLA2alpha mediates TGF-beta-induced epithelial-mesenchymal transition in breast cancer through PI3k/Akt signaling. Cell Death Dis 8(4): e2728. 10.1038/cddis.2017.152
- Lattanzio R, Iezzi M, Sala G, Tinari N, Falasca M, Alberti S, Buglioni S, Mottolese M, Perracchio L, Natali PG, Piantelli M (2019). PLC-gamma-1 phosphorylation status is prognostic of metastatic risk in patients with early-stage Luminal-A and -B breast cancer subtypes. BMC Cancer 19(1): 747. 10.1186/s12885-019-5949-x
- Park JH, Vithayathil S, Kumar S, Sung PL, Dobrolecki LE, Putluri V, Bhat VB, Bhowmik SK, Gupta V, Arora K, Wu D, Tsouko E, Zhang Y, Maity S, Donti TR, Graham BH, Frigo DE, Coarfa C, Yotnda P, Putluri N, Sreekumar A, Lewis MT, Creighton CJ, Wong LC, Kaipparettu BA (2016). Fatty Acid Oxidation-Driven Src Links Mitochondrial Energy Reprogramming and Oncogenic Properties in Triple-Negative Breast Cancer. Cell Rep 14(9): 2154-2165. 10.1016/j.celrep.2016.02.004
- Pascual G, Avgustinova A, Mejetta S, Martin M, Castellanos A, Attolini CS, Berenguer A, Prats N, Toll A, Hueto JA, Bescos C, Di Croce L, Benitah SA (2017). Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 541(7635): 41-45. 10.1038/nature20791
- Nath A, Chan C (2016). Genetic alterations in fatty acid transport and metabolism genes are associated with metastatic progression and poor prognosis of human cancers. Sci Rep 6: 18669. 10.1038/srep18669
- Kim HY, Lee KM, Kim SH, Kwon YJ, Chun YJ, Choi HK (2016). Comparative metabolic and lipidomic profiling of human breast cancer cells with different metastatic potentials. Oncotarget 7(41): 67111-67128. 10.18632/oncotarget.11560
- Nath A, Li I, Roberts LR, Chan C (2015). Elevated free fatty acid uptake via CD36 promotes epithelial-mesenchymal transition in hepatocellular carcinoma. Sci Rep 5: 14752. 10.1038/srep14752
- Hale JS, Otvos B, Sinyuk M, Alvarado AG, Hitomi M, Stoltz K, Wu Q, Flavahan W, Levison B, Johansen ML, Schmitt D, Neltner JM, Huang P, Ren B, Sloan AE, Silverstein RL, Gladson CL, DiDonato JA, Brown JM, McIntyre T, Hazen SL, Horbinski C, Rich JN, Lathia JD (2014). Cancer stem cell-specific scavenger receptor CD36 drives glioblastoma progression. Stem Cells 32(7): 1746-1758. 10.1002/stem.1716
AUTHOR CONTRIBUTIONS
All authors contributed to the manuscript writing, read and approved the submitted version.
ACKNOWLEDGMENTS
The work was supported by a British Heart Foundation Project Grant (PG/15/105/31906) to CM and AN. CM is supported by a British Heart Founda-tion Intermediate Basic Science Research Fellowship (FS/12/38/29640) and by a University of Birmingham Professorial Research Fellowship. AN is supported in part by the NIHR Manchester Biomedical Research Cen-tre.
COPYRIGHT
© 2019

Fatty acids – from energy substrates to key regulators of cell survival, proliferation and effector function by Cucchi et al. is licensed under a Creative Commons Attribution 4.0 International License.


