Reviews:
Cell Stress, Vol. 3, No. 12, pp. 361 - 368; doi: 10.15698/cst2019.12.205
Metabolism remodeling in pancreatic ductal adenocarcinoma

1 Fudan University Shanghai Cancer Center and Cancer Metabolism Laboratory, Institutes of Biomedical Sciences, Shanghai Medical College, Fudan University; Department of Oncology, Shanghai Medical College, Fudan University, Shanghai 200032, People’s Republic of China.
2 Lead Contact.
Keywords: PDAC, KRAS, metabolism, tumor microenvironment.
Abbreviatons:
5-HT – 5-hydroxytryptamine,
αKG – alpha-ketoglutarate,
ADM – acinar-to-ductal metaplasia,
Asp – aspartate,
ACLY – ATP-citrate lyase,
ECM – extracellular matrix,
GAPDH – glyceraldehyde-3-phosphate dehydrogenase,
Gln – glutamine,
HBP – hexosamine biosynthetic pathway,
HIF – hypoxia-inducible factor,
LDH – lactate dehydrogenase,
LDLR – low density lipoprotein receptor,
PanIN – pancreatic intraepithelial neoplasia,
PDAC – pancreatic ductal adenocarcinoma,
PPP – pentose phosphate pathway,
PSC – pancreatic stellate cell,
R5P – ribose-5-phosphate,
ROS – reactive oxygen species,
TAM – tumor-associated macrophage,
TCA – tricarboxylic acid cycle.
Received originally: 10/07/2019 Received in revised form: 21/10/2019
Accepted: 25/10/2019
Published: 04/11/2019
Correspondence:
Miao Yin, Fudan University Shanghai Cancer Center and Cancer Metabolism Laboratory, Institutes of Biomedical Sciences, Shanghai Medical College, Fudan University; Department of Oncology, Shanghai Medical College, Fudan University, Shanghai 200032, People's Repub-lic of China; miaoyin@fudan.edu.cn
Qun-Ying Lei, Fudan University Shanghai Cancer Center and Cancer Metabolism Laboratory, Institutes of Biomedical Sciences, Shanghai Medical College, Fudan University; Department of Oncology, Shanghai Medical College, Fudan University, Shanghai 200032, People's Repub-lic of China; qlei@fudan.edu.cn
Conflict of interest statement: The authors have declared that no conflict of interest ex-ists.
Please cite this article as: Jin-Tao Li, Yi-Ping Wang, Miao Yin and Qun-Ying Lei (2019). Metabolism remodeling in pancreatic ductal adenocarcinoma. Cell Stress 3(12): 316-368. doi: 10.15698/cst2019.12.205
Abstract
Pancreatic ductal adenocarcinoma (PDAC) is predicted to become the second leading cause of death of patients with malignant cancers by 2030. Current options of PDAC treatment are limited and the five-year survival rate is less than 8%, leading to an urgent need to explore innovatively therapeutic strategies. PDAC cells exhibit extensively reprogrammed metabolism to meet their energetic and biomass demands under extremely harsh conditions. The metabolic changes are closely linked to signaling triggered by activation of oncogenes like KRAS as well as inactivation of tumor suppressors. Furthermore, tumor microenvironmental factors including extensive desmoplastic stroma reaction result in series of metabolism remodeling to facilitate PDAC development. In this review, we focus on the dysregulation of metabolism in PDAC and its surrounding microenvironment to explore potential metabolic targets in PDAC therapy.
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is a tumor with early metastatic potential and remarkable resistance to established therapies such as chemotherapy, radiotherapy, and molecular targeted therapy [1]. Given primary localization and cell morphology, PDAC has been generally viewed to be originated from pancreatic ductal cells. Nevertheless, dozens of studies including lineage tracing propose alternative original sources for PDAC cells. For example, acinar-to-ductal metaplasia (ADM) frequently occurres at the early stage of pancreas carcinoma, suggesting an acinar origin of PDAC [2][3][4][5]. In addition, emerging evidences suggest that some rare cell population named as cancer stem cell might be the precursor of PDAC cells for initiation and metastasis [1][6]. Malignant progression of PDAC, starting from the pre-cancerous lesion, pancreatic intraepithelial neoplasia (PanINs), to advanced invasion and metastasis is accompanied by various oncogene activation and tumor suppressor inactivation. KRAS, p16/CDKN2A, TP53, and DPC4/SMAD4 are viewed as driver genes in PDAC development because of their high frequency of mutation during tumorigenesis of PDAC [4]. KRAS activating mutations occur at early PanIN I stage in 95% of cases, followed by the loss of the functional tumor suppressor gene p16/CDKN2A (> 90%). Inactivating mutations in TP53 (75%) and DPC4/SMAD4 (55%) are often observed at late PanIN III stage as shown in Figure 1 [4][7].
–
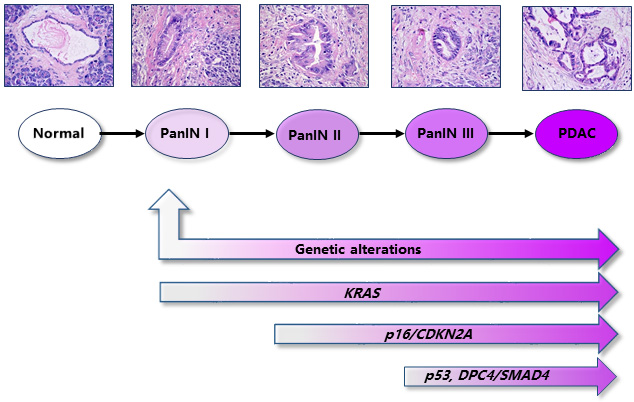 | FIGURE 1: The pathological stages and dysregulated molecular events in PDAC development. The PDAC development process is categorized into pre-cancerous lesion (pancreatic intraepithelial neoplasia, PanINs) and malignant PDAC according to the pathological grade. Simultaneously, aberrant genetic alterations occurre at different stages of PDAC development, including activating mutations of KRAS and inactivation mutations of tumor suppressors, like p16/CDKN2A, p53, and DPC4/SMAD4. |
–
Nutrients in the form of carbohydrates, amino acids, and fatty acids are utilized to support biosynthesis, supply energy and balance oxidative stress. Aberrant alterations of signaling rewire intermediary metabolism to support energetic and biosynthetic demands of PDAC cells. Of note, PDAC cells are surrounded by a microenvironment which is composed of immune cells, stellate cells, and extracellular matrix (ECM) [8][9]. It is therefore necessary to expand our knowledge on the effect of metabolic remodeling of the tumor microenvironment on PDAC development.
–
METABOLIC REPROGRAMMING IN RESPONSE TO ABNORMAL SIGNALING IN PDAC CELLS
Tumor cells prefer to take up more glucose for glycolysis even in presence of abundant oxygen, which is known as Warburg effect [10]. Constitutive activation of KRAS, the most prevalent genetic alteration in PDAC development, plays a key role in metabolic reprogramming, particularly in the glycolytic switch [11][12][13][14]. Analyses of gene expression and metabolic flux show that oncogenic KRAS upregulates expression of glucose transporter -1(GLUT1) to increase glucose influx, and hexokinase (HK) 1 and 2 to speed up glycolytic activity [13][15]. Enhanced glycolysis driven by oncogenic KRAS supports biomass synthesis. The hexosamine biosynthetic pathway (HBP), a side path of glycolysis, is driven by KRAS mutation to provide precursors for protein glycosylation (Figure 2) [15][16]. In addition, KRAS activation leads to enhanced entry of glucose carbon into the pentose phosphate pathway (PPP) [15]. PPP-derived ribose-5-phosphate (R5P) provides materials for DNA and RNA synthesis in proliferating cells. Generally, PPP is divided into two phases: oxidative and non-oxidative. Pancreatic cancer cells with KRAS mutation become dependent on non-oxidative PPP. Consistently, KRAS knockdown decreases expression of enzymes that govern non-oxidative PPP flux, resulting in strong growth inhibition [15]. Of note, most normal cells generate R5P through oxidative PPP. This discrepancy represents a potential metabolic vulnerability in KRAS-driven PDAC [17]. Moreover, KRAS activation coordinates with p16 ablation to upregulate the expression and enzymatic activity of NAD(P)H oxidase 4 (NOX4), leading to enhanced oxidation of NAD(P)H. NAD+, the catalytic product of NAD(P)H, significantly potentiates glycolysis [18].
–
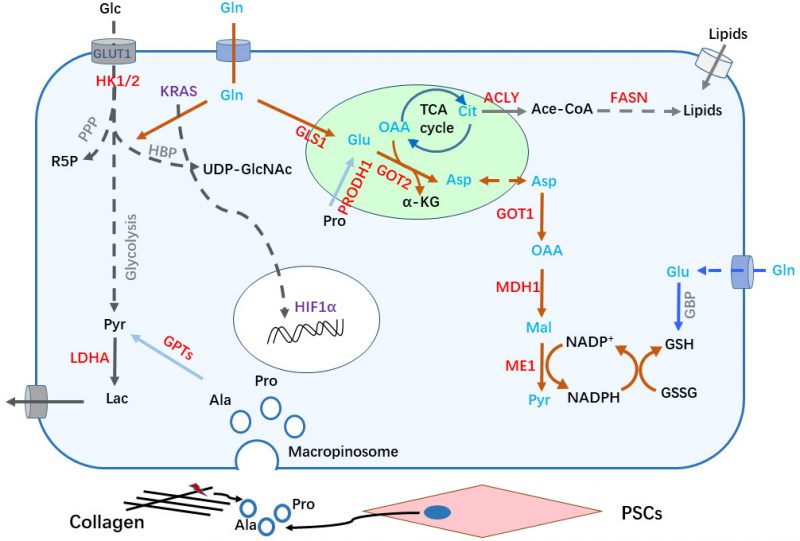 |
FIGURE 2: Metabolic remolding in PDAC cells. (1) KRAS (in cytoplasm) and HIF1α activation in PDAC cells upregulates glucose transporter (GLUT1) and other glycolytic related genes to promote glucose (Glc) uptake and enhance glycolysis flux, including the production of lactate (Lac) and carbon donation into the hexosamine biosynthetic pathway (HBP) and pentose phosphate pathway (PPP). (2) KRAS activation reprograms Gln metabolism to balance cellular redox homeostasis. Gln is sequentially converted to Glu and Asp catalyzed by GLS1 and GOT2 in the mitochondria, Asp is shuttled to the cytoplasm and generates NADPH after a series of reactions to maintain redox homeostasis. (3) The lipid synthesis pathway is activated, citrate is shuttled from the mitochondria into the cytoplasm to produce acetyl CoA (Ace-CoA), thereby enhancing de novo lipid synthesis pathway. Concomitantly, uptake of exogenous lipids is increased to meet the demand of nutrients for rapid proliferation. (4) The tumor microenvironment, including ECM components and stromal cells, also provide various metabolites/nutrients for PDAC cells, such as Ala and Pro derived from collagen degradation or pancreatic stellate cells (PSCs) secretion. The gene expression of molecules labeled in red color are up-regulated. α-KG: alpha ketoglutarate; Ace-CoA: acetyl-Coenzyme A; ACLY: ATP-citrate lyase; Ala: alanine; Asp: aspartate; Cit: citrate; FASN: fatty acid synthase; Glc: glucose; Gln: glutamine; GLS1: glutaminase; Glu: glutamate; GOT: glutamic-oxaloacetic transaminase; GPT: glutamic pyruvic transaminase; GSH: glutathione reduced; GSSG: glutathione oxidized; HBP: hexamine biosynthetic pathway; HIF: hypoxia-inducible factor; Lac: lactate; LDHA: lactate dehydrogenase A; MDH: malate dehydrogenase; ME: malic enzyme; OAA: oxaloacetic acid; PPP: pentose phosphate pathway; Pro: proline; PRODH1: proline dehydrogenase; PSC: pancreatic stellate cell; Pyr: pyruvate; R5P: ribose-5-phosphate; TCA: tricarboxylic acid. |
–
Glutamine (Gln) belongs to the group of non-essential amino acid (NEAA) that contribute to nitrogen and carbon donation for rapidly proliferating cells. Besides, KRAS mutant PDAC cells become addicted to Gln. KRAS directs an alternative metabolic pathway of Gln by transcriptionally regulating expression of key metabolic enzymes including glutamate dehydrogenase (GLUD1) and aspartate transaminase (GOT1), maintaining redox balance in PDAC cells. In this non-canonical pathway, Gln-derived carbon is converted into aspartate (Asp) through a series of reactions in mitochondria. Gln-derived Asp is then released into the cytoplasm and converted to oxaloacetic acid (OAA) and malate, eventually producing NADPH, a reducing equivalent involved in biosynthesis and reduction of reactive oxygen species (ROS) (Figure 2) [19]. Additionally, Nrf2, known as ROS suppressor, facilitates PDAC development [20]. The expression levels of Nrf2-regulated anti-oxidant genes are induced by KRAS [21]. Therefore, low intracellular ROS levels caused by KRAS signaling is vital for PDAC development.
–
p53 mutation is another prominent genetic event in PDAC development. In contrast to KRAS mutations which occur at early stage to initiate PanIN lesions, mutations in p53 are frequently present at late stages and substantially promote PDAC progression. p53 mutation was reported to prevent the glycolytic enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH) from nuclear translocation and increased GAPDH stability in the cytoplasm, supporting glycolysis to avoid apoptosis and autophagy [22]. In addition, p53 mutations result in a decrease of intermediates in the tricarboxylic acid (TCA) cycle, indicating mitochondrial repression in PDAC cells [23]. Recent work demonstrated that differentiation of malignant PDAC cells could be triggered to prevent PDAC development by p53-dependent increase of α-ketoglutarate (αKG) [24].
–
Hypoxia commonly occurs in tumor progression, which induces the expression of hypoxia-inducible factor-1 (HIF-1) and increases its stability. Upregulation of HIF-1α could be detected in PDAC. In preclinical models, hypoxia inducible GFPT2 isoform to accelerate HBP [25]. In addition, hypoxia-inducible factor 1a (HIF1a) upregulates GLUT1 as well as the expression of other glycolysis-related genes to generate cytosolic ATP in PDAC cells (Figure 2) [26][27][28]. However, the effect of HIF1a in PDAC development may be context-dependent. One study showed that HIF1a invalidation supports PDAC formation in a mouse model [29].
–
The hippo pathway was originally identified to function in development, such as controlling organ size [30]. After that, the role of the Hippo pathway in cancer development has been investigated [31]. Particularly, recent studies indicate that Yap1/Tead2 promote PDAC in a KRAS-independent manner [32]. Moreover, accumulating evidences show the mutual interaction between the Hippo pathway and different metabolic pathways [33]. It was reported that glucose metabolism, amino acid metabolism, lipid metabolism and mitochondrial fusion are regulated by YAP and TAZ [34]. YAP phosphorylation and nuclear translocation are modulated by different stress signals and various nutrients/metabolites, including redox status, glucose, lipids, and G protein-coupled receptors (GPCRs). Reciprocally, Hippo signaling also fine-tunes metabolism in the body. Activation of YAP/TAZ upregulates expression of genes encoding both transporters to increase uptake of nutrients/metabolites and rate-limiting enzymes to promote glycolysis and glutamine catabolism [34]. Therefore, it is of great interest to define the effect of Hippo pathway on metabolism reprogramming in PDAC development.
–
In addition to the dysregulation of glucose and Gln metabolism, adaptive changes of various nutrient/metabolites in PDAC development have been uncovered. Preliminary data indicate that decrease of arginine levels could trigger pancreatic cancer cells death. Some clinical trials of bacterial arginine deaminase or bioengineered arginase, aiming to reduce arginine levels, are undergoing in patients with different cancer [35][36]. 5-hydroxytryptamine (5-HT) is known as neurotransmitter which controls critical cognitive and behavioral functions of humans. A recent report demonstrated that 5-HT is elevated in PDAC cells, accompanied by increased tryptophan hydroxylase (TPH1) and decreased mitochondrial enzyme monoamine oxidase A (MAOA) to regulate 5-HT synthesis pathway and degradation pathway, respectively [37]. In addition, treatment with 5-HT receptor inhibitor suppresses growth and reprograms metabolism of pancreatic tumors, prolonging the survival of KPC mice [37]. Moreover, 5-HT activates small GTPase Ras-related C3 botulinum toxin substrate 1 (Rac1) to induce trans-differentiation of acinar cells into ductal, known as ADM [38].
–
LIPID METABOLISM IN PDAC
To sustain uncontrolled cell proliferation, cancer cells need to keep generating various cellular components. Lipids are fundamental materials for structures of cells. Emerging evidence indicates that activated lipid synthesis is required for cancer cell growth. At the initial step of de novo lipid synthesis, ATP-citrate lyase (ACLY) converts citrate to cytoplasmic acetyl-CoA, followed by conversion to malonyl-CoA by acetyl-CoA carboxylase (ACC). Acetyl-CoA and malonyl-CoA are coupled to acyl-carrier protein (ACP) domain of fatty acid synthase (FASN) in an NADPH-dependent manner to synthesize palmitic acid (16-carbon saturated fatty acid) (Figure 2) [39].
–
Lipogenic enzymes including ACLY are frequently overexpressed in PDAC [40] [41]. The growth of PDAC cells is inhibited by the interference of ACLY activity in a xenograft tumor model [42]. In addition, patients with pancreatic cancer expressing high levels of FASN display shorter overall survival period than patients with low FASN expression [43].
–
Cholesterol is an essential structural component of cell membranes. Fatty pancreases were observed in human and are associated with increased risk of pancreas cancer [44]. Furthermore, expression of HMG-CoA (3-hydroxy-3-methylglutaryl-Coenzym-A) reductase and LDLR (low density lipoprotein receptor) is elevated in KRAS-driven PDAC mouse model. Consistently, LDLR silencing reduces ERK signaling activity and inhibits PDAC cell proliferation [45].
–
Hypoxia-inducible HIF-1 also supports lipid synthesis. HIF-1 activation suppresses αKG dehydrogenase (αKGDH), which drives the metabolic shift from the TCA cycle to IDH (isocitrate dehydrogenase)-mediated FA (fatty acid) synthesis [46]. Our study demonstrated that acetate functions as an epigenetic metabolite to promote de novo lipid synthesis under hypoxia conditions [47]. Those data broaden our scope to explore additional function of lipid metabolism in PDAC development.
–
AUTOPHAGY, MITOCHONDRIA, AND PANCREAS CANCER
Classical autophagy is initiated from the formation of a membrane-structured autophagosome which transports damaged cellular components to the lysosome when cells receive stressful stimuli, such as hypoxia, starvation, chemotherapy, and radiation. Processed materials in lysosomes are degraded or recycled to keep cell homeostasis. In tumors, autophagy has an anti- or pro-cancerous function. Enhanced autophagy is detected in PDAC. Nevertheless, opposite evidence shows that elimination of autophagy contributes to PDAC initiation [48]. One interesting study found that KRAS disruption augments autophagy in PDAC [49]. Moreover, our study revealed that acetylation at the K5 residue of lactate dehydrogenase A (LDHA) triggers chaperone-mediated autophagy (CMA) and delivers acetylated LDHA to lysosome for degradation, resulting in alteration of intracellular lactate flux and alleviating the malignant phenotype of PDAC cells in vivo and in vitro [50]. These sophisticated functions of autophagy may explain the poor response to chemotherapy and radiotherapy in current PDAC treatment.
–
Over the past two decades, targeted therapy is one of the most important advances in the treatment of patients with cancer. However, targeting KRAS in PDAC obtains disappointing results. One explanation may be due to the “undruggable” structure of RAS while some recent studies reveal promising advance of RAS inhibitor development [51]. On the other hand, it has been identified that a small population of PDAC cells characterized by stem cell features depend on mitochondrial oxidative phosphorylation and lose response to KRAS signaling [52]. In addition, as mitochondrial respiration is the major resource of intracellular ROS, our study demonstrated that methylation at R248 of malate dehydrogenase 1 (MDH1) is essential to maintain cellular redox homeostasis in PDAC cells [53]. Notably, a novel mechanism that KRAS activation stimulates mitophagy via NIX to sustain PDAC development is disclosed [54]. Altogether, these results indicate that targeting mitochondrial respiration and/or KRAS signaling would significantly improve treatment efficiency of PDAC [51][52][53].
–
METABOLIC REMODLING OF THE PDAC MICROENVIRONMENT
Studies in the recent two decades show that abnormal metabolism remodeling in the tumor microenvironment largely contributes to the poor survival of patients with PDAC.
–
Collagens are the most enriched ECM molecules in the PDAC tumor microenvironment. PDAC cells could uptake cleaved collagen fragments or collagen-derived proline (Pro) through a macropinocytosis-dependent or -independent process (Figure 2). Engulfed collagen fragments are degraded to produce free amino acids in the lysosome, which are entering the TCA cycle and are further metabolized to supply building blocks to promote PDAC cell survival [55]. A further report showed that stimulation of epidermal growth factor receptor (EGFR) – Pak enhances micropinocytosis in PDAC upon nutrient stress [56].
–
Multiple types of stromal cells reside in the PDAC microenvironment. Among them, pancreatic stellate cells (PSCs) are tissue-specific fibroblasts within the pancreas. Reciprocal regulation between PSCs and PDAC cells has been intensively investigated. PSCs-secreted leukemia inhibitory factor (LIF) promotes malignancy of PDAC cells via paracrine [57]. Cancer-associated PSCs also secretes alanine (Ala) to feed PDAC cells and fuel TCA cycle, providing alternative nutrients for cancer cells and decreasing their addiction to glucose as well as other serum-derived nutrients in the austere tumor microenvironment (Figure 2) [8][58][59]. Intriguingly, vitamin D and all-trans retinoic acid (ATRA) were reported to revert stellate cells to a quiescent state, suppressing matrix remodulation and inhibiting cancer cell invasion [60][61].
–
Cancer has been viewed as chronic inflammation without healing. Indeed, various inflammatory cells are involved in tumorigenesis. Tumor-associated macrophages (TAMs) have been reported to promote cancer development. Interestingly, increased glycolysis is observed in TAMs [62]. Furthermore, disruption of PI3Kγ acting as key lipid kinase in macrophages significantly abrogates PDAC invasion and metastasis by enhancing CD8+ T-cell immunosuppression [63]. It is noteworthy that ablation of HIF1a in the pancreatic tissue dramatically boosts malignant progress of KrasG12D-driven PanIN by recruiting a specific subgroup of B cells to infiltrate into the tumor microenvironment [29].
–
Based on these findings, therapeutic strategies against the tumor microenvironment are becoming an attractive opportunity to beat this lethal disease.
–
CONCLUSIONS AND PROSPECTS
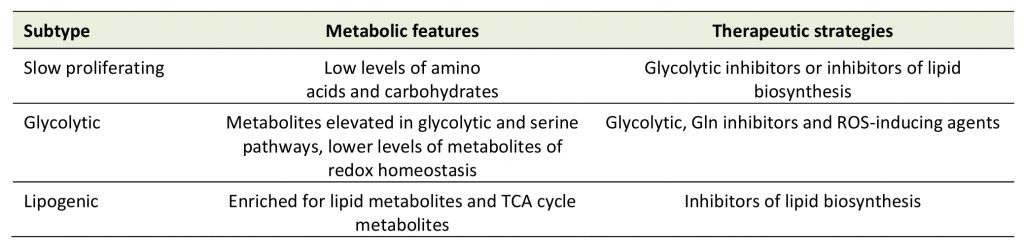
To adapt severely metabolic constraints, PDAC cells rely on specific metabolic reprogramming, offering potentially innovative strategies to treat patients with PDAC in the future. It was reported that PDAC cells could be divided into three different subtypes according to their metabolic profiling, including slow proliferating, glycolytic, and lipogenic subtypes (Table 1) [64]. The glycolytic subtype is more sensitive to glycolytic and glutamine inhibitors, while the lipogenic subtype is more sensitive to inhibitors of lipid biosynthesis. The metabolic plasticity greatly contributes to cancer heterogeneity. A further study on cancer heterogeneity in PDAC revealed that ductal cells are divided into two types based on the features of gene expression profiles via single-cell RNA-sequence analysis in PDAC [65]. Type 1 ductal cells are relative normal and present in both normal and cancer tissues, while type 2 ductal cells are exclusively found in the PDAC region, furthermore, type 2 ductal cells also contain seven subpopulations. Meanwhile, based on single-cell RNA-sequence analysis stromal cells including T cells, macrophages and fibroblasts are highly heterogeneous within the tumor microenvironment? [65][66]. Therefore, targeting cancer metabolism in combination with other targeting agents or cytotoxic compounds would be promising therapeutic strategies beneficial for PDAC patients.
–
Table 1. Subtypes of PDAC based on metabolic features. |
 |
–
REFERENCES
- Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, Reichert M, Beatty GL, Rustgi AK, Vonderheide RH, Leach SD, Stanger BZ (2012). EMT and dissemination precede pancreatic tumor formation. Cell 148(1-2): 349-361. 10.1016/j.cell.2011.11.025
- Parsa I, Longnecker DS, Scarpelli DG, Pour P, Reddy JK, Lefkowitz M (1985). Ductal metaplasia of human exocrine pancreas and its association with carcinoma. Cancer Res 45(3): 1285-1290. 2982487
- Han H, Von Hoff DD (2013). SnapShot: pancreatic cancer. Cancer Cell 23(3): 424-424 e421. 10.1016/j.ccr.2013.03.008
- Hezel AF, Kimmelman AC, Stanger BZ, Bardeesy N, Depinho RA (2006). Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev 20(10): 1218-1249. 10.1101/gad.1415606
- Rooman I, Real FX (2012). Pancreatic ductal adenocarcinoma and acinar cells: a matter of differentiation and development? Gut 61(3): 449-458. 10.1136/gut.2010.235804
- Haeno H, Gonen M, Davis MB, Herman JM, Iacobuzio-Donahue CA, Michor F (2012). Computational modeling of pancreatic cancer reveals kinetics of metastasis suggesting optimum treatment strategies. Cell 148(1-2): 362-375. 10.1016/j.cell.2011.11.060
- Iacobuzio-Donahue CA, Velculescu VE, Wolfgang CL, Hruban RH (2012). Genetic basis of pancreas cancer development and progression: insights from whole-exome and whole-genome sequencing. Clin Cancer Res 18(16): 4257-4265. 10.1158/1078-0432.CCR-12-0315
- Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA (2012). The pancreas cancer microenvironment. Clin Cancer Res 18(16): 4266-4276. 10.1158/1078-0432.CCR-11-3114
- Bapat AA, Hostetter G, Von Hoff DD, Han H (2011). Perineural invasion and associated pain in pancreatic cancer. Nat Rev Cancer 11(10): 695-707. 10.1038/nrc3131
- Warburg O (1956). Origin of cancer cells. Oncologia 9(2): 75-83. 13335077
- Feldmann G, Beaty R, Hruban RH, Maitra A (2007). Molecular genetics of pancreatic intraepithelial neoplasia. J Hepatobiliary Pancreat Surg 14(3): 224-232. 10.1007/s00534-006-1166-5
- Bryant KL, Mancias JD, Kimmelman AC, Der CJ (2014). KRAS: feeding pancreatic cancer proliferation. Trends Biochem Sci 39(2): 91-100. 10.1016/j.tibs.2013.12.004
- Gaglio D, Metallo CM, Gameiro PA, Hiller K, Danna LS, Balestrieri C, Alberghina L, Stephanopoulos G, Chiaradonna F (2011). Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol Syst Biol 7: 523. 10.1038/msb.2011.56
- Dell' Antone P (2012). Energy metabolism in cancer cells: how to explain the Warburg and Crabtree effects? Med Hypotheses 79(3): 388-392. 10.1016/j.mehy.2012.06.002
- Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, Yan H, Wang W, Chen S, Viale A, Zheng H, Paik JH, Lim C, Guimaraes AR, Martin ES, Chang J, Hezel AF, Perry SR, Hu J, Gan B, Xiao Y, Asara JM, Weissleder R, Wang YA, Chin L, Cantley LC, et al. (2012). Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 149(3): 656-670. 10.1016/j.cell.2012.01.058
- Ma Z, Vocadlo DJ, Vosseller K (2013). Hyper-O-GlcNAcylation is anti-apoptotic and maintains constitutive NF-kappaB activity in pancreatic cancer cells. J Biol Chem 288(21): 15121-15130. 10.1074/jbc.M113.470047
- Boros LG, Puigjaner J, Cascante M, Lee WN, Brandes JL, Bassilian S, Yusuf FI, Williams RD, Muscarella P, Melvin WS, Schirmer WJ (1997). Oxythiamine and dehydroepiandrosterone inhibit the nonoxidative synthesis of ribose and tumor cell proliferation. Cancer Res 57(19): 4242-4248. 9331084
- Ju HQ, Ying H, Tian T, Ling J, Fu J, Lu Y, Wu M, Yang L, Achreja A, Chen G, Zhuang Z, Wang H, Nagrath D, Yao J, Hung MC, DePinho RA, Huang P, Xu RH, Chiao PJ (2017). Mutant Kras- and p16-regulated NOX4 activation overcomes metabolic checkpoints in development of pancreatic ductal adenocarcinoma. Nat Commun 8: 14437. 10.1038/ncomms14437
- Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, Kang Y, Fleming JB, Bardeesy N, Asara JM, Haigis MC, DePinho RA, Cantley LC, Kimmelman AC (2013). Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496(7443): 101-105. 10.1038/nature12040
- Chio IIC, Jafarnejad SM, Ponz-Sarvise M, Park Y, Rivera K, Palm W, Wilson J, Sangar V, Hao Y, Ohlund D, Wright K, Filippini D, Lee EJ, Da Silva B, Schoepfer C, Wilkinson JE, Buscaglia JM, DeNicola GM, Tiriac H, Hammell M, Crawford HC, Schmidt EE, Thompson CB, Pappin DJ, Sonenberg N, Tuveson DA (2016). NRF2 Promotes Tumor Maintenance by Modulating mRNA Translation in Pancreatic Cancer. Cell 166(4): 963-976. 10.1016/j.cell.2016.06.056
- Kong B, Qia C, Erkan M, Kleeff J, Michalski CW (2013). Overview on how oncogenic Kras promotes pancreatic carcinogenesis by inducing low intracellular ROS levels. Front Physiol 4: 246. 10.3389/fphys.2013.00246
- Butera G, Pacchiana R, Mullappilly N, Margiotta M, Bruno S, Conti P, Riganti C, Donadelli M (2018). Mutant p53 prevents GAPDH nuclear translocation in pancreatic cancer cells favoring glycolysis and 2-deoxyglucose sensitivity. Biochim Biophys Acta Mol Cell Res 1865(12): 1914-1923. 10.1016/j.bbamcr.2018.10.005
- Schofield HK, Zeller J, Espinoza C, Halbrook CJ, Del Vecchio A, Magnuson B, Fabo T, Daylan AEC, Kovalenko I, Lee HJ, Yan W, Feng Y, Karim SA, Kremer DM, Kumar-Sinha C, Lyssiotis CA, Ljungman M, Morton JP, Galban S, Fearon ER, Pasca di Magliano M (2018). Mutant p53R270H drives altered metabolism and increased invasion in pancreatic ductal adenocarcinoma. JCI Insight 3(2): 97422. 10.1172/jci.insight.97422
- Morris JPt, Yashinskie JJ, Koche R, Chandwani R, Tian S, Chen CC, Baslan T, Marinkovic ZS, Sanchez-Rivera FJ, Leach SD, Carmona-Fontaine C, Thompson CB, Finley LWS, Lowe SW (2019). alpha-Ketoglutarate links p53 to cell fate during tumour suppression. Nature 573(7775): 595-599. 10.1038/s41586-019-1577-5
- Guillaumond F, Leca J, Olivares O, Lavaut MN, Vidal N, Berthezene P, Dusetti NJ, Loncle C, Calvo E, Turrini O, Iovanna JL, Tomasini R, Vasseur S (2013). Strengthened glycolysis under hypoxia supports tumor symbiosis and hexosamine biosynthesis in pancreatic adenocarcinoma. Proc Natl Acad Sci U S A 110(10): 3919-3924. 10.1073/pnas.1219555110
- Baek G, Tse YF, Hu Z, Cox D, Buboltz N, McCue P, Yeo CJ, White MA, DeBerardinis RJ, Knudsen ES, Witkiewicz AK (2014). MCT4 defines a glycolytic subtype of pancreatic cancer with poor prognosis and unique metabolic dependencies. Cell Rep 9(6): 2233-2249. 10.1016/j.celrep.2014.11.025
- Chaika NV, Gebregiworgis T, Lewallen ME, Purohit V, Radhakrishnan P, Liu X, Zhang B, Mehla K, Brown RB, Caffrey T, Yu F, Johnson KR, Powers R, Hollingsworth MA, Singh PK (2012). MUC1 mucin stabilizes and activates hypoxia-inducible factor 1 alpha to regulate metabolism in pancreatic cancer. Proc Natl Acad Sci U S A 109(34): 13787-13792. 10.1073/pnas.1203339109
- Shi M, Cui J, Du J, Wei D, Jia Z, Zhang J, Zhu Z, Gao Y, Xie K (2014). A novel KLF4/LDHA signaling pathway regulates aerobic glycolysis in and progression of pancreatic cancer. Clin Cancer Res 20(16): 4370-4380. 10.1158/1078-0432.CCR-14-0186
- Lee KE, Spata M, Bayne LJ, Buza EL, Durham AC, Allman D, Vonderheide RH, Simon MC (2016). Hif1a Deletion Reveals Pro-Neoplastic Function of B Cells in Pancreatic Neoplasia. Cancer Discov 6(3): 256-269. 10.1158/2159-8290.CD-15-0822
- Fu V, Plouffe SW, Guan KL (2017). The Hippo pathway in organ development, homeostasis, and regeneration. Curr Opin Cell Biol 49: 99-107. 10.1016/j.ceb.2017.12.012
- Zhou X, Lei QY (2016). Regulation of TAZ in cancer. Protein Cell 7(8): 548-561. 10.1007/s13238-016-0288-z
- Kapoor A, Yao W, Ying H, Hua S, Liewen A, Wang Q, Zhong Y, Wu CJ, Sadanandam A, Hu B, Chang Q, Chu GC, Al-Khalil R, Jiang S, Xia H, Fletcher-Sananikone E, Lim C, Horwitz GI, Viale A, Pettazzoni P, Sanchez N, Wang H, Protopopov A, Zhang J, Heffernan T, Johnson RL, Chin L, Wang YA, Draetta G, DePinho RA (2014). Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell 158(1): 185-197. 10.1016/j.cell.2014.06.003
- Ardestani A, Lupse B, Maedler K (2018). Hippo Signaling: Key Emerging Pathway in Cellular and Whole-Body Metabolism. Trends Endocrinol Metab 29(7): 492-509. 10.1016/j.tem.2018.04.006
- Koo JH, Guan KL (2018). Interplay between YAP/TAZ and Metabolism. Cell Metab 28(2): 196-206. 10.1016/j.cmet.2018.07.010
- Bowles TL, Kim R, Galante J, Parsons CM, Virudachalam S, Kung HJ, Bold RJ (2008). Pancreatic cancer cell lines deficient in argininosuccinate synthetase are sensitive to arginine deprivation by arginine deiminase. Int J Cancer 123(8): 1950-1955. 10.1002/ijc.23723
- Huang CC, Tsai ST, Kuo CC, Chang JS, Jin YT, Chang JY, Hsiao JR (2012). Arginine deprivation as a new treatment strategy for head and neck cancer. Oral Oncol 48(12): 1227-1235. 10.1016/j.oraloncology.2012.06.004
- Jiang SH, Li J, Dong FY, Yang JY, Liu DJ, Yang XM, Wang YH, Yang MW, Fu XL, Zhang XX, Li Q, Pang XF, Huo YM, Li J, Zhang JF, Lee HY, Lee SJ, Qin WX, Gu JR, Sun YW, Zhang ZG (2017). Increased Serotonin Signaling Contributes to the Warburg Effect in Pancreatic Tumor Cells Under Metabolic Stress and Promotes Growth of Pancreatic Tumors in Mice. Gastroenterology 153(1): 277-291 e219. 10.1053/j.gastro.2017.03.008
- Saponara E, Visentin M, Baschieri F, Seleznik G, Martinelli P, Esposito I, Buschmann J, Chen R, Parrotta R, Borgeaud N, Bombardo M, Malagola E, Caflisch A, Farhan H, Graf R, Sonda S (2018). Serotonin uptake is required for Rac1 activation in Kras-induced acinar-to-ductal metaplasia in the pancreas. J Pathol 246(3): 352-365. 10.1002/path.5147
- Baenke F, Peck B, Miess H, Schulze A (2013). Hooked on fat: the role of lipid synthesis in cancer metabolism and tumour development. Dis Model Mech 6(6): 1353-1363. 10.1242/dmm.011338
- Swierczynski J, Hebanowska A, Sledzinski T (2014). Role of abnormal lipid metabolism in development, progression, diagnosis and therapy of pancreatic cancer. World J Gastroenterol 20(9): 2279-2303. 10.3748/wjg.v20.i9.2279
- Menendez JA, Lupu R (2007). Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer 7(10): 763-777. 10.1038/nrc2222
- Hatzivassiliou G, Zhao F, Bauer DE, Andreadis C, Shaw AN, Dhanak D, Hingorani SR, Tuveson DA, Thompson CB (2005). ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell 8(4): 311-321. 10.1016/j.ccr.2005.09.008
- Tadros S, Shukla SK, King RJ, Gunda V, Vernucci E, Abrego J, Chaika NV, Yu F, Lazenby AJ, Berim L, Grem J, Sasson AR, Singh PK (2017). De Novo Lipid Synthesis Facilitates Gemcitabine Resistance through Endoplasmic Reticulum Stress in Pancreatic Cancer. Cancer Res 77(20): 5503-5517. 10.1158/0008-5472.CAN-16-3062
- Takahashi M, Hori M, Ishigamori R, Mutoh M, Imai T, Nakagama H (2018). Fatty pancreas: A possible risk factor for pancreatic cancer in animals and humans. Cancer Sci 109(10): 3013-3023. 10.1111/cas.13766
- Guillaumond F, Bidaut G, Ouaissi M, Servais S, Gouirand V, Olivares O, Lac S, Borge L, Roques J, Gayet O, Pinault M, Guimaraes C, Nigri J, Loncle C, Lavaut MN, Garcia S, Tailleux A, Staels B, Calvo E, Tomasini R, Iovanna JL, Vasseur S (2015). Cholesterol uptake disruption, in association with chemotherapy, is a promising combined metabolic therapy for pancreatic adenocarcinoma. Proc Natl Acad Sci U S A 112(8): 2473-2478. 10.1073/pnas.1421601112
- Sun RC, Denko NC (2014). Hypoxic regulation of glutamine metabolism through HIF1 and SIAH2 supports lipid synthesis that is necessary for tumor growth. Cell Metab 19(2): 285-292. 10.1016/j.cmet.2013.11.022
- Gao X, Lin SH, Ren F, Li JT, Chen JJ, Yao CB, Yang HB, Jiang SX, Yan GQ, Wang D, Wang Y, Liu Y, Cai Z, Xu YY, Chen J, Yu W, Yang PY, Lei QY (2016). Acetate functions as an epigenetic metabolite to promote lipid synthesis under hypoxia. Nat Commun 7: 11960. 10.1038/ncomms11960
- New M, Van Acker T, Long JS, Sakamaki JI, Ryan KM, Tooze SA (2017). Molecular Pathways Controlling Autophagy in Pancreatic Cancer. Front Oncol 7: 28. 10.3389/fonc.2017.00028
- Bryant KL, Stalnecker CA, Zeitouni D, Klomp JE, Peng S, Tikunov AP, Gunda V, Pierobon M, Waters AM, George SD, Tomar G, Papke B, Hobbs GA, Yan L, Hayes TK, Diehl JN, Goode GD, Chaika NV, Wang Y, Zhang GF, Witkiewicz AK, Knudsen ES, Petricoin EF, 3rd, Singh PK, Macdonald JM, Tran NL, Lyssiotis CA, Ying H, Kimmelman AC, Cox AD, et al. (2019). Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat Med 25(4): 628-640. 10.1038/s41591-019-0368-8
- Zhao D, Zou SW, Liu Y, Zhou X, Mo Y, Wang P, Xu YH, Dong B, Xiong Y, Lei QY, Guan KL (2013). Lysine-5 acetylation negatively regulates lactate dehydrogenase A and is decreased in pancreatic cancer. Cancer Cell 23(4): 464-476. 10.1016/j.ccr.2013.02.005
- O'Bryan JP (2019). Pharmacological targeting of RAS: Recent success with direct inhibitors. Pharmacol Res 139: 503-511. 10.1016/j.phrs.2018.10.021
- Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sanchez N, Marchesini M, Carugo A, Green T, Seth S, Giuliani V, Kost-Alimova M, Muller F, Colla S, Nezi L, Genovese G, Deem AK, Kapoor A, Yao W, Brunetto E, Kang Y, Yuan M, Asara JM, Wang YA, Heffernan TP, Kimmelman AC, Wang H, Fleming JB, Cantley LC, DePinho RA, Draetta GF (2014). Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 514(7524): 628-632. 10.1038/nature13611
- Wang YP, Zhou W, Wang J, Huang X, Zuo Y, Wang TS, Gao X, Xu YY, Zou SW, Liu YB, Cheng JK, Lei QY (2016). Arginine Methylation of MDH1 by CARM1 Inhibits Glutamine Metabolism and Suppresses Pancreatic Cancer. Mol Cell 64(4): 673-687. 10.1016/j.molcel.2016.09.028
- Humpton TJ, Alagesan B, DeNicola GM, Lu D, Yordanov GN, Leonhardt CS, Yao MA, Alagesan P, Zaatari MN, Park Y, Skepper JN, Macleod KF, Perez-Mancera PA, Murphy MP, Evan GI, Vousden KH, Tuveson DA (2019). Oncogenic KRAS Induces NIX-Mediated Mitophagy to Promote Pancreatic Cancer. Cancer Discov 9(9): 1268-1287. 10.1158/2159-8290.CD-18-1409
- Olivares O, Mayers JR, Gouirand V, Torrence ME, Gicquel T, Borge L, Lac S, Roques J, Lavaut MN, Berthezene P, Rubis M, Secq V, Garcia S, Moutardier V, Lombardo D, Iovanna JL, Tomasini R, Guillaumond F, Vander Heiden MG, Vasseur S (2017). Collagen-derived proline promotes pancreatic ductal adenocarcinoma cell survival under nutrient limited conditions. Nat Commun 8: 16031. 10.1038/ncomms16031
- Lee SW, Zhang Y, Jung M, Cruz N, Alas B, Commisso C (2019). EGFR-Pak Signaling Selectively Regulates Glutamine Deprivation-Induced Macropinocytosis. Dev Cell 50(3): 381-392 e385. 10.1016/j.devcel.2019.05.043
- Shi Y, Gao W, Lytle NK, Huang P, Yuan X, Dann AM, Ridinger-Saison M, DelGiorno KE, Antal CE, Liang G, Atkins AR, Erikson G, Sun H, Meisenhelder J, Terenziani E, Woo G, Fang L, Santisakultarm TP, Manor U, Xu R, Becerra CR, Borazanci E, Von Hoff DD, Grandgenett PM, Hollingsworth MA, Leblanc M, Umetsu SE, Collisson EA, Scadeng M, Lowy AM, et al. (2019). Targeting LIF-mediated paracrine interaction for pancreatic cancer therapy and monitoring. Nature 569(7754): 131-135. 10.1038/s41586-019-1130-6
- Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, Kremer D, Hwang RF, Witkiewicz AK, Ying H, Asara JM, Evans RM, Cantley LC, Lyssiotis CA, Kimmelman AC (2016). Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 536(7617): 479-483. 10.1038/nature19084
- Kamphorst JJ, Nofal M, Commisso C, Hackett SR, Lu W, Grabocka E, Vander Heiden MG, Miller G, Drebin JA, Bar-Sagi D, Thompson CB, Rabinowitz JD (2015). Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res 75(3): 544-553. 10.1158/0008-5472.CAN-14-2211
- Chronopoulos A, Robinson B, Sarper M, Cortes E, Auernheimer V, Lachowski D, Attwood S, Garcia R, Ghassemi S, Fabry B, Del Rio Hernandez A (2016). ATRA mechanically reprograms pancreatic stellate cells to suppress matrix remodelling and inhibit cancer cell invasion. Nat Commun 7: 12630. 10.1038/ncomms12630
- Sherman MH, Yu RT, Engle DD, Ding N, Atkins AR, Tiriac H, Collisson EA, Connor F, Van Dyke T, Kozlov S, Martin P, Tseng TW, Dawson DW, Donahue TR, Masamune A, Shimosegawa T, Apte MV, Wilson JS, Ng B, Lau SL, Gunton JE, Wahl GM, Hunter T, Drebin JA, O'Dwyer PJ, Liddle C, Tuveson DA, Downes M, Evans RM (2014). Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 159(1): 80-93. 10.1016/j.cell.2014.08.007
- Penny HL, Sieow JL, Adriani G, Yeap WH, See Chi Ee P, San Luis B, Lee B, Lee T, Mak SY, Ho YS, Lam KP, Ong CK, Huang RY, Ginhoux F, Rotzschke O, Kamm RD, Wong SC (2016). Warburg metabolism in tumor-conditioned macrophages promotes metastasis in human pancreatic ductal adenocarcinoma. Oncoimmunology 5(8): e1191731. 10.1080/2162402X.2016.1191731
- Kaneda MM, Cappello P, Nguyen AV, Ralainirina N, Hardamon CR, Foubert P, Schmid MC, Sun P, Mose E, Bouvet M, Lowy AM, Valasek MA, Sasik R, Novelli F, Hirsch E, Varner JA (2016). Macrophage PI3Kgamma Drives Pancreatic Ductal Adenocarcinoma Progression. Cancer Discov 6(8): 870-885. 10.1158/2159-8290.CD-15-1346
- Daemen A, Peterson D, Sahu N, McCord R, Du X, Liu B, Kowanetz K, Hong R, Moffat J, Gao M, Boudreau A, Mroue R, Corson L, O'Brien T, Qing J, Sampath D, Merchant M, Yauch R, Manning G, Settleman J, Hatzivassiliou G, Evangelista M (2015). Metabolite profiling stratifies pancreatic ductal adenocarcinomas into subtypes with distinct sensitivities to metabolic inhibitors. Proc Natl Acad Sci U S A 112(32): E4410-4417. 10.1073/pnas.1501605112
- Peng J, Sun BF, Chen CY, Zhou JY, Chen YS, Chen H, Liu L, Huang D, Jiang J, Cui GS, Yang Y, Wang W, Guo D, Dai M, Guo J, Zhang T, Liao Q, Liu Y, Zhao YL, Han DL, Zhao Y, Yang YG, Wu W (2019). Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res 29(9): 725-738. 10.1038/s41422-019-0195-y
- Bernard V, Semaan A, Huang J, San Lucas FA, Mulu FC, Stephens BM, Guerrero PA, Huang Y, Zhao J, Kamyabi N, Sen S, Scheet PA, Taniguchi CM, Kim MP, Tzeng CW, Katz MH, Singhi AD, Maitra A, Alvarez HA (2019). Single-Cell Transcriptomics of Pancreatic Cancer Precursors Demonstrates Epithelial and Microenvironmental Heterogeneity as an Early Event in Neoplastic Progression. Clin Cancer Res 25(7): 2194-2205. 10.1158/1078-0432.CCR-18-1955
ACKNOWLEDGMENTS
This work was supported by MOST (Grant No. 2015CB910401), NSFC (Grant No. 81790253, 81430057, and 81872240) and Innovation Program of Shanghai Municipal Education Commission (No. N173606).
COPYRIGHT
© 2019

Metabolism remodeling in pancreatic ductal adenocarcinoma by Li et al. is licensed under a Creative Commons Attribution 4.0 International License.


