Reviews:
Cell Stress, Vol. 4, No. 6, pp. 114 - 146; doi: 10.15698/cst2020.06.221
Mitochondria in cancer

1 Pole of Pharmacology & Therapeutics, Institut de Recherche Expérimentale et Clinique (IREC), Université catholique de Louvain (UCLouvain), Brussels, Belgium.
Keywords: tumor metabolism, tricarboxylic acid (TCA) cycle, oxidative phosphorylation (OXPHOS), reactive oxygen species (ROS), apoptosis, mitophagy, mitochondrial biogenesis.
Abbreviatons:
2DG – 2-deoxy-D-glucose;
2HG – D-2-hydroxyglutarate;
3BP – 3-bromopyruvate;
α-KG – α-ketoglutarate;
α-TOS – α-tocopheryl succinate; ACO2 – aconitase 2;
AML – acute myeloid leukemia;
AMPK – AMP kinase;
Atg - autophagy-related;
Bcl-2 – B-cell lymphoma 2;
CoQ – coenzyme Q;
CS - citrate synthase;
CSC – cancer stem cell; DRP1 – dynamin-related protein 1;
EMT – epithelial to mesenchymal transition;
EOC – epithelial ovarian carcinoma;
ER – endoplasmic reticulum;
ETC – electron transport chain;
FAO – fatty acid oxidation;
FH – fumarate hydratase;
FoxO1 – forkhead O family protein 1;
FUNDC1 – FUN14 domain-containing 1;
GAPDH – glyceraldehyde-3-phosphate dehydrogenase;
GLUT – glucose transporter;
GSH – glutathione;
GST – glutathione S-transferase;
HIF – hypoxia-inducible factor;
HK – hexokinase;
Hsp70 – heat shock protein 70;
IDH – isocitrate dehydrogenase;
IMM – inner mitochondrial membrane;
IMS – intermembrane space;
KD – ketogenic diet;
KRAS – V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog;
LC3 – microtubule-associated protein light chain 3;
LDH-1 – lactate dehydrogenase-1;
MAVS – mitochondrial anti-viral signaling;
MCL-1 – myeloid leukemia cell differentiation protein-1;
Mdivi-1 – mitochondrial division inhibitor-1;
Mff – mitochondrial fission factor;
Mfn – mitofusin;
mtDNA – mitochondrial DNA;
mtROS - mitochondrial ROS;
mtSNP – single nucleotide polymorphism in mtDNA;
Nrf2 – nuclear factor erythroid-derived 2-like 2;
OMM – outer mitochondrial membrane;
Opa1 – optic atrophy protein 1;
OXPHOS – oxidative phosphorylation;
PDT – photodynamic therapy;
PEP – phosphoenolpyruvate;
PKA – cAMP-activated protein kinase A;
PINK1 – phosphatase and tensin homolog-induced kinase 1;
PTEN – phosphatase and tensin homolog;
ROS – reactive oxygen species;
SCO2 – synthesis of cytochrome oxidase 2;
SDH – succinate dehydrogenase;
Sirt – sirtuin;
SOD – superoxide dismutase;
TCA – tricarboxylic acid (cycle);
TERT – telomerase reverse transcriptase;
TGFβ – transforming growth factor β;
TIGAR – TP53-induced glycolysis and apoptosis regulator;
TNT – tunneling nanotube;
VDAC – voltage-dependent anion channel.
Received originally: 01/01/2020 Received in revised form: 28/04/2020
Accepted: 29/04/2020
Published: 11/05/2020
Correspondence:
Pierre Sonveaux, Pole of Pharmacology & Therapeutics, Institut de Recherche Expérimentale et Clinique (IREC), Université catholique de Louvain (UCLouvain), Avenue Hippocrate 57 box B1.53.09, Brussels, 1200, Belgium. Tel: +32 (0)2 7645267; pierre.sonveaux@uclouvain.be
Conflict of interest statement: The authors declare no conflict of interest.
Please cite this article as: Debora Grasso, Luca X Zampieri, Tânia Capelôa, Justine A. Van de Velde and Pierre Sonveaux (2020). Mitochondria in cancer. Cell Stress 4(6): 114-146. doi: 10.15698/cst2020.06.221
Abstract
The rediscovery and reinterpretation of the Warburg effect in the year 2000 occulted for almost a decade the key functions exerted by mitochondria in cancer cells. Until recent times, the scientific community indeed focused on constitutive glycolysis as a hallmark of cancer cells, which it is not, largely ignoring the contribution of mitochondria to the malignancy of oxidative and glycolytic cancer cells, being Warburgian or merely adapted to hypoxia. In this review, we highlight that mitochondria are not only powerhouses in some cancer cells, but also dynamic regulators of life, death, proliferation, motion and stemness in other types of cancer cells. Similar to the cells that host them, mitochondria are capable to adapt to tumoral conditions, and probably to evolve to ‘oncogenic mitochondria’ capable of transferring malignant capacities to recipient cells. In the wider quest of metabolic modulators of cancer, treatments have already been identified targeting mitochondria in cancer cells, but the field is still in infancy.
INTRODUCTION
The vital role of mitochondria in eukaryotic cells has been demonstrated over a hundred years ago by Otto Warburg, who was the first to perform mitochondrial respiration experiments [1]. In healthy replicative eukaryotic cells, mitochondria regulate important cellular processes, such as proliferation, death, metabolic adaptation and Ca2+ homeostasis. Mitochondria are also the site of important reactions, including fatty acid oxidation (FAO), the tricarboxylic acid (TCA) cycle, oxidative phosphorylation (OXPHOS), the first step of gluconeogenesis, ketogenesis, heme biosynthesis and Fe/S cluster formation [2]. They contain DNA that can vary with evolution, mutate or be partially deleted. Given the numerous functions of mitochondria, it is not surprising that mitochondrial dysfunctions participate in a series of diseases, including cancer. In this review, we focus on mitochondrial functions and their contribution to carcinogenesis and cancer progression.
MITOCHONDRIA PARTICIPATE IN CANCER DEVELOPMENT
There are at least five mechanisms by which mitochondria may be involved in the development of the malignant phenotype over the metabolic reprogramming of cancer cells. First, it is widely demonstrated that a large number of diseases are associated with DNA mutations that affect mitochondria, mainly due to alterations of subunits of the electron transport chain (ETC) [3]. For example, subsets of hepatocellular carcinomas and prostate cancers have been associated with a mutation in the D-loop region of Complex I, and some neurological cancers harbor mutations of succinate dehydrogenase (SDH; Complex II) [4][5][6]. Second, oxidative stress due to reactive oxygen species (ROS) is the most important stimulus for cancer generation and progression towards malignancy [7]. ROS are mainly produced by mitochondria that release superoxide as a byproduct of oxidative respiration [8]. Mitochondrial ROS (mtROS) can be generated either in the TCA cycle or in the ETC [9]. Due to their high reactivity, ROS act as toxic species for cellular macromolecules [10] and, at low concentrations, as intracellular signaling agents regulating metabolic pathways [11][12]. Increased levels of ROS are often found in cancer cells due to increased metabolic activities and altered antioxidant capacities [13]. Third, mitochondria are directly involved in the regulation of cell death, including but not limited to apoptosis and necrosis [14][15]. To induce apoptosis, B-cell lymphoma-2 (Bcl-2) family member proteins interact with mitochondria as they bind to the voltage-dependent anion channel (VDAC) to accelerate its opening and the release of cytochrome c[16]. Thereby, these proteins act as oncogenic or oncosuppressive triggers, participating in cancer progression and therapeutic resistance [17][18]. One of them, myeloid leukemia cell differentiation protein-1 (MCL-1), an anti-apoptotic member of the Bcl-2 family, is frequently overexpressed in human cancer and associated with tumor aggressiveness [19]. MCL-1 and Bcl-xL have been found in different mitochondrial subcompartments. They exert their anti-apoptotic activities by antagonizing the pro-apoptotic members of the Bcl-2 family when located at the outer mitochondrial membrane (OMM) [20], and, when located in the mitochondrial matrix, by regulating mitochondrial homeostasis and bioenergetics by preserving the integrity of the inner mitochondrial membrane (IMM) and promoting the assembly of ATP-synthase oligomers at the ETC [17]. Mitochondria also control necroptosis, a regulated form of necrosis that needs mtROS generation and depends on mitochondrial permeability transition [21]. Fourth, metabolic reprogramming also concerns several mutations in genes encoding TCA cycle enzymes, which promote malignant transformation [22]. Indeed, some TCA cycle intermediates, such as fumarate, succinate, aspartate and D-2-hydroxyglutarate (2HG, a de novo metabolite resulting from mutations of isocitrate dehydrogenases (IDHs)), have important pro-carcinogenic effects when accumulating in cells following genetic mutations and/or cancer-associated modifications of protein expression [23]. Fifth, a distinctive feature of all tumors is sustained cellular proliferation resulting from multiple molecular alterations. One of these alterations is the prevention of telomere erosion by constitutive telomerase expression that ensures the maintenance of telomere length [24]. It has been shown that telomerase reverse transcriptase (TERT) shuttles from the nucleus to mitochondria upon oxidative stress, preserving mitochondrial functions and decreasing oxidative stress, thus protecting mitochondrial DNA (mtDNA) and nuclear DNA (nDNA) from oxidative damage to avoid apoptosis [25][26]. TERT was also found to accumulate in the mitochondria of brain cells in mice upon dietary restriction and rapamycin treatment [27].
MITOCHONDRIA ARE NOT ONLY THE POWERHOUSES OF THE CELL
Despite the fact that mitochondria are well recognized to actively participate in cancer progression, their precise roles in the clinical outcome of cancer patients remain elusive. The interest of scientists for mitochondria has increased over the last 50 years, with discoveries on the impact that these organelles have in multiple vital processes in eukaryotic cells [28].
–
Mitochondria are tubular organelles of ∼0.5 to ∼3 µm in length that undergo a continuous remodeling of their network by fusion and fission events [29]. Textbooks first describe mitochondria as the main site of energy production of cells, and, indeed, mitochondria are a major site of production of ATP and macromolecules. The reactions of the TCA cycle take place in the mitochondrial matrix. Together with CO2 and protons, they generate reducing equivalents (NADH and FADH2) and precursors for the synthesis of lipids, carbohydrates, proteins and nucleotides. Equivalent-reducing electrons fuel the ETC to generate an electrochemical gradient that is required both for ATP production and for the active transport of selective metabolites, such as pyruvate and ATP, across the IMM [30].
–
In addition to this important role, mitochondria are implicated in many other functions related to mitochondrial dynamics and architecture, which influence some of the most important cellular activities. The mitochondrial structure (Figure 1A) is intrinsically connected to mitochondrial functions (ATP production, cell cycle control, programmed cell death control, proliferation and cell signaling) [31]. Mitochondria are indeed composed of two membranes, the OMM and the IMM that delimitate an intermembrane space (IMS) and the mitochondrial matrix inside the organelle. The OMM can be considered as a platform for exchange and signaling, as it is the site where proteins phos-phorylate substrates and regulate the immune response after viral infection trough activation of mitochondrial antiviral signaling (MAVS) proteins [32][33]. The IMM is less permeable and is the site where ETC complexes are located for ATP production and superoxide generation [34]. The matrix is the site of mitochondrial mtDNA replication, transcription and macromolecule biosynthesis, where amphibolic reactions of the TCA cycle take place [35]. The investigation of mitochondrial mechanisms that control metabolic alterations and mitochondrial morphology has produced evidence that, in pathologies like cancer, they can be attractive targets for therapy.
–
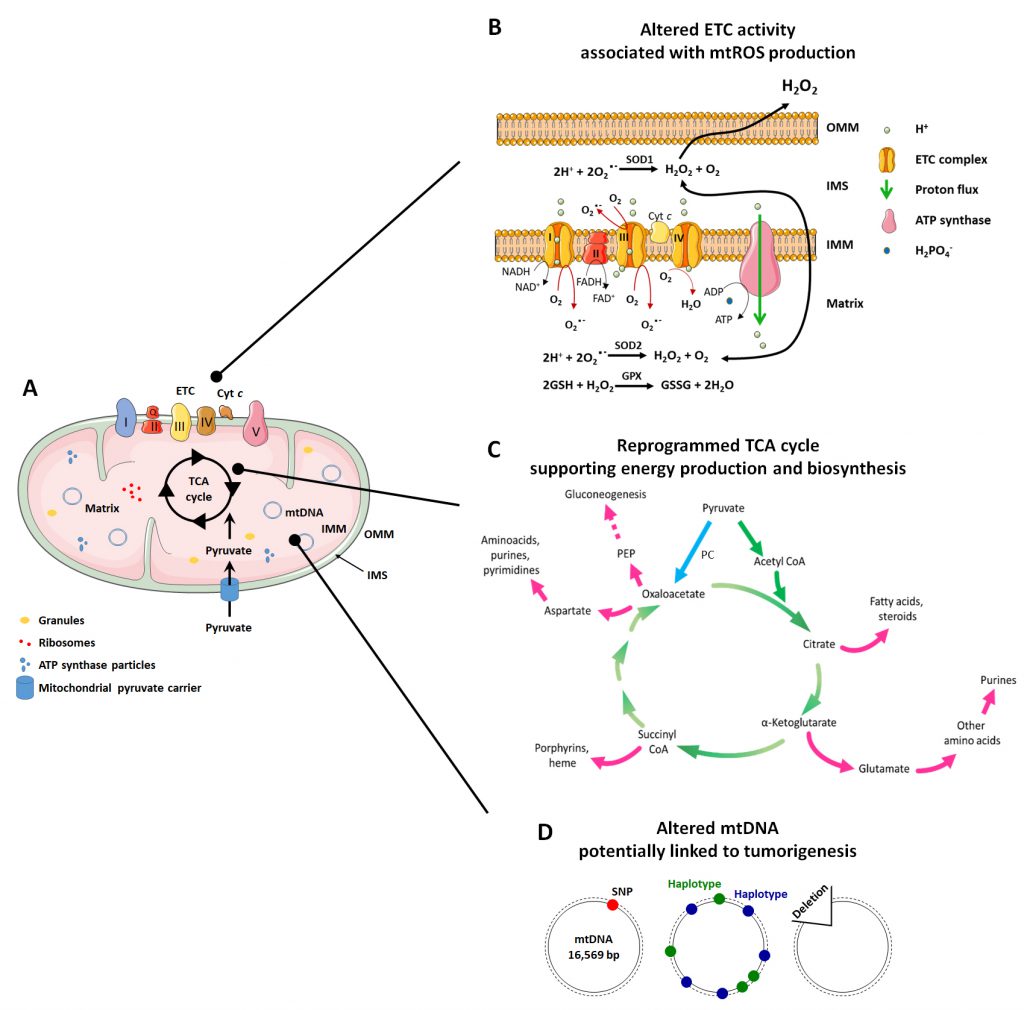 |
FIGURE 1: Cancer is associated with alterations of mitochondrial functions. (A) Mitochondria in normal and in cancer cells are composed of three compartments. They are separated from the cell cytosol by an outer membrane (OMM), an intermembrane space (IMS), and an inner membrane (IMM) that forms invaginations called "crests". The IMM delimitates the mitochondrial matrix, a gelatinous material containing mitochondrial DNA (mtDNA), granules, ribosomes and ATP synthase particles. The mitochondrial matrix hosts the tricarboxylic acid (TCA) cycle, while the IMM hosts the electron transport chain (ETC). (B) In highly metabolically active or hypoxic cancer cells much more than in normal cells under normal conditions, electrons escape during mitochondrial electron transport at Complexes I and III generate superoxide (O2-) from oxygen (O2) in both the IMS and the matrix. O2- is immediately dismutated to H2O2 either spontaneously or under the catalysis of superoxide dismutases SOD1 (in the IMS) or SOD2 (in the matrix). In the matrix, H2O2 can be neutralized by glutathione (GSH). It can also signal to the cytosol. (C) In cancer cells, the TCA cycle not only serve to produce reducing equivalents to fuel the ETC (green arrows), but also to generate biosynthetic intermediates that are necessary for cell proliferation (pink arrows). The most important anaplerotic reaction produces oxaloacetate directly from pyruvate, and is catalyzed by pyruvate carboxylase (PC) (blue arrow). Oxaloacetate can further be converted to phosphoenolpyruvate (PEP) by PEP carboxykinase (PC), contributing to gluconeogenesis. (D) Mitochondrial DNA (mtDNA) variations, including single nucleotide polymorphisms (SNPs), maternally inherited haplotypes and deletions have been studied for their association with cancer. Among these, only large mtDNA deletions seem to be associated with malignancies. Cyt c - cytochrome c; Gpx - glutathione peroxidase; Q - coenzyme Q10. |
THE MITOCHONDRION: A COMPARTMENTALIZED ORGANELLE
The OMM: a platform for signaling
A main function of mitochondria is to ensure that the cell does not undertake processes for which mitochondria are not suitable, thus avoiding a discrepancy between cellular metabolic requirements and the mitochondrial capacity. There are different ways by which mitochondria can communicate with the cell. One of them is through the OMM, which can be compared to a signaling platform. The OMM connects mitochondria to other organelles within the cell, such as the endoplasmic reticulum (ER) and lysosomes, and to the plasma membrane [36][37]. Thanks to VDAC that forms pores composed of different subunits, the OMM is permeable to small ions and proteins [38]. VDAC carries nucleotides, ions and metabolites between the cytosol and the IMS [38], and acts as an intracellular signaling platform for the modulation of metabolism and the control of cell death [39][40]. With respect to metabolic regulation, VDAC also acts as platform for the anchoring of hexokinase 2 (HK2), the embryonic version of the first enzyme of glycolysis, to the OMM to facilitate the use of ATP by HK2 in cancer cells [41]. Because glucose phosphorylation by HK2 traps glucose-6-phosphate within cells, this binding implies that VDAC is involved in the regulation of both glycolysis and mitochondrial respiration. With respect to cell death, the binding of Bcl-2 family members (Bax, Bak, Bok, Bad, Bid or Bim) to VDAC leads to the formation of a pore that results in the release of cytochrome c[16][42], a small hemoprotein free to spread among the different mitochondrial compartments [43]. Cytochrome c, besides being a component of the ETC and, therefore, being necessary for the production of ATP at the ETC, also induces caspase-dependent cell death in response to pro-apoptotic stimuli [44].
–
Mitochondria also exert a control on the innate immune system. The OMM is indeed the site of phosphorylation of a wide range of proteins. For example, protein kinase A (PKA), a tetramer composed of two subunits that bind cAMP and two catalytic subunits, is a mitochondrial resident [45]. Mechanistically, a-kinase-anchoring proteins (AKAPs) are found in the OMM where they allow the binding of the PKA catalytic subunits to the organelle membrane, facilitating PKA localization for protein phosphorylation [32][46]. When cAMP binds to PKA, the two catalytic subunits are dissociated, becoming active and phosphorylating a wide range of target proteins that have the arginine-arginine-X-serine motif exposed, such as splicing factors SRSF1, SRSF2 and SRSF9 [47]. The OMM is also home to antiviral signaling regulators that activate the immune response thanks to MAVS, as recently reviewed in details by Mohanti A et al.[48].
–
OMM signaling is only one of the ways mitochondria communicate with the rest of the cell. Indeed, mitochondria play a vital role in other important signaling pathways. First, they house the production of acetyl-CoA and S-adenosylmethionine that both regulate epigenetics by controlling signal transduction of DNA and histones through acetylation and methylation, respectively [49][50]. Second, mtROS production and release is a response to cellular stress and a signaling factor to notably activate transcription factors hypoxia-inducible factors (HIFs), nuclear factor erythroid-derived 2-like 2 (Nrf2) and downstream gene expression [51]. Third, mitochondria control Ca2+ homeostasis. Ca2+ acts as a signaling molecule between mitochondria and the ER through contact sites termed ‘mitochondrial associated membranes’ (MAMs) [52]. It is one of the most important signals that these organelles use for communication [53]. Ca2+ acts as a bidirectional signaling molecule, as mitochondrial Ca2+ uptake regulates mitochondrial metabolism, while mitochondrial Ca2+ release modulates apoptosis. Indeed, an increased Ca2+ concentration inside mitochondria activates several TCA cycle enzymes [54] and stimulates the production of cAMP [55], which in turn increases ATP production, allowing metabolic adaptation. Conversely, Ca2+ is a signal for programmed cell death, with high levels of Ca2+ inducing the opening of mitochondrial permeability transition pores (MPTPs), triggering the release of cytochrome c and initiating apoptosis [56]. Ca2+ released from ER-mitochondria contact sites can also activate apoptosis through Bcl-2 family members upon a fine regulation of Ca2+ homeostasis [57]. Furthermore, changes of ATP production by mitochondria act as a signal that is transmitted to the cytosol in the form of AMP: AMP activates energy sensor AMP kinase (AMPK), thus decreasing anabolic cell functions in favor of the catabolic reactions used for ATP production [58].
–
Overall, mitochondrial signaling is a dynamic and complex process that affects most cellular functions. By understanding these processes, it may be possible to more effectively treat diseases like cancer.
–
The IMM: the ATP factory
Compared to the OMM, the IMM does not contain porins and is a highly impermeable barrier to ions and molecules that require specific membrane transport proteins for bidirectional exchanges [59]. The IMM has an electrochemical membrane potential of about 180 mV that regulates their passage. It is also more extensive than the OMM, as it is organized in invaginations called ‘mitochondrial cristae’ [60] to allow the arrangement of ETC complexes. The number of invaginations depends on the energy demand of the tissue. In muscles, for example, mitochondria are particularly rich in cristae [61].
–
The IMM further differs from the OMM by its high protein content, with a protein-to-lipid ratio of 80:20 in the IMM and of 50:50 in the OMM [59]. This high protein content is represented by all the complexes responsible for OXPHOS, as well as by transport proteins and proteins that regulate fusion and fission. The respiratory chain consists of a series of multi-enzymatic complexes and hydro- and liposoluble compounds capable of transferring electrons sequentially through the complexes towards the final acceptor, O2. This electron flow creates a motor force that transfers protons from the mitochondrial matrix (their site of production) to the IMS against their concentration gradient, thus generating an electrochemical gradient across the IMM. The various complexes are arranged in increasing order of reduction potentials, in such a way that transported electrons pass from a higher energy state to a lower energy state with consequent energy release, which is used, in part, for ATP synthesis. Another part of the energy produced is used for thermoregulation [62]
–
The respiratory chain is composed of five complexes (three of which are proton pumps) and two carriers that act as co-substrates and electron transporters (Figure 1A and 1B):
–
Complex I – NADH dehydrogenase, also called coenzyme Q (CoQ) reductase. Complex I is composed of 45 subunits. Among them, 14 have catalytic activities and are called ‘essential subunits’, of which seven are hydrophobic (ND1, ND2, ND3, ND4, ND4L, ND5 and ND6) and encoded by the mitochondrial genome, and the other seven are hydrophilic [63]. The remaining subunits are called ‘non-essential subunits’. They are important for the assembly and stability of Complex I and are encoded by the nuclear genome [64]. Complex I receives two hydrogen atoms from NADH, which is oxidized, releasing two electrons and reducing flavin mononucleotide (FMN) to FMNH2. The latter transfers the electrons to the second carrier of the ETC, CoQ, through its Fe/S centers. The energy obtained from the passage of electrons is used by Complex I to transport four protons from the mitochondrial matrix to the IMS, which represents a flow of two protons for each NADH consumed [65]. Complex I dysfunctions have been associated with cancer, as a reduced activity of the complex has been observed in renal oncocytomas [66][67] and thyroid adenomas [68].
–
CoQ. CoQ is a lipophilic ubiquinone carrier embedded in the IMM lipid bilayer. It is able to separate the protons (which are released in the mitochondrial matrix) from the electrons provided by FMNH2[69].
–
Complex II – SDH. Complex II is composed of four nuclear encoded subunits, with two hydrophilic catalytic subunits, SDHA/SDH1 and SDHB/SDH2, and two hydrophobic subunits, SDHC/SDH3 and SDHD/SDH4 [63]. It contains a heme b group and two CoQ-binding sites. It is also part of the TCA cycle. Complex II contributes to electron transfer, but there is no proton pumping towards the IMS. The two electrons produced during the oxidation of succinate to fumarate are directly transferred to CoQ [70]. The reduction activity of Complex II has been shown to be associated with human cancer in renal carcinoma [71] and breast cancer [72], where Complex II activity is lower compared to the corresponding normal tissues.
–
Complex III – Cytochrome bc1 complex, also called Cytochrome c reductase. Complex III is a symmetrical dimer, and each subunit is composed of three catalytic cores (MT-CYB, CYC1 and UQCRFS1) and seven supernumerary subunits [63]. This complex receives electrons from CoQ and passes them to cytochrome c; then, it carries four protons towards the IMS [73]. A higher than physiological activity of Complex III has been detected in breast cancer [72][74].
–
Cytochrome c. Cytochrome c is a hydrophilic heme protein located at the outer surface of the IMM. It transfers electrons between Complexes III and IV [75].
–
Complex IV – Cytochrome c oxidase. Complex IV is composed of 13 or 14 subunits, and it is the only OXPHOS complex containing tissue-specific and developmentally regulated isoforms [63]. This complex transfers four electrons (provided by four molecules of cytochrome c) directly to O2 (provided by the blood), reducing it to two molecules of H2O, which consumes four protons (taken from the mitochondrial matrix) [76]. A reduced activity of Complex IV has been observed in cancer. In renal carcinomas, Complex IV expression was found to be 5-fold lower compared with healthy kidney tissues [71].
–
Complex V – ATP synthase. Complex V is composed of two distinct domains. The F1 domain is extrinsic to the IMM and is found in the matrix, while the Fo domain is intrinsic to the IMM. F1 is composed of nine subunits, while Fo has two subunits [63]. ATP synthase is the complex where ATP is produced from the substrates ADP + H2PO4–+ H+ in a reaction at the end of the OXPHOS process. While electrons traveling across the ETC are finally transferred to O2 at Complex IV, most of the protons that were transferred from to mitochondrial matrix to the IMS return through the ATP synthase complex (a channel protein), nullifying the electro-chemical gradient. The energy released by the return of the protons according to their electrochemical potential is used in the form of mechanical energy to allow the functioning of ATP synthase [77].
–
Recent data have suggested that ETC complexes are organized in supermolecular structures called ‘supercomplexes’ [78]. These structures appear to be organized in different ways and to adopt stoichiometry [79]. For example, Complex I is frequently bound with a dimer of Complex III and with Complex IV [80]. This type of supercomplex is called ‘respirasome’ because it contains all the complexes responsible for the transfer of electrons from NADH down to O2. It has also been shown that Complex I is always associated with supercomplexes, while Complexes III and IV also exist as free oligomeric enzymes [80]. This suggests that the assembly of the complexes in a single structure is necessary for the stabilization and the correct functioning of Complex I in the respiratory chain [81]. Indeed, several studies have shown that loss of Complexes III and IV also causes the loss of Complex I [82][83].
–
ETC activity is associated with cancer in several ways. A growing amount of experimental evidence indicates that OXPHOS affects the production of ATP more significantly in cancer cells than in normal cells [84][85]. In particular, OXPHOS is used for a massive production of ATP by invasive and metastatic cells, as well as by circulating cancer cells [86]. Further evidence shows that cancer stem cells (CSCs) utilize OXPHOS as a preferred form of energy metabolism, and in general they display higher rates of oxygen consumption, ROS production and an overall increase in mitochondrial functions compared to non-stem cancer cells [87]. Moreover, mtROS promote V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (KRAS)-induced anchorage-independent growth [88].
–
As demonstrated by Porporato et al.[89], an ETC overload can increase the migration, invasion, clonogenicity and metastatic potential of cancer cells through a mitochondrial superoxide-dependent mechanism that activates the transforming growth factor β (TGFβ) pathway at the level of src kinase. Despite mitochondrial respiration can be perceived as an intriguing target for cancer treatment, it has been amply demonstrated that tumors are metabolically heterogeneous and influenced by different substrates and factors from the tumor environment [90]. Not only does metabolism differ among the various cancer types, but it also differs between subpopulations of cancer cells within the same tumor [91][92]. Cellular subpopulations with different metabolic phenotypes (elevated OXPHOS or dysfunctional glycolysis) have been identified in melanoma [93], lymphomas [94], pancreatic [95] and breast [96] tumors.
–
The mitochondrial matrix: the fulcrum of metabolism
The mitochondrial matrix is the inner mitochondrial space delimited by the IMM. It mostly hosts mtDNA, ribosomes, enzymes, small organic molecules, nucleotide cofactors and inorganic ions [97]. The mitochondrial matrix is a viscous space due to its high protein content with respect to the IMS, and it has a higher pH than the IMS (7.8 in the matrix; 7.0-7.4 in the space) [98]. It is the site of numerous enzymatic reactions, including those of the TCA cycle, anaplerotic and cataplerotic reactions, the urea cycle (at least in part), transamination reactions and part of ETC reactions (Complex II).
–
The TCA cycle
–
The TCA cycle (also known as Kreb’s cycle or citric acid cycle, Figure 1C) is the most important process that takes place in the mitochondrial matrix. It is composed of a series of chemical reactions capable of processing two-carbon units from carbohydrates, amino acids and fatty acids in acetyl-CoA to generate GTP and the reducing equivalents (NADH and FADH2) that fuel the mitochondrial ETC to generate ATP [97]. Acetyl-CoA is oxidized in a cyclic metabolic pathway to CO2, with a net production of one CoA-SH, two CO2, three NADH, one FADH2, one GTP/ATP and three H+ for each molecule of acetyl-CoA consumed.
–
The first reaction of the cycle is the condensation of acetyl-CoA with oxaloacetate by citrate synthase (CS) to form citrate. Citrate is then converted to its isomer, isocitrate, by mitochondrial aconitase (ACO2) and subsequently decarboxylated to α-ketoglutarate (α-KG) by mitochondrial IDH. In this reaction, a CO2 molecule is released and a NAD+ molecule is reduced to NADH + H+. A second decarboxylation occurs when α-KG is converted to succinyl-CoA by α-KG dehydrogenase, which also produces NADH + H+. The next reactions serves to regenerate oxaloacetate through (1) oxidation of succinyl-CoA in succinate by succinate-CoA synthetase, which produces GTP; (2) oxidation of succinate to fumarate by SDH with the production of a molecule of FADH2; (3) hydration of fumarate to malate by fumarate hydratase (FH); and, finally, (4) oxidation of malate by malate dehydrogenase to regenerate oxaloacetate, which produces NADH + H+[97].
–
The TCA cycle is a focal point of cellular metabolism with a central importance for both energy production and biosynthesis. Therefore, to retain the homeostasis of cellular metabolism, a balance between intermediate production and consumption must be maintained [99]. Anaplerotic reactions are a series of enzymatic reactions that produce metabolic intermediates aimed to replenish the TCA cycle, whereas cataplerotic reactions are biosynthetic reactions that use TCA cycle intermediates as substrates for macromolecule synthesis [99] (Figure 1C). The TCA cycle, in fact, not only produces NADH and FADH2 useful for ATP synthesis at the ETC, but it also provides molecules useful for other metabolic pathways, such as gluconeogenesis and the synthesis of fatty acids and nucleotides [35].
–
The most important anaplerotic reaction produces oxaloacetate directly in mitochondria starting from pyruvate, a reaction which is catalyzed by pyruvate carboxylase (PC) [100]. This reaction is allosterically regulated by acetyl-CoA and aspartate, which signal a deficiency in oxaloacetate [101]. Other anaplerotic reactions produce different intermediates of the TCA cycle, including α-KG produced from glutamate by glutamate-dehydrogenase, succinyl-CoA produced by FAO, and oxaloacetate produced from aspartate by aspartate transaminase [100].
–
When the production of intermediates of the TCA cycle is sufficient for its correct functioning, cataplerotic reactions intervene to balance metabolite concentrations [100]. Hence, citrate can be exported to the cytosol and converted to acetyl-CoA by ATP citrate lyase, initiating fatty acids biosynthesis. In a reversible reaction, α-KG with aspartate produce glutamate and oxaloacetate by the activity of aspartate transaminase, participating in purine synthesis [99]. Oxaloacetate can also be converted to phosphoenolpyruvate (PEP) by PEP carboxykinase (PEPCK), contributing to gluconeogenesis [99].
–
Alterations of the TCA cycle in cancer
–
Mutations in genes encoding TCA cycle enzymes and the abnormal accumulation of TCA cycle intermediates can promote carcinogenesis [102][103]. The main enzymes that were found to be altered in cancer are SDH, FH, IDH, CS and ACO2, which by itself highlights an extensive area open to investigation. Mitochondrial abnormalities induce metabolic reprogramming with cells increasingly relying on glycolysis, which further supports the tumorigenic process [104].
–
The first studies on the implication of SDH mutations in cancer showed that patients with hereditary paragangliomas and pheochromocytomas, two rare neuroendocrine neoplasms, displayed inactivating mutations of subunits of SDH enzymes [105][106]. As a consequence, succinate accumulates in these tumors.
–
Inactivating mutations of FH predispose to hereditary leiomyomatosis, renal cancer and multiple cutaneous and uterine leiomyomas [107][108][109]. Indeed, upon accumulation, fumarate acts as an oncometabolite that inhibits α-KG-dependent dioxygenases involved in DNA and histone demethylation [110][111]. It also promotes the epithelial to mesenchymal transition (EMT) by inhibiting ten eleven translocation (TET)-dependent DNA demethylation of a regulatory region of antimetastatic miR-200, leading to a decreased expression of miR-200 and E-cadherin, and an increased expression of Twist1 and vimentin, among other target genes [112]. Defects of FH further stimulate the nuclear translocation and activity of Nrf2, and the consequent transcription of antioxidant genes through antioxidant response elements (AREs). These genes comprise mitochondrial residents thioredoxin 2, thioredoxin reductase 2, peroxiredoxins and superoxide dismutase 2 (SOD2), as well as transporters and enzymes involved in glutathione (GSH) biosynthesis and redox recycling (see reference [113] for a recent review). Nrf2 activity is mitigated by Kelch-like ECH-associated protein 1 (KEAP1) that interacts with Nrf2 in the cytosol, targeting it for polyubiquitylation followed by proteasomal degradation [114], and by transcription modulators that heterodimerize with Nrf2 in the cell nucleus [115]. Conversely to its induction of antioxidant systems, fumarate can directly react with GSH to produce succinated GSH that mimics the GSH reductase substrate [116]. This causes NADPH consumption without antioxidant effects, thus increasing oxidative stress.
–
IDH presents three isoforms, with NADP+-dependent IDH1 found in the cytoplasm and in peroxisomes, and NADP+-dependent IDH2 and NAD+-dependent IDH3 found in the mitochondrial matrix. IDH1 and IDH2 have been reported to be mutated in 70% of grade II and III gliomas and glioblastomas [117][118], as well as in angio-immunoblastic T-cell lymphomas [119], acute myeloid leukemia (AML) [120][121] and other common cancer types, such as thyroid, colorectal and prostate cancers [122][123]. IDH1 and IDH2 mutations decrease NADPH and GSH levels, leading to enhanced PI3K-AKT-mTOR signaling pathway activity and cancer cell migration [124][125]. Several studies have shown that carcinogenesis and cancer progression are modulated by IDH mutations, and it has been reported that these mutations increase ROS levels in cancer cells [126][127]. Both increased ROS levels and PI3-AKT pathway activation lead to an increased signaling activity in favor of cancer formation and progression [128][129]. Interestingly, mutated IDHs most often gain the function of producing 2HG, the R enantiomer of α-KG [130]. 2HG is an oncometabolite that, when accumulated at a very high concentration, inhibits α-KG-dependent dioxygenases [131], such as histone lysine demethylases, leading to a hypermethylated state of DNA and histones [132]. It also activates HIF-1 by inhibiting HIF-1α degradation through prolylhydroxylases [133].
–
CS is an important enzyme often considered to be the rate-limiting enzyme of the TCA cycle [134]. Its loss causes a metabolic shift from an oxidative to a glycolytic metabolism and induces EMT, increasing tumor malignancy [135]. However, a high expression of CS has been observed in pancreatic, renal and ovarian cancers [136][137][138].
–
A decreased expression of ACO2 has been associated with gastric [139] and prostate [140] cancers. Interestingly, FH-deficient cells also have impaired aconitase activity because fumarate accumulating in these cells exerts a dose-dependent inhibition of ACO2 activity via succination of critical cysteine residues [141].
–
When inactivating mutations occur in TCA cycle enzymes, metabolic intermediates different than the direct substrates of mutated enzymes may accumulate, and they can act as oncometabolites [102][103]. For example, citrate is an oncometabolite that, when accumulating, disrupts the equilibrium of the TCA cycle and promotes cancer development [142]. Indeed, high levels of citrate reduce the activity of pyruvate dehydrogenase, with consequent pyruvate accumulation [142]. In this situation, cells convert pyruvate to lactate with NAD+ regeneration, thus causing a metabolic shift towards glycolysis, a well-known phenomenon in cancer progression [143]. Furthermore, citrate is a substrate of acetyl-CoA carboxylase, by which acetyl-CoA and malonyl-CoA are produced as precursors for lipid and steroid synthesis [144]. On the one hand, lipids are the major structural components of biological membranes and play important functions in cell signaling promoting cell proliferation and transformation [145][146]. On the other hand, acetyl-CoA derived from citrate is a mandatory substrate for histone acetylation, modulating chromatin structure, and, thus, gene transcription [147].
–
Histone acetylation consists of a dynamic and reversible acetyl group transfer process controlled by histone acetyltransferases (HATs) and histone deacetylases (HDACs) [147]. Acetylation of specific histones allows chromatin relaxation, which facilitates the transcription of important genes that regulate cell proliferation, cell cycle transition, differentiation and apoptosis, and have been linked to tumor development, influencing the cellular growth program of cancer cells [148]. The use of HDAC inhibitors has proven to be an effective therapy capable of reversing the transformed cellular phenotype, and several HDAC inhibitors have been approved by the FDA for cancer therapy [149][150]. Of note, in addition to histones, oncoproteins, tumor suppressors and enzymes can be acetylated as well, which controls their activities. It is the case of HIFs, Myc, KRAS, p53, retinoblastoma protein pRb and PTEN, and of enzymes involved in glucose, fatty acid and glutamine metabolism [147].
–
Alterations of mtDNA in cancer
–
Both mtDNA mutations and deletions have been associated with cancer (Figure 1D).
–
Single nucleotide polymorphisms in mtDNA (mtSNPs) were tested for their potential correlation with an increased risk of developing cancer or with a more aggressive progression. While on the one hand a study correlated a decrease in mtDNA copy number with colorectal cancer malignancy [151], on the other hand a similar study excluded any correlation between mtDNA copy number and mtSNPs on cancer progression in colorectal cancer patients [152]. In this study, six mtSNPs (MitoT479C, MitoT491C, MitoT10035C, MitoA13781G, 10398 A/G and 16189 T/C) were analyzed in 536 patients and mtDNA copy number in 274 patients, comparing tumor and healthy tissues. A potential correlation between mtSNPs and the risk of prostate cancer has also been studied. Following the comparison of 350 mtSNPs between 4,086 cancer patients and 3,698 healthy subjects in a multiethnic cohort, the authors reported no association between mtSNPs and the risk of prostate cancer [153].
–
Ethnicities are characterized by different haplogroups, which can be defined as clusters of mtDNA sequence variations that are statistically inherited from the ancestral maternal mitochondrial genome [154]. For example, Europeans present nine major haplogroups (H, U, J, T, K, W, I, V and X) [155]. Differences in the mtDNA sequence can result in alterations of mitochondrial proteins, mostly components of the ETC, that influence their activity, ETC efficiency and ROS production. In oncology, haplogroups have been extensively studied, in particular regarding their possible correlation with the risk of developing specific cancer types. Analyses suggested that some haplogroups are indeed correlated with an increased risk for specific cancer types, whereas they simultaneously reduce the risk of other types of cancer [156]. For example, haplogroup K would statistically be protective against thyroid cancer in Southeastern Europeans, but would increase the risk of breast cancer in European Americans [157]. Another interesting study in a cohort of over 7,700 European individuals found no correlation between haplogroups and breast cancer risk factors, neither in mothers nor in their children [158]. A major difficulty to link haplogroups to cancer risks is due to the way results are analyzed. As a typical example, one study reported that haplogroup N is correlated with an increased risk of breast cancer [159], whereas another similar study reported that haplogroup U and haplogroup K are correlated with a decreased and an increased risk of breast cancer, respectively [160]. However, according to phylotree.org, haplogroup N contains haplogroup U, which in turn includes haplogroup K, making it complex to define what is the actual weight of these alterations on the risk of developing cancer, and making of the comparison of different studies a difficult task. From a molecular point of view, it is still unclear how a same set of mtDNA variations could simultaneously be pro- and anticarcinogenic.
–
Large mtDNA deletions usually result in severe alterations of the ETC and, consequently, in OXPHOS defficiency. Three main studies have analyzed the frequencies of these events in cancers. In the first study, a 4,977 bp mtDNA deletion has been analyzed in more than 1,600 samples of tumor and adjacent healthy tissues [161]. Results suggested that this particular deletion could be a cancer biomarker, especially in breast cancer. The second and third studies focused on the frequency of a 3,400 bp mtDNA deletion in prostate cancer [162][163]. They reported that this deletion is highly frequent in prostate cancer and in the healthy tissue directly adjacent to the lesion, while it was absent in normal prostate epithelium. The 3,400 bp large mtDNA deletion was therefore proposed as a biomarker of prostate cancer.
–
Altogether, to the exception of large mtDNA deletions, the amount of contradictory studies regarding mtDNA variations and their correlations with cancers makes it impossible to conclude anything clinically relevant today. In addition to contradictions, the biological consequences of these alterations are poorly characterized at both molecular and functional levels. Consequently, targets cannot be easily identified and pharmacological approaches cannot be developed. The complete picture is even worse if one considers that the amount of studies with statistical or methodological fallacies, in particular regarding breast cancer, is so high that Salas A et al. wrote an entire article on that topic [164].
MITOCHONDRIAL REACTIVE OXYGEN SPECIES
Mitochondria as ROS producers
The term ‘ROS’ refers to a class of highly reactive molecules containing oxygen and having a very short average half-life. ROS collectively design superoxide (O2°-), the hydroxyl radical (OH) and hydrogen peroxide (H2O2). Mitochondria are considered to be the most important producers of superoxide in cells [34].
–
Mitochondrial superoxide, the proximal ROS, is mainly produced at the ETC from the leakage of electrons at the ubiquinone-binding sites of Complex I (IQ site) and Complex III (IIIQo site) during electron transport [9] (Figure 1B). Superoxide production primarily occurs through the donation of a single electron from totally reduced or partially reduced electron carriers, such as semiquinone, to O2[34]. The kinetics of O2°- production depend on the carrier enzyme average time in a reduced form and on its concentration [34], highlighting that the concentration of enzymes responsible for O2°- production tunes the functions that ROS play within cells. Another important variable is the O2 concentration in cells. Accordingly, altering experimentally the local O2 concentration can increase or decrease the rate of mitochondrial O2 consumption, leading to alterations of O2°- production [34]. Being highly reactive, O2°- is immediately dismutated to H2O2 either spontaneously or under the catalysis of mitochondrial SODs. SOD1 is present in the IMS and in the cytosol where it inactivates superoxide produced at ETC Complex III, whereas SOD2 is localized in the mitochondrial matrix where it inactivates superoxide produced at Complexes I and III [34] (Figure 1B).
–
In addition to ubiquinone-binding sites in ETC Complexes I and III, other ROS-producing sites have been identified in mitochondria: flavin in Complex I (IF site), the electron transferring flavoprotein Q oxidoreductase (ETFQOR) in FAO, glycerol 3-phosphate dehydrogenase, pyruvate dehydrogenase and 2-oxoglutarate dehydrogenase [165]. Although the mechanisms governing the functioning of each of those sites are not yet fully understood, studies have shown that all of them produce ROS in the mitochondrial matrix, while only the IIIQo site and glycerol 3-phosphate dehydrogenase can release O2°- in the IMS [165]. In these two cases, once produced in the IMS, H2O2 can cross the OMM and exercise signaling activities in the cytosol [166] (see Section ‘Antioxidant defenses in mitochondria‘) for more details).
–
Strong evidence exists supporting the signaling role of mtROS as a mode of communication between mitochondria and cellular processes involved in the maintenance of homeostasis and adaptation to stress conditions [167]. mtROS play different signalization roles in cells by promoting cell survival or by enhancing cell death, and for mitochondrial recycling. First, mitochondrial H2O2 regulates posttranscriptional protein modification through oxidation of the thiol groups of cysteines, thus changing the activity of target proteins and, therefore, the final response [168]. For example, the reaction of H2O2 with phosphatases, such as mitogen-activated protein kinase phosphatase (MKP), inhibits their dephosphorylative activity, leading, e.g., to c-Jun N-terminal kinase (JNK) activation [169]. Second, an increased release of mtROS is an adaptive response of cells to hypoxia that activates HIFs [170]. ROS indeed oxidize the iron moiety of HIF prolylhydoxylases (PHDs), thus inhibiting the activity of these enzymes that normally initiate the process of HIF-1α degradation by catalyzing the hydroxylation of this HIF subunit [171]. ROS further oxidize vitamin C, which is a necessary co-factor for PHD recycling though Fe3+ reduction. Notably, HIF-1 is known to upregulate the expression of glycolytic enzymes and transporters to maintain sufficient ATP levels in cells [172] and to enhance the production of vascular endothelial growth factor (VEGF) for the simulation of angiogenesis [173]. Third, mtROS act as a signal for triggering autophagy, which exerts either prosurvival or pro-apoptotic effects [168]. During starvation, mtROS oxidize and inactivate cysteine protease Atg4, which triggers the lipidation of Atg8, an essential step in the process of autophagy, thus promoting autophagy and facilitating the recycling of intracellular molecules [174]. Interestingly, autophagy can further contribute to ROS accumulation due to selective catalase degradation [175]. Fourth, mtROS generate signaling responses at nuclear gene expression level by forming a perinuclear clustering of mitochondria, leading to an accumulation of ROS in the nucleus with subsequent alterations of gene transcription [176].
–
Antioxidant defenses in mitochondria
In order to maintain the balance between ROS production and their harmful consequences, cells harbor specific defense mechanisms. They protect cells from detrimental ROS effects, i.e., damage to lipids, proteins and nucleic acids, and induction of double strand DNA breaks [177]. Superoxide produced in mitochondria is reduced to H2O2 by SOD1 (also known as cumin-zinc superoxide dismutase, CuZnSOD) in the IMS and in the cytosol, and by SOD2 (also known as manganese superoxide dismutase, MnSOD) in the mitochondrial matrix [34] (Figure 1B). Of note, SOD3 is also a member of the CuZnSOD family, but is extracellular [178]. After the conversion of superoxide to H2O2 by SODs, the subsequent conversion of two H2O2 to O2+ two H2O is catalyzed by peroxidases: catalase, located in peroxisomes, the thioredoxin system, and GSH peroxidase. The thioredoxin system coupled to the peroxiredoxins catalyzes the reduction of H2O2 to H2O in the presence of NADPH, and consists of thioredoxin peroxidases, thioredoxin reductases and the substrates thioredoxin an peroxiredoxin [179] (Figure 2). Mitochondria have their own specific thioredoxin reductase (ThxR2) for redox regulation and peroxiredoxin for the reduction of peroxides [180]. Finally, in the cytosol, GSH peroxidase catalyzes the conversion of H2O2 to H2O using GSH as a substrate, which is reduced to GSSG and then regenerated with the use of NADPH by GSH reductase [180]. Thus, GSH synthesis (via glutaminolysis and the serine pathway) and NADPH production actively support H2O2 inactivation [181].
–
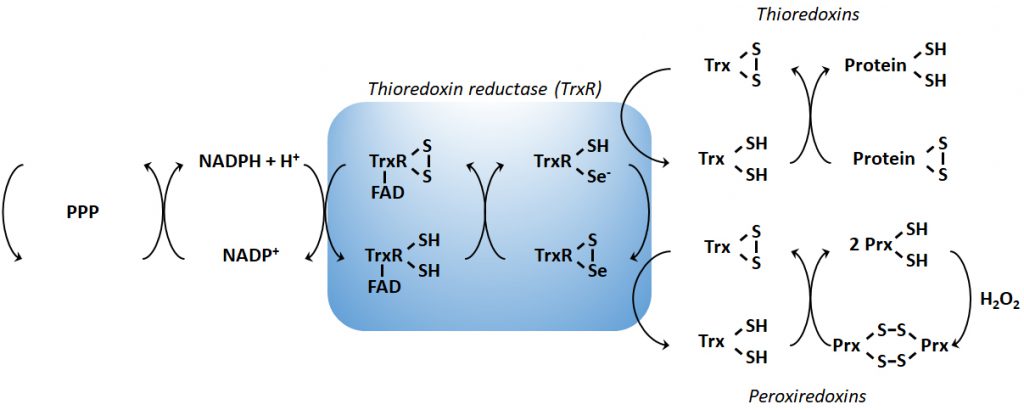 |
FIGURE 2: The arsenal of mitochondrial antioxidant defenses comprises the thioredoxin and peroxiredoxin pathways. The image depicts redox reactions catalyzed by the thioredoxin and peroxiredoxin systems, comprising thioredoxin reductases (TrxR, of which ThxR2 is expressed in mitochondria), thioredoxins (Trx), peroxiredoxins (Prx) and NADPH. The electron source is NADPH, which mostly originates from the pentose phosphate pathway (PPP). Oxidized thioredoxins (Trx-S2) are reduced by NADPH and selenoenzymes TrxRs. Electrons are sequentially transferred from NADPH to FAD, to the N-terminal redox active disulfide in one subunit of TrxR, and, finally, to the C-terminal active site of another subunit. Reduced thioredoxins (Trx-SH2) catalyze disulfide bond reduction in many proteins, including Prxs, thus ensuring oxidative damage repair in proteins as well as H2O2 detoxification. |
METABOLIC AND MITOCHONDRIAL CONTROL OF CELL DEATH
There is a clear connection between metabolism and cell death mediated by various signal transduction pathways. In cancer cells, p53 coordinates a common central pathway [182]. p53 is the most important pro-apoptotic protein and is mutated/inactivated in ∼50% of tumors [183]. However, while p53 has a central role in tumor suppression, it is also involved in the modulation of cancer metabolism. Two well-known targets of p53 that regulate cell metabolism are TP53-induced glycolysis and apoptosis regulator (TIGAR) [184] and synthesis of cytochrome oxidase 2 (SCO2), a cytochrome oxidase 2 (COX2) assembly protein [185]. While TIGAR decreases the glycolytic flux by dephosphorylating fructose-2,6-bisphosphate [184], SCO2 promotes ETC assembly and OXPHOS [186]. p53 is thus able to repress glycolysis and to promote OXPHOS and FAO [182].
–
Nutrient availability is commonly altered during tumor growth [187]. Abnormal tumor perfusion and, consequently, nutrient restriction, impact cell death. For instance, a common microenvironmental alteration in tumors is hypoxia, which can act as a signal for p53 activation and cell death induction [188]. In addition, the metabolic status of the cell acts as a signal for p53 induction: when cellular ATP levels decline, the resulting decrease in the ATP/AMP ratio activates AMPK, and AMPK phosphorylates/activates p53 on serine 15, thus initiating an AMPK-dependent cell-cycle arrest [189][190].
–
AMPK acts in most cases as a tumor suppressor, not only by inducing a cell cycle arrest, but also by inhibiting the synthesis of most cellular macromolecules. AMPK activation indeed inhibits mammalian target of rapamycin complex 1 (mTORC1) by phosphorylating its upstream regulator tuberous sclerosis complex 2 (TSC2), thus inhibiting cell growth [191]. ATP levels and AMPK thereby provide an important connection between p53-mediated regulation of energy metabolism and programmed cell death [191]. Of note, adenosine signaling can further induce apoptosis by stimulating adenosine receptor A2B, which activates a caspase- and p53-upregulated modulator of apoptosis (PUMA)-dependent apoptotic response involving a downregulation of Bcl-2 expression [182].
–
Mitochondria are another important crossroad between metabolism and cell death. They control cell death through apoptosis (most notably by regulating the release of cytochrome c through VDAC and the mitochondrial transition pore), and some forms of necrosis [14][192]. Mitochondria control the intrinsic apoptotic pathway through OMM permeability, which is tightly regulated by Bcl-2 proteins [193][194]. A release of mitochondrial Ca2+ is critically involved in the initiation and effectuation of apoptotic cell death. In this context, AKT activation confers resistance to apoptosis by stimulating Bcl-2 protein expression [195], and the binding of HK2 to VDAC on the mitochondrial surface further represses apoptosis [196]. In addition to circumstances where mitochondrial integrity is altered, cell death is directly related to the metabolic activity of mitochondria. For example, inhibiting mitochondrial OXPHOS in renal cell carcinoma cells resistant to glucose starvation was reported to induce cell death under glucose deprivation [197].
–
Different from classical types of cell death, autophagy is considered to exert a dual function in cancer, as it is both a tumor suppressor and a protector of cancer cell survival [198]. Autophagy is indirectly modulated by metabolic enzymes. For example, lactate dehydrogenase-1 (LDH-1), catalyzing the conversion of lactate and NAD+ to pyruvate, NADH and H+, recently emerged as a modulator of autophagy [199]. LDH-1 indeed interacts with proton pump vacuolar (V)-ATPase at the surface of lysosomes, which it fuels with protons. Another example concerns glycolytic enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH) [200]. During glucose deprivation, GAPDH is phosphorylated by AMPK and is translocated to the cell nucleus where it interacts with the NAD+-dependent deacetylase sirtuin 1 (Sirt1) [201]. Both AMPK-dependent phosphorylation and the nuclear translocation of GAPDH mediate rapid Sirt1 activation in the nucleus, leading to the transcriptional induction of the autophagic program [201].
–
Other sirtuins also exert their activities at the interface between cell metabolism and death. Sirt2 has dual effects on mitophagy. In normal metabolic conditions, it can translocate from the cytosol to mitochondria, where it forms a complex with heat shock protein 70 (Hsp70), preventing Hsp70 acetylation and, thereby, inhibiting mitophagy [202]. However, Sirt2 also exists as a heterodimer with forkhead O family protein 1 (FoxO1) in the cytoplasm of cancer cells: upon serum starvation or oxidative stress, the complex is disrupted, resulting in FoxO1 acetylation and FoxO1 activation of autophagy-related 7 (Atg7) through protein-protein interaction, which triggers autophagic cell death [203]. Comparatively, Sirt3 is a mitochondrial resident. Under hypoxia, it promotes mitophagy and prevents mtROS-induced apoptosis by facilitating the binding of Parkin to VDAC1 [204]. The exact mechanism underlying this effect is not well characterized. Sirt4-7 could also regulate mitophagy, but molecular details are lacking. Sirt4 would promote mitochondrial fusion, thereby limiting mitophagy [205], while Sirt5 would indirectly prevent mitophagy by limiting ammonia production [206]. The expression of both Sirt6 and Sirt7 was shown to be important to preserve functional autophagy in cancer cells [207][208].
MITOCHONDRIAL DYNAMICS
Mitochondria are dynamic organelles that move through the cell, divide, fuse and undergo a regulated turnover through mitophagy [209][210] (Figure 3). They can also be exchanged between cells. These mitochondrial dynamics reflect metabolic alterations.
–
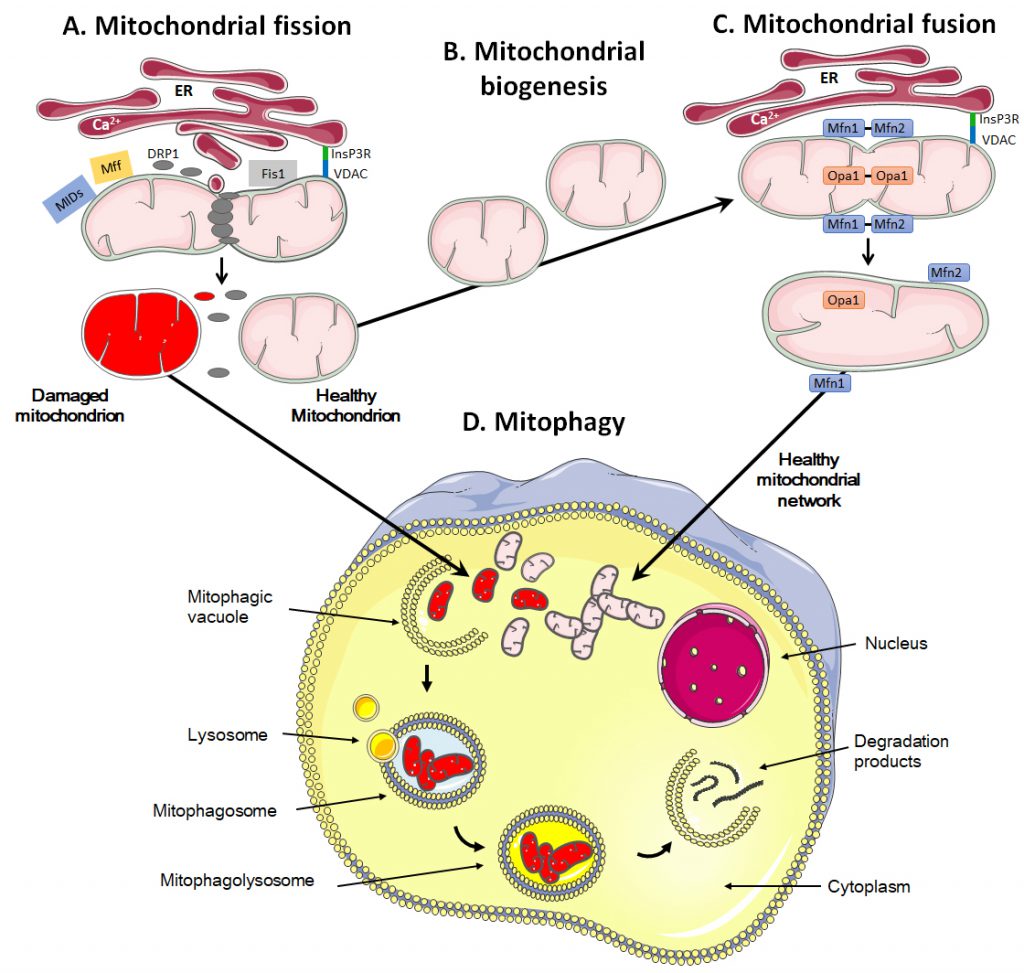 |
FIGURE 3: A high mitochondrial turnover rate is characteristic of many cancer cells. Mitochondrial quality control involves fission and mitophagy to eliminate defective mitochondria, whereas repopulation and functionalization depends on mitochondrial biogenesis and fusion. (A) During fission, the mitochondrion is marked and anchored by the endoplasmic reticulum (ER), notably through the binding of inositol 1,4,5-trisphosphate receptor (InsP3R) at the ER surface to voltage-dependent anion channel (VDAC) at the mitochondrial surface. This leads to the recruitment of dynamin-related protein 1 (DRP1), mitochondrial receptor protein 1 (Fis1), mitochondrial fission factor (Mff) and mitochondrial dynamic proteins (MIDs), allowing oligomerization and constriction to yield two daughter mitochondria. (B) During mitochondrial biogenesis, a mitochondrion self-replicates. (C) During fusion, mitofusins Mfn1 and Mfn2 are located on the outer mitochondrial membrane, allowing the exchange of calcium for signaling and creating antiparallel connections between two fusing mitochondria. Optic atrophy 1 (Opa1) together with Mnf1 participate in the fusion of the inner mitochondrial membrane. Fusion allows the formation of mitochondrial networks. (D) The mitophagic process consists in the engulfment of damaged mitochondria in a vacuole, called ‘mitophagic vacuole'. The subsequent fusion of the mitophagosome with lysosomes, forming a mitophagolysosome, allows the degradation of mitochondria in macromolecules that are delivered to the cytosol. Mitophagy can be non-selective or selective, using canonical and non-canonical pathways. It prevents the accumulation of damaged mitochondria that could harm or even kill the cell if apoptosis and/or the production of reactive oxygen species would derail. |
–
Mitochondrial fission and fusion
Mitochondrial fission is the process during which mitochondria divide into two or more independent structures, allowing the creation of new mitochondria. Conversely, mitochondrial fusion characterizes the merging of several neighboring mitochondria, which mixes the content of partially damaged mitochondria as a form of complementation [211]. Both processes require highly regulated mechanisms to allow the formation of new, functional organelles and to avoid a loss of intramitochondrial content.
–
During fission, mitochondria are divided into smaller pieces, an essential process to increase their number in dividing cells. Fission also promotes mitochondrial removal through mitophagy, facilitates the movement of mitochondria through the cytoskeletal network and regulates apoptosis and Ca2+ homeostasis [211][212][213]. The process is initiated by actin and the ER that first mark the site of division on the OMM [214][215] (Figure 3A). Once the contact between ER and mitochondria is established through VDAC at the mitochondrial surface and the inositol 1,4,5-trisphosphate receptor InsP3R at the ER surface, the ER releases Ca2+ into the mitochondrion to trigger actin polymerization at the constriction site [216]. This provides a site for the recruitment and assembly of dynamin-related protein 1 (DRP1), a cytosolic GTPase that translocates to the OMM upon activation [217]. DRP1 then recruits membrane-anchored receptor protein Fis1 [218], tail-anchored mitochondrial fission factor (Mff) and anchored mitochondrial dynamic proteins (MiDs) [219]. Together, these proteins spirally surround the mitochondrion, constricting and breaking it in two pieces.
–
High DRP1 expression has been associated with different types of cancers, including glioblastomas, thyroid, lung and breast tumors [220][221][222][223], as well as with an increased metastatic potential of cancer cells [224][225]. Interestingly, silencing DRP1 has been shown to reduce the metastatic capacity of breast cancer cells due to inhibition of lamellipodia formation, an important mechanism that drives cell migration [224]. DRP1 was also found to be overexpressed in squamous cell carcinomas (SCCs), and loss of DRP1 in this tumor type causes mitochondrial elongation with subsequent inhibition of cell proliferation and a G2 arrest [226]. Mechanistically, DRP1 expression was found to positively correlate with the expression of cell cycle genes that regulate mitosis in epithelial ovarian carcinoma (EOC) [227]. Elevated DRP1 expression promotes mitosis, thus supporting cell proliferation in the development of primary and relapsed EOC. DRP1 is also linked to cancer cell metabolism, since depletion of DRP1 in HeLa cells has been shown to decrease the activity of ETC complexes, mitochondrial respiration, mitochondrial membrane conductance and ATP synthesis [228][229]. An intriguing link between mitochondrial fission and cancer comes from the functional connection between DRP1 and cellular stress, where DRP1 has been proposed as a transcriptional target of p53 [230] and where oncogenic RAS/MAPK signaling upregulates DRP1 mRNA levels [223]. The multiple ways by which DRP1 is involved in cancer suggest that this protein plays important roles that could be independent of its principal role of ‘separating’ mitochondria.
–
Numerous studies have addressed the metabolism of CSCs (see references [231][232] for reviews). While their metabolic activities vary across tumor types, mitochondrial fission has been singled out to be important for stemness maintenance [233]. Accordingly, silencing DRP1 or its pharmacological inhibition in brain tumor-initiating cells reduced their tumorigenicity and triggered apoptosis [220]. Similarly, the pharmacological inhibition of DRP1 by mdivi-1 not only caused a defect in tumor sphere formation by breast CSCs, but also inhibited their migration and survival [234]. In another recent example, Civenni et al.[235] observed that depleting Mff in prostate CSCs rapidly exhausted their tumorigenic potential, which was associated with the induction of CSC senescence. Interestingly, the high rate of mitochondrial fission in CSCs has been proposed to allow mother stem cells to keep an intact mitochondrial content, whereas daughter cells committed to differentiate would inherit a pool of intact and deficient mitochondria [236]. This asymmetric segregation would participate in the perpetuation of cancer stemness despite frequent cell divisions [233].
–
During fusion, for example following mitochondrial replication (also known as mitochondrial biogenesis, Figure 3B), the OMM and the IMM merge, and the contents of the IMS and mitochondrial matrix are mixed (Figure 3C). Molecularly, fusion is controlled by three dynamin family GTPases, mitofusin (Mfn) 1 and 2 in the OMM and optic atrophy protein 1 (Opa1) in the IMM [237][238]. Opa1 displays two long isoforms (L-Opa1) and three short isoforms (S-Opa1) [239]. The fusion of the OMMs is for the most part synchronized with the fusion of the IMMs. Fusion proteins are regulated through ubiquitination of OMM proteins and proteolytic cleavage of IMM proteins [240]. When two mitochondria are in close proximity, Mfn1 and Mfn2 start to dimerize with homo- and heterotypic interactions, which creates antiparallel connections between mitochondria, allowing the fusion of the OMM through mixing the lipid bilayers [241]. Further interaction of the two fusing mitochondria with the ER is believed to facilitate the process, as Mfn2 has also been found in the ER where it promotes the interaction between mitochondria and ER and allows the exchange of Ca2+ for signaling [242]. Fusion of IMMs is then ensured by Opa1 together with Mnf1 [243].
–
In cancer cells, mitochondrial fusion appears to have the opposite effect than fission in terms of tumor growth, metastatic capacity and metabolic activities. Indeed, lower levels of Mfn1/2 have been found in mouse medulloblastoma cancer cells compared to non-transformed cells [244], and inhibition of DRP1 by mitochondrial division inhibitor-1 (Mdivi-1) stimulated fusion and initiated mtDNA replication [245]. Moreover, Mfn2 overexpression reduced lung cancer growth [222], as well as the migration and invasion of breast cancer cell lines [224]. Hyperfused mitochondria are found during the G1/S phase of the cell cycle that is associated with a greater oxidative capacity and higher ATP production [245]. Interestingly, Mfn2 gene therapy, which involves the insertion of genetic material (DNA) into the cells to restore Mfn2 gene expression, has been reported to reduce the proliferation of A549 human lung cancer cells by promoting apoptosis [222].
–
Fusion and fission also allow a subcellular specialization of mitochondria [246]. Mitochondria indeed distribute subcellularly depending on where their metabolic function is required (local demand of ATP, Ca2+ buffering and other functions). For example, at the axonal level in neurons, mitochondria appear fragmented because they move along the cytoskeleton (interacting with desmin, vimentin and tubulin), while at the dendritic level in the region of synapses, they appear elongated because in this region the demand/consumption of ATP is higher for pumping back the ions derived from synaptic vesicles [247]. Fission facilitates the distribution of mitochondria within the cell, and apoptosis through the release of cytochrome c[248]. Conversely, fusion benefits the cell thanks to mitochondrial complementation, which allows damaged mitochondria or mitochondria with altered mtDNA to fuse with healthy mitochondria in order to compensate the deleterious effects of dysfunctional organelles [249].
–
Mitophagy and mitochondrial transfer
The quality control of mitochondrial dynamics also includes mitophagy, i.e., a degradation process that removes dysfunctional or damaged mitochondria (Figure 3D) [250]. This process is vital to guarantee the physiological functions of cells and tissues and to avoid the onset of diseases like cancer [251]. It also regulates the number of mitochondria in response to the metabolic needs of cells and during some stages of cell development, such as the differentiation of erythrocytes [252].
–
Mitochondrial removal most often uses the molecular machinery of macro-autophagy, a specific type of autophagy characterized by the formation of mitophagosomes, i.e., double-membrane structures that form vesicles around mitochondria [253][254]. It can occur either in a selective way or in a non-selective way where autophagosomes sequester mitochondria together with cytosolic components and other organelles [255].
–
Selective mitophagy starts with the evaluation of healthy and damaged mitochondria by PTEN-induced kinase 1 (PINK1) (Figure 4). PINK1 is recruited by mitochondria as it contains a mitochondria-targeting sequence [252]. If the mitochondrion is healthy (polarized), PINK1 is transported from the cytosol towards the mitochondrial matrix by translocases of the OMM (TOM) and of the IMM (TIM) (Figure 4A). When crossing the IMM membrane, it is cleaved and released back to the cytosol by mitochondrial protease presenilin-associated rhomboid-like protein (PARL) and matrix processing peptidase (MPP) [252][256]. If, on the contrary, the mitochondrion is dysfunctional, the IMM becomes depolarized, PINK 1 is not cleaved and cannot be transported to the IMS. It accumulates in the OMM (Figure 4B). There, it can recruit and activate Parkin by phosphorylating OMM-resident ubiquitins [257][258] and Parkin itself [259]. Parkin is a cytosolic E3 ubiquitin ligase [260] that, once activated, starts to ubiquitinate proteins in the OMM, including Mfn1, Mfn2, VDAC1, TOM20 and mitochondrial Rho GTPase 1, initializing mitophagy [261][262][263][264]. In the cascade, autophagy receptors (such as optineurin) and autophagy initiators (such as unc-51 like autophagy activating kinase 1 [ULK1]) are recruited and activated, as recently detailed in reference [255]. Protein ubiquitination further activates microtubule-associated protein light chain 3 (LC3) [265], which is normally present in the cell cytosol under the form of LC3-I. When activated, LC3 is conjugated to phosphatidylenolamine to form LC3-II [265], which is recruited to autophagosomes during the formation of their double membrane. Besides this canonical pathway, alternatives routes have also been described involving a direct interaction between an activated form of OMM protein FUN14 domain containing 1 (FUNDC1) and LC3, or BH3 only domain proteins (BNIP3 and NIX), beclins and ULK1 [255]. Interestingly, a study by Soubannier et al.[266] further showed that mitochondria-derived vesicles can be formed in early oxidative stress response, and these vesicles are directed to lysosomes independently of LC3 as a form of mitophagy complementation.
–
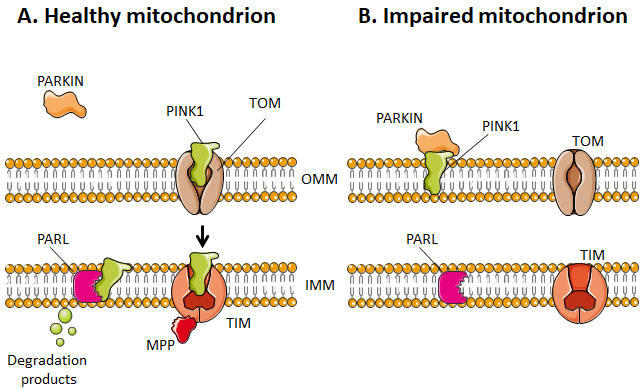 |
FIGURE 4: Dysfunctional mitochondria are targeted to mitophagy. (A) Healthy mitochondria have a polarized outer mitochondrial membrane (OMM), which allows PTEN-induced kinase 1 (PINK1) to cross the membrane and be degraded by presenilin-associated rhomboid-like protein (PARL) at the inner mitochondrial membrane (IMM). (B) Impaired mitochondria, instead, have a depolarized OMM, which hinders the entry of PINK1 and, therefore, its degradation. PINK1 can thus bind to parkin to initiate mitophagy. MPP- matrix-processing peptidase; TIM - translocase of inner mitochondrial membrane; TOM - translocase of outer mitochondrial membrane. |
–
Dysfunctional mitophagy is associated with tumor initiation and progression in many types of cancers [267][268]. In response to stresses such as hypoxia and nutrient starvation, mitophagy is activated to reduce the overall mitochondrial mass, thus preserving valuable nutrients and preventing excessive mtROS generation [269][270]. Even if the exact molecular mechanism is not yet well understood, part of the response could be mediated by receptors that dispose of C-terminal transmembrane domains localized at the OMM, including BNIP3, NIX and FUNDC1 [255]. Parkin has been suspected to be a tumor suppressor: its expression increases oxidative metabolism, limits the Warburg Effect and regulates levels of cyclin D1, cyclin E and cyclin-dependent kinase 4 (CDK4) in cancers [267]. Interestingly, mitophagy appears to have a dual role in cancer treatment resistance, as both its inhibition (in colorectal CSCs) and induction (in preclinical AML models) increased the sensitivity to chemotherapeutic drugs, such as doxorubicin [271]. Mitophagy could also participate in the radioresistance of head and neck cancer cells to γ rays [272].
–
While mitophagy coupled to mitochondrial biogenesis is a major pathway to maintain a pool of functional mitochondria in cancer cells [273], cancer cells can also acquire intact mitochondria from nonmalignant cells present in the tumor microenvironment. This mitochondrial transfer involves the formation of intercellular tunneling nanotubes (TNTs) and larger microtubes that have been observed in several types of cancer cells [274] based on the initial finding that mitochondria-deficient ρ0 cells could acquire mtDNA from host cells [275]. mtDNA transfer was later found to involve full mitochondria transfer between nonmalignant donor cells and malignant receiver cells [276]. In AML, ROS have been identified to drive the formation of TNTs that support full mitochondria exchange between bone marrow stromal cells and AML blasts, which was further shown to contribute to the metabolic capacity of cancer cells [277]. While the molecular mechanisms of TNT formation have been extensively reviewed elsewhere [278], it is in our opinion important to further mention that mitochondrial transfer can also transfer malignant capabilities between cancer cells. In one example, the experimental transfer of mitochondria from an invasive (T24 cells) to a less invasive (RT4 cells) bladder cancer cell line resulted increased invasiveness of RT4 cells [279]. In another example, mtDNA transfer between poorly and highly metastatic lung cancer cells was found to simultaneously transfer the high metastatic traits [280]. In yet another example, Pasquier et al.[281] showed that MCF7 breast cancer cells that received mitochondria from endothelial cells became more resistant to doxorubicin chemotherapy. Together, even if TNT formation is extremely difficult to document in vivo, these observations support a potential clinical significance of intercellular mitochondrial transfer.
–
Mitochondrial dynamics reflect metabolic alterations
Due to frequent fission and fusion events, different forms of mitochondria can be found inside a cell. They can morphologically resemble to small vesicles, short bars or reticular nets, which are a snapshot of shapes resulting from a constantly changing balance. Their organization further depends on cellular types and specific stress conditions [211].
–
Mitochondrial dynamics have been closely related to the activities that they perform, and changes in mitochondrial morphology have been linked to alterations that occur in cancer [282]. Many studies have demonstrated the existence of a link between energy substrates, oxygen supply and the mitochondrial architecture [283][284]. Cellular metabolic dysfunctions have been associated with increased mitochondrial fragmentation, whereas hyperfused mitochondria better resist to metabolic insults and, given to the merge, can protect cellular integrity [285]. In β cells of the pancreas, Molina et al.[286] have shown that mitochondria that are in a nutrient-rich environment are separated and subject to fission, whereas mitochondria in a starved environment characterized by a severe deficiency of nutrient availability below cellular needs tend to create nets and remain elongated for a long duration. Moreover, starvation induces the accumulation of fatty acid droplets inside mitochondria, shifting cellular metabolism towards FAO for ATP production [287]. Mechanistically, nutrient starvation induces mitochondrial elongation through cAMP-activated PKA that inhibits mitochondrial fission and protects the organelles against autophagosomal degradation [288][289]. In cancer, existing data suggest that a hyperfused state of mitochondria supports the survival of cancer cells not only by maintaining the production of ATP, but also by compensating for damaged mitochondria, sustaining intramitochondrial exchanges of fatty acids and avoiding metabolic reprogramming towards autophagy [287][288][289].
THERAPEUTIC STRATEGIES TARGETING MITOCHONDRIA IN CANCER CELLS
Given the key functions that mitochondria exert in cancer cells, several strategies have been imagined and tested that considered mitochondria as anticancer targets (Table 1). We here offer a brief overview of major approaches aiming to modulate mitochondrial anaplerosis, mitochon-drial turnover, the TCA cycle, the ETC, mtROS and mitochondria-driven apoptosis.
–
TABLE 1. Therapeutic strategies targeting mitochondria in cancer with clinical perspectives. |
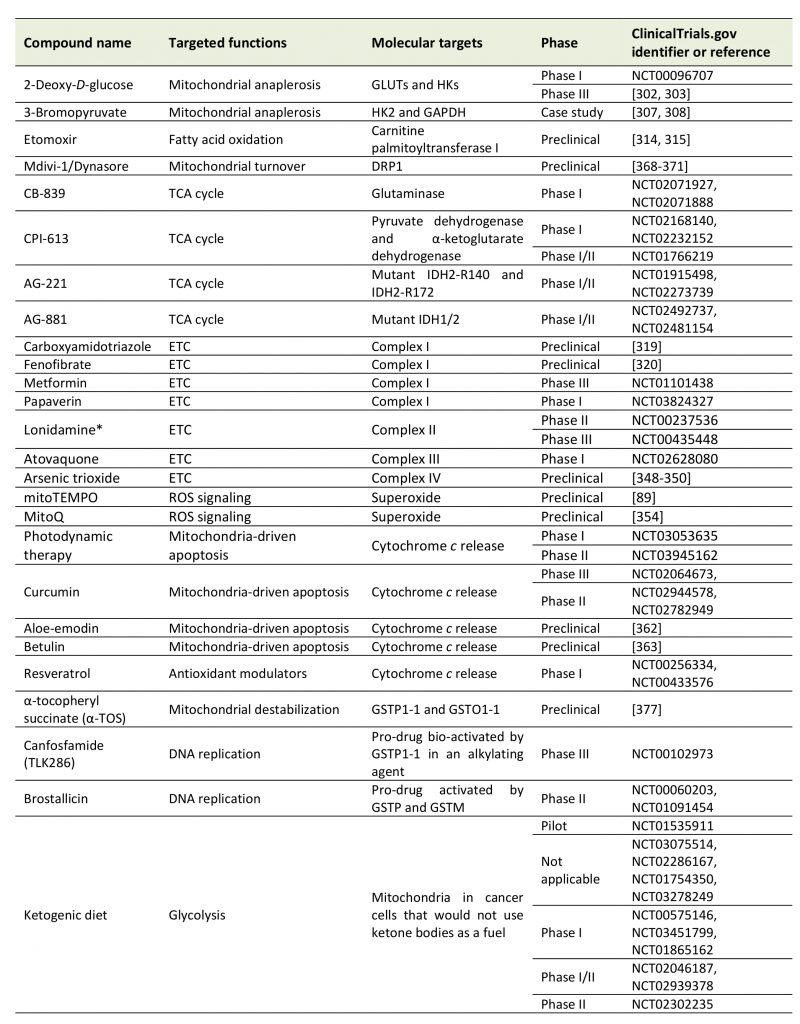 |
* withdrawn from clinical studies. DRP1 - dynamin-related protein 1; ETC - electron transport chain; GLUT - glucose transporter; GST- glutathione S-transferase; HK - hexokinase; IDH - isocitrate dehydrogenase; ROS - reactive oxygen species; TCA - tricarboxylic acid (cycle). |
–
Targeting mitochondrial anaplerosis
Targeting pathways supporting mitochondrial anaplerosis, i.e., glycolysis, glutaminolysis and FAO, has been considered as an anticancer approach. While these anaplerotic pathways may in part support cancer cell survival and proliferation independently of mitochondrial metabolism, their inhibition results in TCA cycle fuel deprivation.
–
2-Deoxy-D-glucose (2DG) is a competitor of glucose for glucose transporters (GLUTs) and HKs, which are often overexpressed in cancer cells, offering some anticancer selectivity for the treatment [290]. By interfering with glucose uptake and phosphorylation, 2DG reduces the availability of pyruvate for mitochondria, thus impairing mitochondrial anaplerosis and OXPHOS. In addition to its anti-metabolic activity, preclinical studies reported that 2DG exerts additional anticancer effects that comprise an anti-angiogenic activity [291], inhibition of cancer metastasis [292] and inhibition of the viral replication of Kaposi’s sarcoma-associated herpes virus [293]. It can also improve the effects of autophagy inhibition [294][295][296]. However, clinical trials were generally inconclusive, as 2DG as a standalone treatment did not show significant anticancer activity at tolerated doses for patients [297]. Additional in vitro and in vivo preclinical studies nevertheless suggested its possible application in combination with conventional chemotherapy, including cisplatin [298][299] and doxorubicin [300][301]. Promising results have also been obtained in combination with radiotherapy for the treatment of glioblastoma, and a Phase III clinical is currently ongoing [302][303].
–
3-Bromopyruvate (3BP) is an alkylating agent that, among other effects, has been reported to inhibit glycolysis. 3BP indeed inhibits HK2 [304] and GAPDH [305] among a larger list of targets [306]. However, even if promising case studies have been published supporting that 3BP exerts anticancer effects in humans [307][308], to our knowledge no clinical trial has been completed to date. Although a promising compound, 3BP faces several limitations for its clinical utility, including a rapid deactivation by GSH and a burning sensation when administered intravenously. In order to limit side effects and potentiate its anticancer properties, 3BP can be formulated in liposomes, PEG-liposomes or other targeted or untargeted nanocarriers [309].
–
Glutaminolysis has been shown to support cancer cells growth, in particular in human pancreatic ductal adenocarcinoma (PDAC) [310][311] and triple negative breast cancer [312]. Glutaminase 1 (GLS1) has been proposed as a target for anticancer treatment, however there are no pharmacological treatments available today.
–
Targeting FAO is another potential anticancer strategy. A special focus has been set on carnitine palmitoyltransferase I (CPT1), an enzyme located at the OMM that converts acyl-CoA (the end product of FAO) to acyl-carnitine that crosses mitochondrial membranes in order to fuel the TCA cycle in the mitochondrial matrix [313]. The irreversible CPT1 inhibitor etomoxir has been shown to improve the effectiveness of radiotherapy [314], and it may be used to treat cachexia, a syndrome associated to elevated FAO in cancer patients [315]. Etomoxir has been tested in clinical trials for type 2 diabetes and congestive heart failure, which revealed a safe to use profile of the drug [316][317]. However, to our knowledge, no clinical trial in cancer patients has been initiated today.
–
Targeting the TCA cycle
Whether and how to selectively target the TCA cycle in cancer cells has been extensively explored in the past years and recently reviewed in details by Anderson et al.[35]. Importantly, several Phase I and II clinical trials have been conducted involving drugs capable of inhibiting deregulated pathways related to the TCA cycle. They include CB-839, a specific inhibitor of glutaminase (NCT02071927 and NCT02071888); CPI-613, a lipoate analog inhibiting pyruvate dehydrogenase and α-KG dehydrogenase that was recently tested in Phase I and II clinical trials as a single agent or in combination with standard chemotherapy to treat diverse types of cancers (NCT02168140, NCT02232152 and NCT01766219); and enasidinib/AG-221, an orally available inhibitor of mutant IDH2-R140 and IDH2-R172 that undergoes Phase I/II clinical trials as a single agent for the treatment of AML, angio-immunoblastic T-cell lymphoma and glioma (NCT01915498 and NCT02273739). In addition, AG-881 is a promising orally available dual inhibitor of mutant IDH1 and mutant IDH2 that was in Phase I/II clinical trial until this year, recruiting AML patients with mutant IDH1/2, as well as glioma patients (NCT02492737 and NCT02481154).
–
Targeting the ETC
As reviewed in the section ‘Antioxidant defenses in mitochondria‘, cancer cell mitochondria can also display alterations in the functions of ETC complexes that constitute attractive anticancer targets.
–
Complex I
–
A multiplicity of compounds have been tested in vitro and in vivo to inhibit Complex I [318], which include carboxyamidotriazole [319] and fenofibrate [320]. However, the most advanced drugs targeting Complex I are biguanides metformin and phenformin. These two compounds are FDA-approved drugs widely used for diabetes treatment [321]. Interestingly, they can also impair the proliferation of several cancer cell lines [322][323], and in vivo studies further demonstrated that metformin and phenformin inhibit tumor growth and metastasis formation in several animal models [324][325][326][327][328]. While these two drugs were believed to primarily act through AMPK activation, two studies published in the year 2000 demonstrated that metformin primarily inhibits ETC Complex I in cancer cells, consequently inhibiting OXPHOS and activating AMPK [329][330]. The mode of action of these biguanides involves inhibition of ubiquinone reduction [331]. This finding opened new perspectives for the use of biguanides in the field of cancer. In fact, metformin is currently tested in several clinical trials for cancer patients [332]. Until now, most promising results have been obtained in Phase III clinical trial NCT01101438, which tested the efficacy of metformin for the treatment of breast cancer, other nonmetastatic cancers and cancers with a smaller degree of malignity [333]. After receiving metformin twice a day for five years after diagnosis, patients experienced a significant improvement of progression-free survival. Papaverin, a non-narcotic opioid usually used for the treatment of vasospasms and erectile dysfunction by inhibiting phosphodiesterase 10A [334], has also been reported as a Complex I inhibitor in cancer cells [335]. As a consequence, Benej et al.[335] reported that papaverin can, through oxygen sparing, radiosensitize lung and breast cancer cells and tumors in vivo. With the additional description of anticancer effects of papaverin on prostate [336], breast [337] and hepatocarcinoma [338] cancer cells, as well as on glioma xenografts in mice [339], a Phase I clinical trial is ongoing where papaverin hydrochloride is tested as a radiosensitizer in non-small cell lung cancer patients (NCT03824327).
–
Complex II
–
Comparatively to Complex I, there are currently no well characterized Complex II inhibitors. Lonidamine has recently been shown to inhibit Complex II in isolated mitochondria and in DB-1 melanoma cells [340][341]. However, the compound did not demonstrate any benefit in two randomized Phase III clinical trials in combination with chemotherapy, and was therefore withdrawn from clinical studies [340].
–
Complex III
–
Atovaquone is an FDA-approved drug used to treat pneumocystis, pneumonia and malaria [342]. This drug is a ubiquinone analogue that acts as a Complex III inhibitor in parasites, cancer cell lines and breast CSCs, diminishing the oxygen consumption rate and reducing tumor hypoxia at pharmacologically acceptable concentrations [343][344][345][346][347]. Atovaquone is currently tested in a Phase I clinical trial (NCT02628080) for its effects on hypoxia in non-small cell lung carcinoma in a pre-operative window of opportunity study. While recruitment has been completed, no results have been published yet regarding this trial.
–
Complex IV
–
Arsenic trioxide has been described as a Complex IV inhibitor and is FDA-approved for the treatment of acute pro-myelocytic leukemia. In the past years, it has been explored in other types of cancer. Preclinically, because it inhibits cell respiration, arsenic trioxide was shown to decrease hypoxia in Lewis lung carcinoma and transplantable mouse liver tumors, leading to an improvement of the response of mice to radiotherapy [348]. Nitric oxide and hydrocortisone are other compounds that, among other effects, can inhibit Complex IV and increase the efficacy of radiotherapy preclinically [349][350].
–
Complex V
–
To our knowledge, no studies involving in vivo or clinical trials have been reported with drugs targeting ATP synthase in cancer models.
–
Targeting mitochondrial ROS production
The use of mitochondria-targeted antioxidants like mitoTEMPO and mitoQ [89] has proven to be a good strategy to repress the migratory, invasive and metastatic phenotypes of cancer cells. Both drugs repress mtROS-induced activation of the TGFβ pathway [89]. MitoTEMPO is a superoxide scavenger able to mimic mitochondrial SODs [351]. It can easily pass through lipid bilayers and selectively accumulates in negatively charged mitochondria thanks to a positively charged triphenylphosphonium moiety. MitoQ is a very similar compound, with an ubiquinone covalently bound to triphenylphosphonium. Similar to mitoTEMPO, it can rapidly cross biological membranes and concentrate up to 100-fold in mitochondria [352]. It can access the membrane core of mitochondria, acting as a chain-breaking antioxidant, which further allows the recycling of MitoQ to its ubiquinol form via reduction by ETC Complex II [353]. Besides its antimetastatic potential, MitoQ as a mitochondria-targeted antioxidant can successfully decrease KRAS-induced pancreatic tumorigenesis in vivo[354].
–
Restoring mitochondria-driven apoptosis
Strategies that stimulate cytochrome c release can be used to induce apoptotic cancer cell death. Several studies [355][356][357] have indeed proposed that photodynamic therapy (PDT) can induce damage in cancer cells by disturbing the mitochondrial membrane potential, thus triggering the release of cytochrome c and activating caspase-dependent cell death. Several photosensitizers used in PDT have been approved for clinical applications or are under clinical trials [358].
–
Additionally, the use of natural compounds to induce cancer cell apoptosis has shown interesting results. For examples, resveratrol [359][360], curcumin [361], aloe-emodin [362] and betulin [363] were shown to stimulate apoptosis in various cancer cell lines by increasing cytochrome c release from mitochondria. Resveratrol is tested in Phase I clinical trials with colon cancer patients (NCT00256334, NCT00433576). Encouraging preclinical results obtained with FDA-approved curcumin in cancer were an incentive to launch several clinical trials in cancer patients (NCT02064673, NCT02944578, NCT02782949). The non-toxicity towards non-cancer cells demonstrated by these natural compounds added to the low toxicity induced in normal cells by PDT suggests that targeting cancer cells by restoring cytochrome c-driven apoptosis deserves further research efforts.
–
Targeting mitochondrial turnover
Mitochondrial fission, fusion and mitophagy have been investigated as potential anticancer targets. However, few studies specifically investigated mitophagy compared to more general autophagy.
–
Mdivi-1 can block fission by inhibiting the GTPase activity of DRP1 [364], thus preventing mitophagy. For treatments longer than 24 h, mdivi-1 has a cytostatic effect [365], which may be an additional advantage over mitochondrial impairment [366] in cancer therapy. Dynasore is another GTPase inhibitor of DRP1 [367]. Inhibition of mitochondrial fission by dynasore has been shown to suppress cancer cell proliferation and to induces apoptosis in A549 lung cancer cells [368]. It inhibits migration and/or invasion in different cancer cell lines, including bladder cancer cell line T24 [369], lung cancer cell line H1080 [370] and osteosarcoma cancer cell lines MNNG/HOS [371], MG-63 [371], and U2OS [370]. In vivo in mice, dynasore showed additive effects when combined to cisplatin [371].
–
Inducing mitochondrial fusion can also have anticancer effects, as illustrated by S3, a small natural molecule that has been shown to promote fusion hence inhibiting mitophagy [372]. Its mechanisms of action remain to be elucidated, which will necessitate in vivo studies. Of note, inhibiting mitophagy may be one of the mechanisms of action of Temsirolimus, a mTOR inhibitor already in use for other clinical applications than cancer [373][374].
–
Other mitochondrial modulators
In addition to mitochondrial inhibitors, other compounds can indirectly modulate mitochondrial functions. Among them is α-tocopheryl succinate (α-TOS), an analogue of α-tocopherol (vitamin E). α-tocopherol is a potent inhibitor of cytosolic GSH S-transferases GSTP1-1 [375] and GSTO1-1 [376], i.e., enzymes that normally detoxify endogenous and exogenous compounds by catalyzing the conjugation of electrophilic centers to GSH. Its analogue α-TOS selectively induces apoptosis in cancer cells by destabilizing mitochondria [377]. This activity, however, seems to be independent of GST inhibition, and would rather primarily involve ETC Complex I [378] or Complex II [379] inhibition.
–
Interestingly, the catalytic activity of GSTs could also be used to bio-activate prodrugs, allowing their selective accumulation in cancer cells with high expression of some GST isoenzymes. For example, canfosfamide/TLK286, a modified GSH analogue and nitrogen mustard prodrug, is bio-activated by GSTP1-1 in an alkylating metabolite capable of covalently binding DNA [380]. This compound reached Phase III clinical trials with good tolerability [381][382][383]. Brostallicin is another example of a GST-activated prodrug [384][385][386]. It is currently in Phase II clinical trials.
–
Ketogenic diets (K.Ds) based on a high fat and low carbohydrate alimentation have the objective to limit glucose availability for tumors [387]. They were initially developed as a treatment for rare metabolic diseases, such as GLUT1 and pyruvate dehydrogenase (PDH) complex deficiencies [388][389]. KDs have been reported to decrease blood glucose levels and to increase ketone body use, leading to a shift from glycolysis to respiration. Their anticancer activity is based on the assumption that cancer cells with altered mitochondria should not be able to use ketone bodies [390][391]. Several studies associating mitochondrial modulators to KDs have been performed to treat cancer patients, which have recently been reviewed by Weber et al.[387]. The most significant findings pertain to case reports of patients with glioblastoma [392][393][394]. While anticancer activities have been reported, many clinical trials are still ongoing. The results of these studies are expected to confirm whether KDs, and which type of KD, could be used as a nutritional support to improve the outcome of some types of cancers.
CONCLUSIONS
Introductory statements in the scientific literature too often describe cancer cells as being constitutively glycolytic. This reductionist view is based on the rediscovery of the Warburg effect that has strongly increased our knowledge of cancer biochemistry since the year 2000, with the drawback that studying other metabolic pathways has been under pursued for almost a decade. If indeed the Warburg phenotype provides a biosynthetic advantage for cancer cell proliferation [143][395][396], it is evident that not all cancer cells simultaneously proliferate in experimental and clinical tumors. Hence, tumor metabolism is generally characterized by the same heterogeneity as the phenotypic heterogeneity of the cancer cells that compose the tumor. Good examples are the metabolic cooperativity that exists between oxidative and glycolytic cancer cells in many tumor types [397][398] and the observation that, in a given tumor, metastatic progenitor cells [89] and CSCs [87] have a different metabolic behavior than the bulk population of cancer cells. It is nevertheless noticeable that mutations in genes encoding TCA cycle enzymes [111][121][399] or ETC subunits [67][105][106] logically limit this metabolic plasticity, but they are rare events in rare tumor types.
–
This review paper aimed to contribute to re-center mitochondria in the overall metabolic map of cancers. We therefore attempted to provide a comprehensive overview of the many functions that these organelles exert in cancer cells, not only as powerhouses, but also as dynamic signaling organelles controlling cell survival and death, motility, stemness and resistance to treatment. However, key questions have not yet been answered by the scientific community. In our opinion, a significant issue is to determine whether metabolic alterations are a cause or a consequence of the malignant process. Elements in the literature support both possibilities. For example, it is clear that anaerobic glycolysis coupled to lactic fermentation [170][172][400] and increased mitophagy [401] are adaptive survival pathways to hypoxia, hence consequences of hypoxia. The situation is less clear for aerobic glycolysis, where an allosteric control of pyruvate kinase M2 by fructose-1,6-bisphosphate (that activates the enzyme) and alanine (that inhibits the enzyme) may dictate progression in the cell cycle [402], or, vice versa, the cell cycle could impose metabolic cycles to cancer cells. These cycles would be characterized by alternations of energy production and biosynthesis. For mitochondria in particular, experiments of mitochondrial transfer have clearly demonstrated that these organelles can carry and transfer malignant information, such as the capacity to metastasize [280]. Identifying whether specific metabolic alterations drive or merely follow the phenotypic evolution of cancer cells is not trivial, as in the first case targeting these changes may block phenotypic progression, whereas in the second case metabolic plasticity could rapidly overcome therapeutic interventions.
–
In the last section of this review, we briefly described the most advanced therapeutic compounds targeting mitochondria in cancer. For experts in the field, it is obvious that the list is short and that most of the drugs do not have unique targets. It probably reflects that the field is still in its infancy, in the sense that several important fundamental discoveries are still to be made that would identify precise mitochondrial alterations in cancer allowing specific anticancer interventions. In other words, the molecular definition of ‘oncogenic mitochondria’, i.e., mitochondria that carry and can transfer malignant information, should be a priority for basic research. Because they carry their own genetic material and are subjected to environmental changes within cells (such as pH, pO2, the availability of metabolites and exposure to treatments) and as metabolic sensors of the extracellular microenvironment, it is possible that mitochondria could undergo Darwinian selection during tumor progression. In support of this hypothesis, several papers already identified mutations in mtDNA with functional effects [403], for example in renal oncocytomas [404] and pancreatic cancers [405]. The development of mtDNA editing tools [406] is expected to provide experimental strategies to track, characterize and repair oncogenic mitochondria, which will further require a deep understanding of mitochondrial epigenetics in cancer.
REFERENCES
- Warburg O, Wind F, Negelein E (1927). The metabolism of tumors in the body. J Gen Physiol 8(6): 519-530. 10.1085/jgp.8.6.519
- Reichert AS, Neupert W (2004). Mitochondriomics or what makes us breathe. Trends Genet 20(11): 555-562. 10.1016/j.tig.2004.08.012
- Wallace DC (1999). Mitochondrial diseases in man and mouse. Science 283(5407): 1482-1488. 10.1126/science.283.5407.1482
- Nomoto S, Yamashita K, Koshikawa K, Nakao A, Sidransky D (2002). Mitochondrial D-loop mutations as clonal markers in multicentric hepatocellular carcinoma and plasma. Clin Cancer Res 8(2): 481-487. 11839667
- Eng C, Kiuru M, Fernandez MJ, Aaltonen LA (2003). A role for mitochondrial enzymes in inherited neoplasia and beyond. Nat Rev Cancer 3(3): 193-202. 10.1038/nrc1013
- Jeronimo C, Nomoto S, Caballero OL, Usadel H, Henrique R, Varzim G, Oliveira J, Lopes C, Fliss MS, Sidransky D (2001). Mitochondrial mutations in early stage prostate cancer and bodily fluids. Oncogene 20(37): 5195-5198. 10.1038/sj.onc.1204646
- Kumari S, Badana AK, G MM, G S, Malla R (2018). Reactive Oxygen Species: A Key Constituent in Cancer Survival. Biomark Insights 13: 1177271918755391. 10.1177/1177271918755391
- Nohl H, Kozlov AV, Gille L, Staniek K (2003). Cell respiration and formation of reactive oxygen species: facts and artefacts. Biochem Soc Trans 31(Pt 6): 1308-1311. 10.1042/bst0311308
- Galvan DL, Green NH, Danesh FR (2017). The hallmarks of mitochondrial dysfunction in chronic kidney disease. Kidney Int 92(5): 1051-1057. 10.1016/j.kint.2017.05.034
- Buechter DD (1988). Free radicals and oxygen toxicity. Pharm Res 5(5): 253-260. 10.1023/a:1015914418014
- Gius D, Spitz DR (2006). Redox signaling in cancer biology. Antioxid Redox Signal 8(7-8): 1249-1252. 10.1089/ars.2006.8.1249
- Waris G, Ahsan H (2006). Reactive oxygen species: role in the development of cancer and various chronic conditions. J Carcinog 5: 14. 10.1186/1477-3163-5-14
- Sena LA, Chandel NS (2012). Physiological roles of mitochondrial reactive oxygen species. Mol Cell 48(2): 158-167. 10.1016/j.molcel.2012.09.025
- Arakawa S, Nakanomyo I, Kudo-Sakamoto Y, Akazawa H, Komuro I, Shimizu S (2015). Identification of a novel compound that inhibits both mitochondria-mediated necrosis and apoptosis. Biochem Biophys Res Commun 467(4): 1006-1011. 10.1016/j.bbrc.2015.10.022
- Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, Blagosklonny MV, El-Deiry WS, Golstein P, Green DR, Hengartner M, Knight RA, Kumar S, Lipton SA, Malorni W, Nunez G, Peter ME, Tschopp J, Yuan J, Piacentini M, Zhivotovsky B, Melino G, Nomenclature Committee on Cell D (2009). Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ 16(1): 3-11. 10.1038/cdd.2008.150
- Shimizu S, Narita M, Tsujimoto Y (1999). Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature 399(6735): 483-487. 10.1038/20959
- Perciavalle RM, Stewart DP, Koss B, Lynch J, Milasta S, Bathina M, Temirov J, Cleland MM, Pelletier S, Schuetz JD, Youle RJ, Green DR, Opferman JT (2012). Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat Cell Biol 14(6): 575-583. 10.1038/ncb2488
- Wuilleme-Toumi S, Robillard N, Gomez P, Moreau P, Le Gouill S, Avet-Loiseau H, Harousseau JL, Amiot M, Bataille R (2005). Mcl-1 is overexpressed in multiple myeloma and associated with relapse and shorter survival. Leukemia 19(7): 1248-1252. 10.1038/sj.leu.2403784
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, Mc Henry KT, Pinchback RM, Ligon AH, Cho YJ, Haery L, Greulich H, Reich M, Winckler W, Lawrence MS, Weir BA, Tanaka KE, Chiang DY, Bass AJ, Loo A, Hoffman C, Prensner J, Liefeld T, Gao Q, Yecies D, Signoretti S, et al. (2010). The landscape of somatic copy-number alteration across human cancers. Nature 463(7283): 899-905. 10.1038/nature08822
- Andersen JL, Kornbluth S (2012). Mcl-1 rescues a glitch in the matrix. Nat Cell Biol 14(6): 563-565. 10.1038/ncb2511
- Christofferson DE, Yuan J (2010). Necroptosis as an alternative form of programmed cell death. Curr Opin Cell Biol 22(2): 263-268. 10.1016/j.ceb.2009.12.003
- Gaude E, Frezza C (2014). Defects in mitochondrial metabolism and cancer. Cancer Metab 2: 10. 10.1186/2049-3002-2-10
- Sullivan LB, Gui DY, Vander Heiden MG (2016). Altered metabolite levels in cancer: implications for tumour biology and cancer therapy. Nat Rev Cancer 16(11): 680-693. 10.1038/nrc.2016.85
- Dong CK, Masutomi K, Hahn WC (2005). Telomerase: regulation, function and transformation. Crit Rev Oncol Hematol 54(2): 85-93. 10.1016/j.critrevonc.2004.12.005
- Muzza M, Colombo C, Cirello V, Perrino M, Vicentini L, Fugazzola L (2016). Oxidative stress and the subcellular localization of the telomerase reverse transcriptase (TERT) in papillary thyroid cancer. Mol Cell Endocrinol 431: 54-61. 10.1016/j.mce.2016.05.005
- Singhapol C, Pal D, Czapiewski R, Porika M, Nelson G, Saretzki GC (2013). Mitochondrial telomerase protects cancer cells from nuclear DNA damage and apoptosis. PLoS One 8(1): e52989. 10.1371/journal.pone.0052989
- Miwa S, Saretzki G (2017). Telomerase and mTOR in the brain: the mitochondria connection. Neural Regen Res 12(3): 358-361. 10.4103/1673-5374.202922
- McBride HM, Neuspiel M, Wasiak S (2006). Mitochondria: more than just a powerhouse. Curr Biol 16(14): R551-560. 10.1016/j.cub.2006.06.054
- Chan DC (2006). Mitochondrial fusion and fission in mammals. Annu Rev Cell Dev Biol 22: 79-99. 10.1146/annurev.cellbio.22.010305.104638
- Kaback HR (1980). Electrochemical ion gradients and active transport. Ann N Y Acad Sci 339: 53-60. 10.1111/j.1749-6632.1980.tb15968.x
- Vafai SB, Mootha VK (2012). Mitochondrial disorders as windows into an ancient organelle. Nature 491(7424): 374-383. 10.1038/nature11707
- Harada H, Becknell B, Wilm M, Mann M, Huang LJ, Taylor SS, Scott JD, Korsmeyer SJ (1999). Phosphorylation and inactivation of BAD by mitochondria-anchored protein kinase A. Mol Cell 3(4): 413-422. 10.1016/s1097-2765(00)80469-4
- Seth RB, Sun L, Ea CK, Chen ZJ (2005). Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122(5): 669-682. 10.1016/j.cell.2005.08.012
- Murphy MP (2009). How mitochondria produce reactive oxygen species. Biochem J 417(1): 1-13. 10.1042/BJ20081386
- Anderson NM, Mucka P, Kern JG, Feng H (2018). The emerging role and targetability of the TCA cycle in cancer metabolism. Protein Cell 9(2): 216-237. 10.1007/s13238-017-0451-1
- Elbaz-Alon Y, Rosenfeld-Gur E, Shinder V, Futerman AH, Geiger T, Schuldiner M (2014). A dynamic interface between vacuoles and mitochondria in yeast. Dev Cell 30(1): 95-102. 10.1016/j.devcel.2014.06.007
- Ellenrieder L, Rampelt H, Becker T (2017). Connection of protein transport and organelle contact sites in mitochondria. J Mol Biol 429(14): 2148-2160. 10.1016/j.jmb.2017.05.023
- Bayrhuber M, Meins T, Habeck M, Becker S, Giller K, Villinger S, Vonrhein C, Griesinger C, Zweckstetter M, Zeth K (2008). Structure of the human voltage-dependent anion channel. Proc Natl Acad Sci U S A 105(40): 15370-15375. 10.1073/pnas.0808115105
- Shimizu S, Matsuoka Y, Shinohara Y, Yoneda Y, Tsujimoto Y (2001). Essential role of voltage-dependent anion channel in various forms of apoptosis in mammalian cells. J Cell Biol 152(2): 237-250. 10.1083/jcb.152.2.237
- Shoshan-Barmatz V, Israelson A, Brdiczka D, Sheu SS (2006). The voltage-dependent anion channel (VDAC): function in intracellular signalling, cell life and cell death. Curr Pharm Des 12(18): 2249-2270. 10.2174/138161206777585111
- Arora KK, Pedersen PL (1988). Functional significance of mitochondrial bound hexokinase in tumor cell metabolism. Evidence for preferential phosphorylation of glucose by intramitochondrially generated ATP. J Biol Chem 263(33): 17422-17428. 10.2174/138161206777585111
- Shimizu S, Ide T, Yanagida T, Tsujimoto Y (2000). Electrophysiological study of a novel large pore formed by Bax and the voltage-dependent anion channel that is permeable to cytochrome c. J Biol Chem 275(16): 12321-12325. 10.1074/jbc.275.16.12321
- Bertini I, Cavallaro G, Rosato A (2006). Cytochrome c: occurrence and functions. Chem Rev 106(1): 90-115. 10.1021/cr050241v
- Liu X, Kim CN, Yang J, Jemmerson R, Wang X (1996). Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 86(1): 147-157. 10.1016/s0092-8674(00)80085-9
- Soberg K, Moen LV, Skalhegg BS, Laerdahl JK (2017). Evolution of the cAMP-dependent protein kinase (PKA) catalytic subunit isoforms. PLoS One 12(7): e0181091. 10.1371/journal.pone.0181091
- Merrill RA, Strack S (2014). Mitochondria: a kinase anchoring protein 1, a signaling platform for mitochondrial form and function. Int J Biochem Cell Biol 48: 92-96. 10.1016/j.biocel.2013.12.012
- Kvissel AK, Orstavik S, Eikvar S, Brede G, Jahnsen T, Collas P, Akusjarvi G, Skalhegg BS (2007). Involvement of the catalytic subunit of protein kinase A and of HA95 in pre-mRNA splicing. Exp Cell Res 313(13): 2795-2809. 10.1016/j.yexcr.2007.05.014
- Mohanty A, Tiwari-Pandey R, Pandey NR (2019). Mitochondria: the indispensable players in innate immunity and guardians of the inflammatory response. J Cell Commun Signal 13(3): 303-318. 10.1007/s12079-019-00507-9
- Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB (2009). ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324(5930): 1076-1080. 10.1126/science.1164097
- Shyh-Chang N, Locasale JW, Lyssiotis CA, Zheng Y, Teo RY, Ratanasirintrawoot S, Zhang J, Onder T, Unternaehrer JJ, Zhu H, Asara JM, Daley GQ, Cantley LC (2013). Influence of threonine metabolism on S-adenosylmethionine and histone methylation. Science 339(6116): 222-226. 10.1126/science.1226603
- Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT (1998). Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci U S A 95(20): 11715-11720. 10.1073/pnas.95.20.11715
- Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T (1998). Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280(5370): 1763-1766. 10.1126/science.280.5370.1763
- Szabadkai G, Duchen MR (2008). Mitochondria: the hub of cellular Ca2+ signaling. Physiology 23: 84-94. 10.1152/physiol.00046.2007
- Cardenas C, Miller RA, Smith I, Bui T, Molgo J, Muller M, Vais H, Cheung KH, Yang J, Parker I, Thompson CB, Birnbaum MJ, Hallows KR, Foskett JK (2010). Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 142(2): 270-283. 10.1016/j.cell.2010.06.007
- Di Benedetto G, Scalzotto E, Mongillo M, Pozzan T (2013). Mitochondrial Ca(2)(+) uptake induces cyclic AMP generation in the matrix and modulates organelle ATP levels. Cell Metab 17(6): 965-975. 10.1016/j.cmet.2013.05.003
- Bravo-Sagua R, Rodriguez AE, Kuzmicic J, Gutierrez T, Lopez-Crisosto C, Quiroga C, Diaz-Elizondo J, Chiong M, Gillette TG, Rothermel BA, Lavandero S (2013). Cell death and survival through the endoplasmic reticulum-mitochondrial axis. Curr Mol Med 13(2): 317-329. 10.2174/156652413804810781
- Pinton P, Giorgi C, Siviero R, Zecchini E, Rizzuto R (2008). Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 27(50): 6407-6418. 10.1038/onc.2008.308
- Hardie DG, Lin SC (2017). AMP-activated protein kinase – not just an energy sensor. F1000Res 6: 1724. 10.12688/f1000research.11960.1
- Alberts B, Johnson A, Lewis J, Raff M, Roberts K, and Walter P (2002). Molecular biology of the cell. Garland, New York.
- Mannella CA (2006). Structure and dynamics of the mitochondrial inner membrane cristae. Biochim Biophys Acta 1763(5-6): 542-548. 10.1016/j.bbamcr.2006.04.006
- Nielsen J, Gejl KD, Hey-Mogensen M, Holmberg HC, Suetta C, Krustrup P, Elemans CPH, Ortenblad N (2017). Plasticity in mitochondrial cristae density allows metabolic capacity modulation in human skeletal muscle. J Physiol 595(9): 2839-2847. 10.1113/JP273040
- Seebacher F (2009). Responses to temperature variation: integration of thermoregulation and metabolism in vertebrates. J Exp Biol 212(18): 2885-2891. 10.1242/jeb.024430
- Signes A, Fernandez-Vizarra E (2018). Assembly of mammalian oxidative phosphorylation complexes I-V and supercomplexes. Essays Biochem 62(3): 255-270. 10.1042/EBC20170098
- Stroud DA, Surgenor EE, Formosa LE, Reljic B, Frazier AE, Dibley MG, Osellame LD, Stait T, Beilharz TH, Thorburn DR, Salim A, Ryan MT (2016). Accessory subunits are integral for assembly and function of human mitochondrial complex I. Nature 538(7623): 123-126. 10.1038/nature19754
- Vinothkumar KR, Zhu J, Hirst J (2014). Architecture of mammalian respiratory complex I. Nature 515(7525): 80-84. 10.1038/nature13686
- Simonnet H, Demont J, Pfeiffer K, Guenaneche L, Bouvier R, Brandt U, Schagger H, Godinot C (2003). Mitochondrial complex I is deficient in renal oncocytomas. Carcinogenesis 24(9): 1461-1466. 10.1093/carcin/bgg109
- Mayr JA, Meierhofer D, Zimmermann F, Feichtinger R, Kogler C, Ratschek M, Schmeller N, Sperl W, Kofler B (2008). Loss of complex I due to mitochondrial DNA mutations in renal oncocytoma. Clin Cancer Res 14(8): 2270-2275. 10.1158/1078-0432.CCR-07-4131
- Zimmermann FA, Mayr JA, Neureiter D, Feichtinger R, Alinger B, Jones ND, Eder W, Sperl W, Kofler B (2009). Lack of complex I is associated with oncocytic thyroid tumours. Br J Cancer 100(9): 1434-1437. 10.1038/sj.bjc.6605028
- Kroger A, Klingenberg M (1973). The kinetics of the redox reactions of ubiquinone related to the electron-transport activity in the respiratory chain. Eur J Biochem 34(2): 358-368. 10.1111/j.1432-1033.1973.tb02767.x
- Van Vranken JG, Na U, Winge DR, Rutter J (2015). Protein-mediated assembly of succinate dehydrogenase and its cofactors. Crit Rev Biochem Mol Biol 50(2): 168-180. 10.3109/10409238.2014.990556
- Meierhofer D, Mayr JA, Foetschl U, Berger A, Fink K, Schmeller N, Hacker GW, Hauser-Kronberger C, Kofler B, Sperl W (2004). Decrease of mitochondrial DNA content and energy metabolism in renal cell carcinoma. Carcinogenesis 25(6): 1005-1010. 10.1093/carcin/bgh104
- Owens KM, Kulawiec M, Desouki MM, Vanniarajan A, Singh KK (2011). Impaired OXPHOS complex III in breast cancer. PLoS One 6(8): e23846. 10.1371/journal.pone.0023846
- Sousa JS, D’Imprima E, Vonck J (2018). Mitochondrial respiratory chain complexes. Subcell Biochem 87: 167-227. 10.1007/978-981-10-7757-9_7
- Ohashi Y, Kaneko SJ, Cupples TE, Young SR (2004). Ubiquinol cytochrome c reductase (UQCRFS1) gene amplification in primary breast cancer core biopsy samples. Gynecol Oncol 93(1): 54-58. 10.1016/j.ygyno.2004.01.019
- Margoliash E, Ferguson-Miller S, Tulloss J, Kang CH, Feinberg BA, Brautigan DL, Morrison M (1973). Separate intramolecular pathways for reduction and oxidation of cytochrome c in electron transport chain reactions. Proc Natl Acad Sci U S A 70(11): 3245-3249. 10.1073/pnas.70.11.3245
- Belevich I, Verkhovsky MI, Wikstrom M (2006). Proton-coupled electron transfer drives the proton pump of cytochrome c oxidase. Nature 440(7085): 829-832. 10.1038/nature04619
- Mitchell P (2011). Chemiosmotic coupling in oxidative and photosynthetic phosphorylation. 1966. Biochim Biophys Acta 1807(12): 1507-1538. 10.1016/j.bbabio.2011.09.018
- Schagger H, Pfeiffer K (2000). Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J 19(8): 1777-1783. 10.1093/emboj/19.8.1777
- Acin-Perez R, Fernandez-Silva P, Peleato ML, Perez-Martos A, Enriquez JA (2008). Respiratory active mitochondrial supercomplexes. Mol Cell 32(4): 529-539. 10.1016/j.molcel.2008.10.021
- Schagger H, Pfeiffer K (2001). The ratio of oxidative phosphorylation complexes I-V in bovine heart mitochondria and the composition of respiratory chain supercomplexes. J Biol Chem 276(41): 37861-37867. 10.1074/jbc.M106474200
- Acin-Perez R, Bayona-Bafaluy MP, Fernandez-Silva P, Moreno-Loshuertos R, Perez-Martos A, Bruno C, Moraes CT, Enriquez JA (2004). Respiratory complex III is required to maintain complex I in mammalian mitochondria. Mol Cell 13(6): 805-815. 10.1016/j.molcel.2008.10.021
- D’Aurelio M, Gajewski CD, Lenaz G, Manfredi G (2006). Respiratory chain supercomplexes set the threshold for respiration defects in human mtDNA mutant cybrids. Hum Mol Genet 15(13): 2157-2169. 10.1093/hmg/ddl141
- Vempati UD, Han X, Moraes CT (2009). Lack of cytochrome c in mouse fibroblasts disrupts assembly/stability of respiratory complexes I and IV. J Biol Chem 284(7): 4383-4391. 10.1074/jbc.M805972200
- Moreno-Sanchez R, Marin-Hernandez A, Saavedra E, Pardo JP, Ralph SJ, Rodriguez-Enriquez S (2014). Who controls the ATP supply in cancer cells? Biochemistry lessons to understand cancer energy metabolism. Int J Biochem Cell Biol 50: 10-23. 10.1016/j.biocel.2014.01.025
- Zu XL, Guppy M (2004). Cancer metabolism: facts, fantasy, and fiction. Biochem Biophys Res Commun 313(3): 459-465. 10.1016/j.bbrc.2003.11.136
- LeBleu VS, O’Connell JT, Gonzalez Herrera KN, Wikman H, Pantel K, Haigis MC, de Carvalho FM, Damascena A, Domingos Chinen LT, Rocha RM, Asara JM, Kalluri R (2014). PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol 16(10): 992-1003, 1001-1015. 10.1038/ncb3039
- Snyder V, Reed-Newman TC, Arnold L, Thomas SM, Anant S (2018). Cancer stem cell metabolism and potential therapeutic targets. Front Oncol 8: 203. 10.3389/fonc.2018.00203
- Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger GR, Chandel NS (2010). Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A 107(19): 8788-8793. 10.1073/pnas.1003428107
- Porporato PE, Payen VL, Perez-Escuredo J, De Saedeleer CJ, Danhier P, Copetti T, Dhup S, Tardy M, Vazeille T, Bouzin C, Feron O, Michiels C, Gallez B, Sonveaux P (2014). A mitochondrial switch promotes tumor metastasis. Cell Rep 8(3): 754-766. 10.1016/j.celrep.2014.06.043
- Martinez-Outschoorn UE, Pavlides S, Howell A, Pestell RG, Tanowitz HB, Sotgia F, Lisanti MP (2011). Stromal-epithelial metabolic coupling in cancer: integrating autophagy and metabolism in the tumor microenvironment. Int J Biochem Cell Biol 43(7): 1045-1051. 10.1016/j.biocel.2011.01.023
- Viale A, Corti D, Draetta GF (2015). Tumors and mitochondrial respiration: a neglected connection. Cancer Res 75(18): 3685-3686. 10.1158/0008-5472.CAN-15-0491
- Hao W, Chang CP, Tsao CC, Xu J (2010). Oligomycin-induced bioenergetic adaptation in cancer cells with heterogeneous bioenergetic organization. J Biol Chem 285(17): 12647-12654. 10.1074/jbc.M109.084194
- Barbi de Moura M, Vincent G, Fayewicz SL, Bateman NW, Hood BL, Sun M, Suhan J, Duensing S, Yin Y, Sander C, Kirkwood JM, Becker D, Conrads TP, Van Houten B, Moschos SJ (2012). Mitochondrial respiration–an important therapeutic target in melanoma. PLoS One 7(8): e40690. 10.1371/journal.pone.0040690
- Jitschin R, Hofmann AD, Bruns H, Giessl A, Bricks J, Berger J, Saul D, Eckart MJ, Mackensen A, Mougiakakos D (2014). Mitochondrial metabolism contributes to oxidative stress and reveals therapeutic targets in chronic lymphocytic leukemia. Blood 123(17): 2663-2672. 10.1182/blood-2013-10-532200
- Cheng G, Zielonka J, McAllister D, Tsai S, Dwinell MB, Kalyanaraman B (2014). Profiling and targeting of cellular bioenergetics: inhibition of pancreatic cancer cell proliferation. Br J Cancer 111(1): 85-93. 10.1038/bjc.2014.272
- Diers AR, Vayalil PK, Oliva CR, Griguer CE, Darley-Usmar V, Hurst DR, Welch DR, Landar A (2013). Mitochondrial bioenergetics of metastatic breast cancer cells in response to dynamic changes in oxygen tension: effects of HIF-1alpha. PLoS One 8(6): e68348. 10.1371/journal.pone.0068348
- Voet D, Voet JG, and Pratt CW (2016). Fundamentals of biochemistry: life at the molecular level. John Wiley & Sons, Hoboken.
- Porcelli AM, Ghelli A, Zanna C, Pinton P, Rizzuto R, Rugolo M (2005). pH difference across the outer mitochondrial membrane measured with a green fluorescent protein mutant. Biochem Biophys Res Commun 326(4): 799-804. 10.1016/j.bbrc.2004.11.105
- Owen OE, Kalhan SC, Hanson RW (2002). The key role of anaplerosis and cataplerosis for citric acid cycle function. J Biol Chem 277(34): 30409-30412. 10.1074/jbc.R200006200
- Nelson DL, Cox MM, and Lehninger AL (2017). Lehninger principles of biochemistry. W.H. Freeman and Company; Macmillan Higher Education, New York.
- Jitrapakdee S, St Maurice M, Rayment I, Cleland WW, Wallace JC, Attwood PV (2008). Structure, mechanism and regulation of pyruvate carboxylase. Biochem J 413(3): 369-387. 10.1042/BJ20080709
- Raimundo N, Baysal BE, Shadel GS (2011). Revisiting the TCA cycle: signaling to tumor formation. Trends Mol Med 17(11): 641-649. 10.1016/j.molmed.2011.06.001
- Menendez JA, Alarcon T, Joven J (2014). Gerometabolites: the pseudohypoxic aging side of cancer oncometabolites. Cell Cycle 13(5): 699-709. 10.4161/cc.28079
- Ahn CS, Metallo CM (2015). Mitochondria as biosynthetic factories for cancer proliferation. Cancer Metab 3(1): 1. 10.1186/s40170-015-0128-2
- Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, van der Mey A, Taschner PE, Rubinstein WS, Myers EN, Richard CW, 3rd, Cornelisse CJ, Devilee P, Devlin B (2000). Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 287(5454): 848-851. 10.1126/science.287.5454.848
- Niemann S, Muller U (2000). Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat Genet 26(3): 268-270. 10.1038/81551.
- Gunnala V, Pereira N, Irani M, Lilienthal D, Pirog EC, Soslow R, Caputo TA, Elias R, Kligman I, Rosenwaks Z (2018). Novel fumarate hydratase mutation in siblings with early onset uterine leiomyomas and hereditary leiomyomatosis and renal cell cancer syndrome. Int J Gynecol Pathol 37(3): 256-261. 10.1097/PGP.0000000000000423
- Mann ML, Ezzati M, Tarnawa ED, Carr BR (2015). Fumarate Hydratase Mutation in a Young Woman With Uterine Leiomyomas and a Family History of Renal Cell Cancer. Obstet Gynecol 126(1): 90-92. 10.1097/AOG.0000000000000702
- Lehtonen HJ, Kiuru M, Ylisaukko-Oja SK, Salovaara R, Herva R, Koivisto PA, Vierimaa O, Aittomaki K, Pukkala E, Launonen V, Aaltonen LA (2006). Increased risk of cancer in patients with fumarate hydratase germline mutation. J Med Genet 43(6): 523-526. 10.1136/jmg.2005.036400
- Laukka T, Mariani CJ, Ihantola T, Cao JZ, Hokkanen J, Kaelin WG, Jr., Godley LA, Koivunen P (2016). Fumarate and succinate regulate expression of hypoxia-inducible genes via TET enzymes. J Biol Chem 291(8): 4256-4265. 10.1074/jbc.M115.688762
- Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, Liu L, Liu Y, Yang C, Xu Y, Zhao S, Ye D, Xiong Y, Guan KL (2012). Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev 26(12): 1326-1338. 10.1101/gad.191056.112
- Sciacovelli M, Goncalves E, Johnson TI, Zecchini VR, da Costa AS, Gaude E, Drubbel AV, Theobald SJ, Abbo SR, Tran MG, Rajeeve V, Cardaci S, Foster S, Yun H, Cutillas P, Warren A, Gnanapragasam V, Gottlieb E, Franze K, Huntly B, Maher ER, Maxwell PH, Saez-Rodriguez J, Frezza C (2016). Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature 537(7621): 544-547. 10.1038/nature19353
- Cho HY, Kleeberger SR (2020). Mitochondrial biology in airway pathogenesis and the role of NRF2. Arch Pharm Res 43(3): 297-320. 10.1007/s12272-019-01182-5
- Itoh K, Wakabayashi N, Katoh Y, Ishii T, O’Connor T, Yamamoto M (2003). Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells 8(4): 379-391. 10.1046/j.1365-2443.2003.00640.x
- Raghunath A, Sundarraj K, Nagarajan R, Arfuso F, Bian J, Kumar AP, Sethi G, Perumal E (2018). Antioxidant response elements: Discovery, classes, regulation and potential applications. Redox Biol 17: 297-314. 10.1016/j.redox.2018.05.002
- Sullivan LB, Martinez-Garcia E, Nguyen H, Mullen AR, Dufour E, Sudarshan S, Licht JD, Deberardinis RJ, Chandel NS (2013). The proto-oncometabolite fumarate binds glutathione to amplify ROS-dependent signaling. Mol Cell 51(2): 236-248. 10.1016/j.molcel.2013.05.003
- Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, Olivi A, McLendon R, Rasheed BA, Keir S, Nikolskaya T, Nikolsky Y, Busam DA, Tekleab H, Diaz LA, Jr., Hartigan J, Smith DR, Strausberg RL, Marie SK, Shinjo SM, Yan H, Riggins GJ, Bigner DD, Karchin R, Papadopoulos N, Parmigiani G, et al. (2008). An integrated genomic analysis of human glioblastoma multiforme. Science 321(5897): 1807-1812. 10.1126/science.1164382
- Cohen AL, Holmen SL, Colman H (2013). IDH1 and IDH2 mutations in gliomas. Curr Neurol Neurosci Rep 13(5): 345. 10.1007/s11910-013-0345-4
- Cairns RA, Iqbal J, Lemonnier F, Kucuk C, de Leval L, Jais JP, Parrens M, Martin A, Xerri L, Brousset P, Chan LC, Chan WC, Gaulard P, Mak TW (2012). IDH2 mutations are frequent in angioimmunoblastic T-cell lymphoma. Blood 119(8): 1901-1903. 10.1182/blood-2011-11-391748
- Abbas S, Lugthart S, Kavelaars FG, Schelen A, Koenders JE, Zeilemaker A, van Putten WJ, Rijneveld AW, Lowenberg B, Valk PJ (2010). Acquired mutations in the genes encoding IDH1 and IDH2 both are recurrent aberrations in acute myeloid leukemia: prevalence and prognostic value. Blood 116(12): 2122-2126. 10.1182/blood-2009-11-250878
- Paschka P, Schlenk RF, Gaidzik VI, Habdank M, Kronke J, Bullinger L, Spath D, Kayser S, Zucknick M, Gotze K, Horst HA, Germing U, Dohner H, Dohner K (2010). IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J Clin Oncol 28(22): 3636-3643. 10.1200/JCO.2010.28.3762
- Kang MR, Kim MS, Oh JE, Kim YR, Song SY, Seo SI, Lee JY, Yoo NJ, Lee SH (2009). Mutational analysis of IDH1 codon 132 in glioblastomas and other common cancers. Int J Cancer 125(2): 353-355. 10.1002/ijc.24379
- Yen KE, Bittinger MA, Su SM, Fantin VR (2010). Cancer-associated IDH mutations: biomarker and therapeutic opportunities. Oncogene 29(49): 6409-6417. 10.1038/onc.2010.444
- Zhu H, Zhang Y, Chen J, Qiu J, Huang K, Wu M, Xia C (2017). IDH1 R132H IDH1 R132H mutation enhances cell migration by activating AKT-mTOR signaling pathway, but sensitizes cells to 5-FU treatment as NADPH and GSH are reduced. PLoS One 12(1): e0169038. 10.1371/journal.pone.0169038
- Engelman JA (2009). Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer 9(8): 550-562. 10.1038/nrc2664
- Shi J, Zuo H, Ni L, Xia L, Zhao L, Gong M, Nie D, Gong P, Cui D, Shi W, Chen J (2014). An IDH1 mutation inhibits growth of glioma cells via GSH depletion and ROS generation. Neurol Sci 35(6): 839-845. 10.1007/s10072-013-1607-2
- Reitman ZJ, Yan H (2010). Isocitrate dehydrogenase 1 and 2 mutations in cancer: alterations at a crossroads of cellular metabolism. J Natl Cancer Inst 102(13): 932-941. 10.1093/jnci/djq187
- Trachootham D, Alexandre J, Huang P (2009). Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov 8(7): 579-591. 10.1038/nrd2803
- Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, Egia A, Sasaki AT, Thomas G, Kozma SC, Papa A, Nardella C, Cantley LC, Baselga J, Pandolfi PP (2008). Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest 118(9): 3065-3074. 10.1172/JCI34739
- Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S, Losman JA, Joensuu P, Bergmann U, Gross S, Travins J, Weiss S, Looper R, Ligon KL, Verhaak RG, Yan H, Kaelin WG, Jr. (2012). Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 483(7390): 484-488. 10.1038/nature10898
- Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT, Liu LX, Jiang WQ, Liu J, Zhang JY, Wang B, Frye S, Zhang Y, Xu YH, Lei QY, Guan KL, Zhao SM, Xiong Y (2011). Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 19(1): 17-30. 10.1016/j.ccr.2010.12.014
- Chowdhury R, Yeoh KK, Tian YM, Hillringhaus L, Bagg EA, Rose NR, Leung IK, Li XS, Woon EC, Yang M, McDonough MA, King ON, Clifton IJ, Klose RJ, Claridge TD, Ratcliffe PJ, Schofield CJ, Kawamura A (2011). The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep 12(5): 463-469. 10.1038/embor.2011.43
- Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P, Yu W, Li Z, Gong L, Peng Y, Ding J, Lei Q, Guan KL, Xiong Y (2009). Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science 324(5924): 261-265. 10.1126/science.1170944
- Krebs HA (1970). Rate control of the tricarboxylic acid cycle. Adv Enzyme Regul 8: 335-353. 10.1016/0065-2571(70)90028-2
- Lin CC, Cheng TL, Tsai WH, Tsai HJ, Hu KH, Chang HC, Yeh CW, Chen YC, Liao CC, Chang WT (2012). Loss of the respiratory enzyme citrate synthase directly links the Warburg effect to tumor malignancy. Sci Rep 2: 785. 10.1038/srep00785
- Schlichtholz B, Turyn J, Goyke E, Biernacki M, Jaskiewicz K, Sledzinski Z, Swierczynski J (2005). Enhanced citrate synthase activity in human pancreatic cancer. Pancreas 30(2): 99-104. 10.1097/01.mpa.0000153326.69816.7d
- Simonnet H, Alazard N, Pfeiffer K, Gallou C, Beroud C, Demont J, Bouvier R, Schagger H, Godinot C (2002). Low mitochondrial respiratory chain content correlates with tumor aggressiveness in renal cell carcinoma. Carcinogenesis 23(5): 759-768. 10.1093/carcin/23.5.759
- Chen L, Liu T, Zhou J, Wang Y, Wang X, Di W, Zhang S (2014). Citrate synthase expression affects tumor phenotype and drug resistance in human ovarian carcinoma. PLoS One 9(12): e115708. 10.1371/journal.pone.0115708
- Wang P, Mai C, Wei YL, Zhao JJ, Hu YM, Zeng ZL, Yang J, Lu WH, Xu RH, Huang P (2013). Decreased expression of the mitochondrial metabolic enzyme aconitase (ACO2) is associated with poor prognosis in gastric cancer. Med Oncol 30(2): 552. 10.1007/s12032-013-0552-5
- Singh KK, Desouki MM, Franklin RB, Costello LC (2006). Mitochondrial aconitase and citrate metabolism in malignant and nonmalignant human prostate tissues. Mol Cancer 5: 14. 10.1186/1476-4598-5-14
- Ternette N, Yang M, Laroyia M, Kitagawa M, O’Flaherty L, Wolhulter K, Igarashi K, Saito K, Kato K, Fischer R, Berquand A, Kessler BM, Lappin T, Frizzell N, Soga T, Adam J, Pollard PJ (2013). Inhibition of mitochondrial aconitase by succination in fumarate hydratase deficiency. Cell Rep 3(3): 689-700. 10.1016/j.celrep.2013.02.013
- Icard P, Poulain L, Lincet H (2012). Understanding the central role of citrate in the metabolism of cancer cells. Biochim Biophys Acta 1825(1): 111-116. 10.1016/j.bbcan.2011.10.007
- Vander Heiden MG, Cantley LC, Thompson CB (2009). Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324(5930): 1029-1033. 10.1126/science.1160809
- Wakil SJ, Stoops JK, Joshi VC (1983). Fatty acid synthesis and its regulation. Annu Rev Biochem 52(537-579. 10.1146/annurev.bi.52.070183.002541
- Li JN, Mahmoud MA, Han WF, Ripple M, Pizer ES (2000). Sterol regulatory element-binding protein-1 participates in the regulation of fatty acid synthase expression in colorectal neoplasia. Exp Cell Res 261(1): 159-165. 10.1006/excr.2000.5054
- Bauer DE, Hatzivassiliou G, Zhao F, Andreadis C, Thompson CB (2005). ATP citrate lyase is an important component of cell growth and transformation. Oncogene 24(41): 6314-6322. 10.1038/sj.onc.1208773
- Zhao D, Li FL, Cheng ZL, Lei QY (2014). Impact of acetylation on tumor metabolism. Mol Cell Oncol 1(3): e963452. 10.4161/23723548.2014.963452
- Cai L, Sutter BM, Li B, Tu BP (2011). Acetyl-CoA induces cell growth and proliferation by promoting the acetylation of histones at growth genes. Mol Cell 42(4): 426-437. 10.1016/j.molcel.2011.05.004
- Spiegel S, Milstien S, Grant S (2012). Endogenous modulators and pharmacological inhibitors of histone deacetylases in cancer therapy. Oncogene 31(5): 537-551. 10.1038/onc.2011.267
- Rius M, Lyko F (2012). Epigenetic cancer therapy: rationales, targets and drugs. Oncogene 31(39): 4257-4265. 10.1038/onc.2011.601
- Cui H, Huang P, Wang Z, Zhang Y, Zhang Z, Xu W, Wang X, Han Y, Guo X (2013). Association of decreased mitochondrial DNA content with the progression of colorectal cancer. BMC Cancer 13: 110. 10.1186/1471-2407-13-110
- Mohideen AM, Dicks E, Parfrey P, Green R, Savas S (2015). Mitochondrial DNA polymorphisms, its copy number change and outcome in colorectal cancer. BMC Res Notes 8: 272. 10.1186/s13104-015-1250-5
- Giorgi EE, Li Y, Caberto CP, Beckman KB, Lum-Jones A, Haiman CA, Le Marchand L, Stram DO, Saxena R, Cheng I (2016). No association between the mitochondrial genome and prostate cancer risk: the multiethnic cohort. Cancer Epidemiol Biomarkers Prev 25(6): 1001-1003. 10.1158/1055-9965.EPI-16-0111
- Goncalves VF (2019). Mitochondrial genetics. Adv Exp Med Biol 1158: 247-255. 10.1007/978-981-13-8367-0_13
- Torroni A, Huoponen K, Francalacci P, Petrozzi M, Morelli L, Scozzari R, Obinu D, Savontaus ML, Wallace DC (1996). Classification of European mtDNAs from an analysis of three European populations. Genetics 144(4): 1835-1850. 8978068
- Dong J, Wong LJ, Mims MP (2018). Mitochondrial inheritance and cancer. Transl Res 202: 24-34. 10.1016/j.trsl.2018.06.004
- Bai RK, Leal SM, Covarrubias D, Liu A, Wong LJ (2007). Mitochondrial genetic background modifies breast cancer risk. Cancer Res 67(10): 4687-4694. 10.1158/0008-5472.CAN-06-3554
- Riley V, Erzurumluoglu AM, Rodriguez S, Bonilla C (2018). Mitochondrial DNA haplogroups and breast cancer risk factors in the Avon longitudinal study of parents and children (ALSPAC). Genes 9(8): 395. 10.3390/genes9080395
- Shen L, Wei J, Chen T, He J, Qu J, He X, Jiang L, Qu Y, Fang H, Chen G, Lu J, Bai Y (2011). Evaluating mitochondrial DNA in patients with breast cancer and benign breast disease. J Cancer Res Clin Oncol 137(4): 669-675. 10.1007/s00432-010-0912-x
- Ma L, Fu Q, Xu B, Zhou H, Gao J, Shao X, Xiong J, Gu Q, Wen S, Li F, Shen L, Chen G, Fang H, Lyu J (2018). Breast cancer-associated mitochondrial DNA haplogroup promotes neoplastic growth via ROS-mediated AKT activation. Int J Cancer 142(9): 1786-1796. 10.1002/ijc.31207
- Nie H, Chen G, He J, Zhang F, Li M, Wang Q, Zhou H, Lyu J, Bai Y (2016). Mitochondrial common deletion is elevated in blood of breast cancer patients mediated by oxidative stress. Mitochondrion 26: 104-112. 10.1016/j.mito.2015.12.001
- Maki J, Robinson K, Reguly B, Alexander J, Wittock R, Aguirre A, Diamandis EP, Escott N, Skehan A, Prowse O, Thayer RE, Froberg MK, Wilson MJ, Maragh S, Jakupciak JP, Wagner PD, Srivastava S, Dakubo GD, Parr RL (2008). Mitochondrial genome deletion aids in the identification of false- and true-negative prostate needle core biopsy specimens. Am J Clin Pathol 129(1): 57-66. 10.1309/UJJTH4HFEPWAQ78Q
- Robinson K, Creed J, Reguly B, Powell C, Wittock R, Klein D, Maggrah A, Klotz L, Parr RL, Dakubo GD (2010). Accurate prediction of repeat prostate biopsy outcomes by a mitochondrial DNA deletion assay. Prostate Cancer Prostatic Dis 13(2): 126-131. 10.1038/pcan.2009.64
- Salas A, Garcia-Magarinos M, Logan I, Bandelt HJ (2014). The saga of the many studies wrongly associating mitochondrial DNA with breast cancer. BMC Cancer 14: 659. 10.1186/1471-2407-14-659
- Brand MD (2010). The sites and topology of mitochondrial superoxide production. Exp Gerontol 45(7-8): 466-472. 10.1016/j.exger.2010.01.003
- Bienert GP, Schjoerring JK, Jahn TP (2006). Membrane transport of hydrogen peroxide. Biochim Biophys Acta 1758(8): 994-1003. 10.1016/j.bbamem.2006.02.015
- Shadel GS, Horvath TL (2015). Mitochondrial ROS signaling in organismal homeostasis. Cell 163(3): 560-569. 10.1016/j.cell.2015.10.001
- Finkel T (2012). Signal transduction by mitochondrial oxidants. J Biol Chem 287(7): 4434-4440. 10.1074/jbc.R111.271999
- Tonks NK (2005). Redox redux: revisiting PTPs and the control of cell signaling. Cell 121(5): 667-670. 10.1016/j.cell.2005.05.016
- Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, Schumacker PT (2000). Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J Biol Chem 275(33): 25130-25138. 10.1074/jbc.M001914200
- Movafagh S, Crook S, Vo K (2015). Regulation of hypoxia-inducible factor-1a by reactive oxygen species: new developments in an old debate. J Cell Biochem 116(5): 696-703. 10.1002/jcb.25074
- Semenza GL, Roth PH, Fang HM, Wang GL (1994). Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J Biol Chem 269(38): 23757-23763. 8089148
- Palazon A, Tyrakis PA, Macias D, Velica P, Rundqvist H, Fitzpatrick S, Vojnovic N, Phan AT, Loman N, Hedenfalk I, Hatschek T, Lovrot J, Foukakis T, Goldrath AW, Bergh J, Johnson RS (2017). An HIF-1alpha/VEGF-A axis in cytotoxic T cells regulates tumor progression. Cancer Cell 32(5): 669-683 e665. 10.1016/j.ccell.2017.10.003
- Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z (2007). Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J 26(7): 1749-1760. 10.1038/sj.emboj.7601623
- Yu L, Wan F, Dutta S, Welsh S, Liu Z, Freundt E, Baehrecke EH, Lenardo M (2006). Autophagic programmed cell death by selective catalase degradation. Proc Natl Acad Sci U S A 103(13): 4952-4957. 10.1073/pnas.0511288103
- Al-Mehdi AB, Pastukh VM, Swiger BM, Reed DJ, Patel MR, Bardwell GC, Pastukh VV, Alexeyev MF, Gillespie MN (2012). Perinuclear mitochondrial clustering creates an oxidant-rich nuclear domain required for hypoxia-induced transcription. Sci Signal 5(231): ra47. 10.1126/scisignal.2002712
- Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M (2006). Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact 160(1): 1-40. 10.1016/j.cbi.2005.12.009
- Nguyen NH, Tran GB, Nguyen CT (2020). Anti-oxidative effects of superoxide dismutase 3 on inflammatory diseases. J Mol Med 98(1): 59-69. 10.1007/s00109-019-01845-2
- Pannala VR, Dash RK (2015). Mechanistic characterization of the thioredoxin system in the removal of hydrogen peroxide. Free Radic Biol Med 78: 42-55. 10.1016/j.freeradbiomed.2014.10.508
- Lu J, Holmgren A (2014). The thioredoxin antioxidant system. Free Radic Biol Med 66: 75-87. 10.1016/j.freeradbiomed.2013.07.036
- Sabharwal SS, Schumacker PT (2014). Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles’ heel? Nat Rev Cancer 14(11): 709-721. 10.1038/nrc3803
- Long JS, Crighton D, O’Prey J, Mackay G, Zheng L, Palmer TM, Gottlieb E, Ryan KM (2013). Extracellular adenosine sensing-a metabolic cell death priming mechanism downstream of p53. Mol Cell 50(3): 394-406. 10.1016/j.molcel.2013.03.016
- Beroud C, Soussi T (2003). The UMD-p53 database: new mutations and analysis tools. Hum Mutat 21(3): 176-181. 10.1002/humu.10187
- Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, Gottlieb E, Vousden KH (2006). TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 126(1): 107-120. 10.1016/j.cell.2006.05.036
- Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, Hurley PJ, Bunz F, Hwang PM (2006). p53 regulates mitochondrial respiration. Science 312(5780): 1650-1653. 10.1126/science.1126863
- Du W, Amarachintha S, Wilson AF, Pang Q (2016). SCO2 mediates oxidative stress-induced glycolysis to oxidative phosphorylation switch in hematopoietic stem cells. Stem Cells 34(4): 960-971. 10.1002/stem.2260
- Hanahan D, Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144(5): 646-674. 10.1016/j.cell.2011.02.013
- Madan E, Gogna R, Bhatt M, Pati U, Kuppusamy P, Mahdi AA (2011). Regulation of glucose metabolism by p53: emerging new roles for the tumor suppressor. Oncotarget 2(12): 948-957. 10.18632/oncotarget.389
- Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB (2005). AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell 18(3): 283-293. 10.1016/j.molcel.2005.03.027
- He G, Zhang YW, Lee JH, Zeng SX, Wang YV, Luo Z, Dong XC, Viollet B, Wahl GM, Lu H (2014). AMP-activated protein kinase induces p53 by phosphorylating MDMX and inhibiting its activity. Mol Cell Biol 34(2): 148-157. 10.1128/MCB.00670-13
- Hardie DG, Alessi DR (2013). LKB1 and AMPK and the cancer-metabolism link – ten years after. BMC Biol 11: 36. 10.1186/1741-7007-11-36
- Liu Y, Zhang Y (2015). CHCHD2 connects mitochondrial metabolism to apoptosis. Mol Cell Oncol 2(4): e1004964. 10.1080/23723556.2015.1004964
- Okada H, Mak TW (2004). Pathways of apoptotic and non-apoptotic death in tumour cells. Nat Rev Cancer 4(8): 592-603. 10.1038/nrc1412
- Orrenius S, Gogvadze V, Zhivotovsky B (2015). Calcium and mitochondria in the regulation of cell death. Biochem Biophys Res Commun 460(1): 72-81. 10.1016/j.bbrc.2015.01.137
- Du K, Montminy M (1998). CREB is a regulatory target for the protein kinase Akt/PKB. J Biol Chem 273(49): 32377-32379. 10.1074/jbc.273.49.32377
- Mathupala SP, Ko YH, Pedersen PL (2006). Hexokinase II: cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene 25(34): 4777-4786. 10.1038/sj.onc.1209603
- Isono T, Chano T, Yonese J, Yuasa T (2016). Therapeutic inhibition of mitochondrial function induces cell death in starvation-resistant renal cell carcinomas. Sci Rep 6: 25669. 10.1038/srep25669
- Lee JG, Shin JH, Shim HS, Lee CY, Kim DJ, Kim YS, Chung KY (2015). Autophagy contributes to the chemo-resistance of non-small cell lung cancer in hypoxic conditions. Respir Res 16: 138. 10.1186/s12931-015-0285-4
- Brisson L, Banski P, Sboarina M, Dethier C, Danhier P, Fontenille MJ, Van Hee VF, Vazeille T, Tardy M, Falces J, Bouzin C, Porporato PE, Frederick R, Michiels C, Copetti T, Sonveaux P (2016). Lactate dehydrogenase B controls lysosome activity and autophagy in cancer. Cancer Cell 30(3): 418-431. 10.1016/j.ccell.2016.08.005
- Tristan C, Shahani N, Sedlak TW, Sawa A (2011). The diverse functions of GAPDH: views from different subcellular compartments. Cell Signal 23(2): 317-323. 10.1016/j.cellsig.2010.08.003
- Chang C, Su H, Zhang D, Wang Y, Shen Q, Liu B, Huang R, Zhou T, Peng C, Wong CC, Shen HM, Lippincott-Schwartz J, Liu W (2015). AMPK-Dependent phosphorylation of GAPDH triggers Sirt1 activation and is necessary for autophagy upon glucose starvation. Mol Cell 60(6): 930-940. 10.1016/j.molcel.2015.10.037
- Sun F, Jiang X, Wang X, Bao Y, Feng G, Liu H, Kou X, Zhu Q, Jiang L, Yang Y (2019). Vincristine ablation of Sirt2 induces cell apoptosis and mitophagy via Hsp70 acetylation in MDA-MB-231 cells. Biochem Pharmacol 162: 142-153. 10.1016/j.bcp.2018.10.021
- Zhao Y, Yang J, Liao W, Liu X, Zhang H, Wang S, Wang D, Feng J, Yu L, Zhu WG (2010). Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat Cell Biol 12(7): 665-675. 10.1038/ncb2069
- Qiao A, Wang K, Yuan Y, Guan Y, Ren X, Li L, Chen X, Li F, Chen AF, Zhou J, Yang JM, Cheng Y (2016). Sirt3-mediated mitophagy protects tumor cells against apoptosis under hypoxia. Oncotarget 7(28): 43390-43400. 10.18632/oncotarget.9717
- Lang A, Anand R, Altinoluk-Hambuchen S, Ezzahoini H, Stefanski A, Iram A, Bergmann L, Urbach J, Bohler P, Hansel J, Franke M, Stuhler K, Krutmann J, Scheller J, Stork B, Reichert AS, Piekorz RP (2017). SIRT4 interacts with OPA1 and regulates mitochondrial quality control and mitophagy. Aging 9(10): 2163-2189. 10.18632/aging.101307
- Polletta L, Vernucci E, Carnevale I, Arcangeli T, Rotili D, Palmerio S, Steegborn C, Nowak T, Schutkowski M, Pellegrini L, Sansone L, Villanova L, Runci A, Pucci B, Morgante E, Fini M, Mai A, Russo MA, Tafani M (2015). SIRT5 regulation of ammonia-induced autophagy and mitophagy. Autophagy 11(2): 253-270. 10.1080/15548627.2015.1009778
- Huang N, Liu Z, Zhu J, Cui Z, Li Y, Yu Y, Sun F, Pan Q, Yang Q (2017). Sirtuin 6 plays an oncogenic role and induces cell autophagy in esophageal cancer cells. Tumour Biol 39(6): 1010428317708532. 10.1177/1010428317708532
- Ding M, Jiang CY, Zhang Y, Zhao J, Han BM, Xia SJ (2020). SIRT7 depletion inhibits cell proliferation and androgen-induced autophagy by suppressing the AR signaling in prostate cancer. J Exp Clin Cancer Res 39(1): 28. 10.1186/s13046-019-1516-1
- Mao K, Wang K, Liu X, Klionsky DJ (2013). The scaffold protein Atg11 recruits fission machinery to drive selective mitochondria degradation by autophagy. Dev Cell 26(1): 9-18. 10.1016/j.devcel.2013.05.024
- Chan DC (2012). Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet 46: 265-287. 10.1146/annurev-genet-110410-132529
- Youle RJ, van der Bliek AM (2012). Mitochondrial fission, fusion, and stress. Science 337(6098): 1062-1065. 10.1126/science.1219855
- Burte F, Carelli V, Chinnery PF, Yu-Wai-Man P (2015). Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat Rev Neurol 11(1): 11-24. 10.1038/nrneurol.2014.228
- Suen DF, Norris KL, Youle RJ (2008). Mitochondrial dynamics and apoptosis. Genes Dev 22(12): 1577-1590. 10.1101/gad.1658508
- Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK (2011). ER tubules mark sites of mitochondrial division. Science 334(6054): 358-362. 10.1126/science.1207385
- Manor U, Bartholomew S, Golani G, Christenson E, Kozlov M, Higgs H, Spudich J, Lippincott-Schwartz J (2015). A mitochondria-anchored isoform of the actin-nucleating spire protein regulates mitochondrial division. Elife 4: e08828. 10.7554/eLife.08828
- Steffen J, Koehler CM (2018). ER-mitochondria contacts: actin dynamics at the ER control mitochondrial fission via calcium release. J Cell Biol 217(1): 15-17. 10.1083/jcb.201711075
- Lee H, Yoon Y (2016). Mitochondrial fission and fusion. Biochem Soc Trans 44(6): 1725-1735. 10.1042/BST20160129
- Yoon Y, Krueger EW, Oswald BJ, McNiven MA (2003). The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol Cell Biol 23(15): 5409-5420. 10.1128/mcb.23.15.5409-5420.2003
- Gandre-Babbe S, van der Bliek AM (2008). The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol Biol Cell 19(6): 2402-2412. 10.1091/mbc.E07-12-1287
- Xie Q, Wu Q, Horbinski CM, Flavahan WA, Yang K, Zhou W, Dombrowski SM, Huang Z, Fang X, Shi Y, Ferguson AN, Kashatus DF, Bao S, Rich JN (2015). Mitochondrial control by DRP1 in brain tumor initiating cells. Nat Neurosci 18(4): 501-510. 10.1038/nn.3960
- Ferreira-da-Silva A, Valacca C, Rios E, Populo H, Soares P, Sobrinho-Simoes M, Scorrano L, Maximo V, Campello S (2015). Mitochondrial dynamics protein Drp1 is overexpressed in oncocytic thyroid tumors and regulates cancer cell migration. PLoS One 10(3): e0122308. 10.1371/journal.pone.0122308
- Rehman J, Zhang HJ, Toth PT, Zhang Y, Marsboom G, Hong Z, Salgia R, Husain AN, Wietholt C, Archer SL (2012). Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. FASEB J 26(5): 2175-2186. 10.1096/fj.11-196543
- Serasinghe MN, Wieder SY, Renault TT, Elkholi R, Asciolla JJ, Yao JL, Jabado O, Hoehn K, Kageyama Y, Sesaki H, Chipuk JE (2015). Mitochondrial division is requisite to RAS-induced transformation and targeted by oncogenic MAPK pathway inhibitors. Mol Cell 57(3): 521-536. 10.1016/j.molcel.2015.01.003
- Zhao J, Zhang J, Yu M, Xie Y, Huang Y, Wolff DW, Abel PW, Tu Y (2013). Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene 32(40): 4814-4824. 10.1038/onc.2012.494
- Kashatus JA, Nascimento A, Myers LJ, Sher A, Byrne FL, Hoehn KL, Counter CM, Kashatus DF (2015). Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol Cell 57(3): 537-551. 10.1016/j.molcel.2015.01.002
- Kitamura S, Yanagi T, Imafuku K, Hata H, Abe R, Shimizu H (2017). Drp1 regulates mitochondrial morphology and cell proliferation in cutaneous squamous cell carcinoma. J Dermatol Sci 88(3): 298-307. 10.1016/j.jdermsci.2017.08.004
- Tanwar DK, Parker DJ, Gupta P, Spurlock B, Alvarez RD, Basu MK, Mitra K (2016). Crosstalk between the mitochondrial fission protein, Drp1, and the cell cycle is identified across various cancer types and can impact survival of epithelial ovarian cancer patients. Oncotarget 7(37): 60021-60037. 10.18632/oncotarget.11047
- Liesa M, Shirihai OS (2013). Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab 17(4): 491-506. 10.1016/j.cmet.2013.03.002
- Rambold AS, Pearce EL (2018). Mitochondrial dynamics at the interface of immune cell Metabolism and function. Trends Immunol 39(1): 6-18. 10.1016/j.it.2017.08.006
- Li J, Donath S, Li Y, Qin D, Prabhakar BS, Li P (2010). miR-30 regulates mitochondrial fission through targeting p53 and the dynamin-related protein-1 pathway. PLoS Genet 6(1): e1000795. 10.1371/journal.pgen.1000795
- Sancho P, Barneda D, Heeschen C (2016). Hallmarks of cancer stem cell metabolism. Br J Cancer 114(12): 1305-1312. 10.1038/bjc.2016.152
- Corbet C (2017). Stem cell metabolism in cancer and healthy tissues: pyruvate in the limelight. Front Pharmacol 8: 958. 10.3389/fphar.2017.00958
- Cuyas E, Verdura S, Folguera-Blasco N, Bastidas-Velez C, Martin AG, Alarcon T, Menendez JA (2018). Mitostemness. Cell Cycle 17(8): 918-926. 10.1080/15384101.2018.1467679
- Peiris-Pages M, Bonuccelli G, Sotgia F, Lisanti MP (2018). Mitochondrial fission as a driver of stemness in tumor cells: mDIVI1 inhibits mitochondrial function, cell migration and cancer stem cell (CSC) signalling. Oncotarget 9(17): 13254-13275. 10.18632/oncotarget.24285
- Civenni G, Bosotti R, Timpanaro A, Vazquez R, Merulla J, Pandit S, Rossi S, Albino D, Allegrini S, Mitra A, Mapelli SN, Vierling L, Giurdanella M, Marchetti M, Paganoni A, Rinaldi A, Losa M, Mira-Cato E, D’Antuono R, Morone D, Rezai K, D’Ambrosio G, Ouafik L, Mackenzie S, Riveiro ME, Cvitkovic E, Carbone GM, Catapano CV (2019). Epigenetic control of mitochondrial fission enables self-renewal of stem-like tumor cells in human prostate cancer. Cell Metab 30(2): 303-318 e306. 10.1016/j.cmet.2019.05.004
- Katajisto P, Dohla J, Chaffer CL, Pentinmikko N, Marjanovic N, Iqbal S, Zoncu R, Chen W, Weinberg RA, Sabatini DM (2015). Stem cells. Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science 348(6232): 340-343. 10.1126/science.1260384
- Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC (2003). Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol 160(2): 189-200. 10.1083/jcb.200211046
- Delettre C, Lenaers G, Griffoin JM, Gigarel N, Lorenzo C, Belenguer P, Pelloquin L, Grosgeorge J, Turc-Carel C, Perret E, Astarie-Dequeker C, Lasquellec L, Arnaud B, Ducommun B, Kaplan J, Hamel CP (2000). Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet 26(2): 207-210. 10.1038/79936
- Guillery O, Malka F, Landes T, Guillou E, Blackstone C, Lombes A, Belenguer P, Arnoult D, Rojo M (2008). Metalloprotease-mediated OPA1 processing is modulated by the mitochondrial membrane potential. Biol Cell 100(5): 315-325. 10.1042/BC20070110
- Saita S, Ishihara T, Maeda M, Iemura S, Natsume T, Mihara K, Ishihara N (2016). Distinct types of protease systems are involved in homeostasis regulation of mitochondrial morphology via balanced fusion and fission. Genes Cells 21(5): 408-424. 10.1111/gtc.12351
- Koshiba T, Detmer SA, Kaiser JT, Chen H, McCaffery JM, Chan DC (2004). Structural basis of mitochondrial tethering by mitofusin complexes. Science 305(5685): 858-862. 10.1126/science.1099793
- de Brito OM, Scorrano L (2008). Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456(7222): 605-610. 10.1038/nature07534
- Cipolat S, Martins de Brito O, Dal Zilio B, Scorrano L (2004). OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci U S A 101(45): 15927-15932. 10.1073/pnas.0407043101
- Malhotra A, Dey A, Prasad N, Kenney AM (2016). Sonic hedgehog signaling drives mitochondrial fragmentation by suppressing mitofusins in cerebellar granule neuron precursors and medulloblastoma. Mol Cancer Res 14(1): 114-124. 10.1158/1541-7786.MCR-15-0278
- Mitra K, Wunder C, Roysam B, Lin G, Lippincott-Schwartz J (2009). A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc Natl Acad Sci U S A 106(29): 11960-11965. 10.1073/pnas.0904875106
- Kuznetsov AV, Margreiter R (2009). Heterogeneity of mitochondria and mitochondrial function within cells as another level of mitochondrial complexity. Int J Mol Sci 10(4): 1911-1929. 10.3390/ijms10041911
- Schwarz TL (2013). Mitochondrial trafficking in neurons. Cold Spring Harb Perspect Biol 5(6): a011304 10.1101/cshperspect.a011304
- Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL, Youle RJ (2001). The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell 1(4): 515-525. 10.1016/s1534-5807(01)00055-7
- Nakada K, Inoue K, Ono T, Isobe K, Ogura A, Goto YI, Nonaka I, Hayashi JI (2001). Inter-mitochondrial complementation: mitochondria-specific system preventing mice from expression of disease phenotypes by mutant mtDNA. Nat Med 7(8): 934-940. 10.1038/90976
- Lemasters JJ (2005). Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res 8(1): 3-5. 10.1089/rej.2005.8.3
- Vara-Perez M, Felipe-Abrio B, Agostinis P (2019). Mitophagy in cancer: a tale of adaptation. Cells 8(5): 493. 10.3390/cells8050493
- Jin SM, Youle RJ (2012). PINK1- and Parkin-mediated mitophagy at a glance. J Cell Sci 125(Pt 4): 795-799. 10.1242/jcs.093849
- Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cecconi F, Choi AM, Chu CT, Codogno P, Colombo MI, Cuervo AM, Debnath J, Deretic V, Dikic I, Eskelinen EL, Fimia GM, Fulda S, Gewirtz DA, Green DR, Hansen M, Harper JW, Jaattela M, Johansen T, Juhasz G, Kimmelman AC, Kraft C, Ktistakis NT, Kumar S, Levine B, Lopez-Otin C, et al. (2017). Molecular definitions of autophagy and related processes. EMBO J 36(13): 1811-1836. 10.15252/embj.201796697
- Pickrell AM, Youle RJ (2015). The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 85(2): 257-273. 10.1016/j.neuron.2014.12.007
- Chen G, Kroemer G, Kepp O (2020). Mitophagy: an emerging role in aging and age-associated diseases. Front Cell Dev Biol 8: 200. 10.3389/fcell.2020.00200
- Greene AW, Grenier K, Aguileta MA, Muise S, Farazifard R, Haque ME, McBride HM, Park DS, Fon EA (2012). Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep 13(4): 378-385. 10.1038/embor.2012.14
- Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, Youle RJ (2014). PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol 205(2): 143-153. 10.1083/jcb.201402104
- Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T, Endo T, Fon EA, Trempe JF, Saeki Y, Tanaka K, Matsuda N (2014). Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510(7503): 162-166. 10.1038/nature13392
- Kondapalli C, Kazlauskaite A, Zhang N, Woodroof HI, Campbell DG, Gourlay R, Burchell L, Walden H, Macartney TJ, Deak M, Knebel A, Alessi DR, Muqit MM (2012). PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol 2(5): 120080. 10.1098/rsob.120080
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392(6676): 605-608. 10.1038/33416
- Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AH, Taanman JW (2010). Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet 19(24): 4861-4870. 10.1093/hmg/ddq419
- Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W (2010). PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 12(2): 119-131. 10.1038/ncb2012
- Yoshii SR, Kishi C, Ishihara N, Mizushima N (2011). Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J Biol Chem 286(22): 19630-19640. 10.1074/jbc.M110.209338
- Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, Rice S, Steen J, LaVoie MJ, Schwarz TL (2011). PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 147(4): 893-906. 10.1016/j.cell.2011.10.018
- Tanida I, Ueno T, Kominami E (2008). LC3 and autophagy. Methods Mol Biol 445: 77-88. 10.1007/978-1-59745-157-4_4
- Soubannier V, McLelland GL, Zunino R, Braschi E, Rippstein P, Fon EA, McBride HM (2012). A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr Biol 22(2): 135-141. 10.1016/j.cub.2011.11.057
- Bernardini JP, Lazarou M, Dewson G (2017). Parkin and mitophagy in cancer. Oncogene 36(10): 1315-1327. 10.1038/onc.2016.302
- O’Flanagan CH, O’Neill C (2014). PINK1 signalling in cancer biology. Biochim Biophys Acta 1846(2): 590-598. 10.1016/j.bbcan.2014.10.006
- Zhang H, Gao P, Fukuda R, Kumar G, Krishnamachary B, Zeller KI, Dang CV, Semenza GL (2007). HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell 11(5): 407-420. 10.1016/j.ccr.2007.04.001
- Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, Lippincott-Schwartz J (2010). Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 141(4): 656-667. 10.1016/j.cell.2010.04.009
- Yan C, Li TS (2018). Dual role of mitophagy in cancer drug resistance. Anticancer Res 38(2): 617-621. 10.21873/anticanres.12266
- Grasso D, Medeiros HCD, Zampieri LX, Bol V, Danhier P, van Gisbergen MW, Bouzin C, Brusa D, Gregoire V, Smeets H, Stassen APM, Dubois LJ, Lambin P, Dutreix M, Sonveaux P (2020). Fitter mitochondria are associated with radioresistance in human head and neck SQD9 cancer cells. Front Pharmacol 11: 263. 10.3389/fphar.2020.00263
- Srivastava S (2017). The Mitochondrial Basis of Aging and Age-Related Disorders. Genes 8(12): 398. 10.3390/genes8120398
- Herst PM, Dawson RH, Berridge MV (2018). Intercellular communication in tumor biology: a role for mitochondrial transfer. Front Oncol 8: 344. 10.3389/fonc.2018.00344
- Tan AS, Baty JW, Dong LF, Bezawork-Geleta A, Endaya B, Goodwin J, Bajzikova M, Kovarova J, Peterka M, Yan B, Pesdar EA, Sobol M, Filimonenko A, Stuart S, Vondrusova M, Kluckova K, Sachaphibulkij K, Rohlena J, Hozak P, Truksa J, Eccles D, Haupt LM, Griffiths LR, Neuzil J, Berridge MV (2015). Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab 21(1): 81-94. 10.1016/j.cmet.2014.12.003
- Dong LF, Kovarova J, Bajzikova M, Bezawork-Geleta A, Svec D, Endaya B, Sachaphibulkij K, Coelho AR, Sebkova N, Ruzickova A, Tan AS, Kluckova K, Judasova K, Zamecnikova K, Rychtarcikova Z, Gopalan V, Andera L, Sobol M, Yan B, Pattnaik B, Bhatraju N, Truksa J, Stopka P, Hozak P, Lam AK, Sedlacek R, Oliveira PJ, Kubista M, Agrawal A, Dvorakova-Hortova K, et al. (2017). Horizontal transfer of whole mitochondria restores tumorigenic potential in mitochondrial DNA-deficient cancer cells. Elife 6: e22187. 10.7554/eLife.22187
- Marlein CR, Zaitseva L, Piddock RE, Robinson SD, Edwards DR, Shafat MS, Zhou Z, Lawes M, Bowles KM, Rushworth SA (2017). NADPH oxidase-2 derived superoxide drives mitochondrial transfer from bone marrow stromal cells to leukemic blasts. Blood 130(14): 1649-1660. 10.1182/blood-2017-03-772939
- Vignais ML, Caicedo A, Brondello JM, Jorgensen C (2017). Cell connections by tunneling nanotubes: effects of mitochondrial trafficking on target cell metabolism, homeostasis, and response to therapy. Stem Cells Int 2017: 6917941. 10.1155/2017/6917941
- Lu J, Zheng X, Li F, Yu Y, Chen Z, Liu Z, Wang Z, Xu H, Yang W (2017). Tunneling nanotubes promote intercellular mitochondria transfer followed by increased invasiveness in bladder cancer cells. Oncotarget 8(9): 15539-15552. 10.18632/oncotarget.14695
- Ishikawa K, Takenaga K, Akimoto M, Koshikawa N, Yamaguchi A, Imanishi H, Nakada K, Honma Y, Hayashi J (2008). ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 320(5876): 661-664. 10.1126/science.1156906
- Pasquier J, Guerrouahen BS, Al Thawadi H, Ghiabi P, Maleki M, Abu-Kaoud N, Jacob A, Mirshahi M, Galas L, Rafii S, Le Foll F, Rafii A (2013). Preferential transfer of mitochondria from endothelial to cancer cells through tunneling nanotubes modulates chemoresistance. J Transl Med 11: 94. 10.1186/1479-5876-11-94
- Maycotte P, Marin-Hernandez A, Goyri-Aguirre M, Anaya-Ruiz M, Reyes-Leyva J, Cortes-Hernandez P (2017). Mitochondrial dynamics and cancer. Tumour Biol 39(5): 1010428317698391. 10.1177/1010428317698391
- Rossignol R, Gilkerson R, Aggeler R, Yamagata K, Remington SJ, Capaldi RA (2004). Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res 64(3): 985-993. 10.1158/0008-5472.can-03-1101
- Kim H, Scimia MC, Wilkinson D, Trelles RD, Wood MR, Bowtell D, Dillin A, Mercola M, Ronai ZA (2011). Fine-tuning of Drp1/Fis1 availability by AKAP121/Siah2 regulates mitochondrial adaptation to hypoxia. Mol Cell 44(4): 532-544. 10.1016/j.molcel.2011.08.045
- Wai T, Langer T (2016). Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol Metab 27(2): 105-117. 10.1016/j.tem.2015.12.001
- Molina AJ, Wikstrom JD, Stiles L, Las G, Mohamed H, Elorza A, Walzer G, Twig G, Katz S, Corkey BE, Shirihai OS (2009). Mitochondrial networking protects beta-cells from nutrient-induced apoptosis. Diabetes 58(10): 2303-2315. 10.2337/db07-1781
- Rambold AS, Cohen S, Lippincott-Schwartz J (2015). Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev Cell 32(6): 678-692. 10.1016/j.devcel.2015.01.029
- Gomes LC, Di Benedetto G, Scorrano L (2011). During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol 13(5): 589-598. 10.1038/ncb2220
- Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J (2011). Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci U S A 108(25): 10190-10195. 10.1073/pnas.1107402108
- Zhang D, Li J, Wang F, Hu J, Wang S, Sun Y (2014). 2-Deoxy-D-glucose targeting of glucose metabolism in cancer cells as a potential therapy. Cancer letters 355(2): 176-183. 10.1016/j.canlet.2014.09.003
- Merchan JR, Kovacs K, Railsback JW, Kurtoglu M, Jing Y, Pina Y, Gao N, Murray TG, Lehrman MA, Lampidis TJ (2010). Antiangiogenic activity of 2-deoxy-D-glucose. PLoS One 5(10): e13699. 10.1371/journal.pone.0013699
- Park GB, Chung YH, Kim D (2017). 2-Deoxy-D-glucose suppresses the migration and reverses the drug resistance of colon cancer cells through ADAM expression regulation. Anticancer Drugs 28(4): 410-420. 10.1097/CAD.0000000000000472
- Leung HJ, Duran EM, Kurtoglu M, Andreansky S, Lampidis TJ, Mesri EA (2012). Activation of the unfolded protein response by 2-deoxy-D-glucose inhibits Kaposi’s sarcoma-associated herpesvirus replication and gene expression. Antimicrob Agents Chemother 56(11): 5794-5803. 10.1128/AAC.01126-12
- Xi H, Kurtoglu M, Liu H, Wangpaichitr M, You M, Liu X, Savaraj N, Lampidis TJ (2011). 2-Deoxy-D-glucose activates autophagy via endoplasmic reticulum stress rather than ATP depletion. Cancer Chemother Pharmacol 67(4): 899-910. 10.1007/s00280-010-1391-0
- DiPaola RS, Dvorzhinski D, Thalasila A, Garikapaty V, Doram D, May M, Bray K, Mathew R, Beaudoin B, Karp C, Stein M, Foran DJ, White E (2008). Therapeutic starvation and autophagy in prostate cancer: a new paradigm for targeting metabolism in cancer therapy. Prostate 68(16): 1743-1752. 10.1002/pros.20837
- Wu H, Zhu H, Liu DX, Niu TK, Ren X, Patel R, Hait WN, Yang JM (2009). Silencing of elongation factor-2 kinase potentiates the effect of 2-deoxy-D-glucose against human glioma cells through blunting of autophagy. Cancer Res 69(6): 2453-2460. 10.1158/0008-5472.CAN-08-2872
- Raez LE, Papadopoulos K, Ricart AD, Chiorean EG, Dipaola RS, Stein MN, Rocha Lima CM, Schlesselman JJ, Tolba K, Langmuir VK, Kroll S, Jung DT, Kurtoglu M, Rosenblatt J, Lampidis TJ (2013). A phase I dose-escalation trial of 2-deoxy-D-glucose alone or combined with docetaxel in patients with advanced solid tumors. Cancer Chemother Pharmacol 71(2): 523-530. 10.1007/s00280-012-2045-1
- Zhang L, Su J, Xie Q, Zeng L, Wang Y, Yi D, Yu Y, Liu S, Li S, Xu Y (2015). 2-Deoxy-d-glucose sensitizes human ovarian cancer cells to cisplatin by increasing ER stress and decreasing ATP stores in acidic vesicles. J Biochem Mol Toxicol 29(12): 572-578. 10.1002/jbt.21730
- Jalota A, Kumar M, Das BC, Yadav AK, Chosdol K, Sinha S (2016). Synergistic increase in efficacy of a combination of 2-deoxy-D-glucose and cisplatin in normoxia and hypoxia: switch from autophagy to apoptosis. Tumour Biol 37(9): 12347-12358. 10.1007/s13277-016-5089-8
- Mustafa EH, Mahmoud HT, Al-Hudhud MY, Abdalla MY, Ahmad IM, Yasin SR, Elkarmi AZ, Tahtamouni LH (2015). 2-Deoxy-D-glucose synergizes with doxorubicin or L-buthionine sulfoximine to reduce adhesion and migration of breast cancer cells. Asian Pac J Cancer Prev 16(8): 3213-3222. 10.7314/apjcp.2015.16.8.3213
- Wang SY, Wei YH, Shieh DB, Lin LL, Cheng SP, Wang PW, Chuang JH (2015). 2-Deoxy-d-glucose can complement doxorubicin and sorafenib to suppress the growth of papillary thyroid carcinoma cells. PLoS One 10(7): e0130959. 10.1371/journal.pone.0130959
- Singh D, Banerji AK, Dwarakanath BS, Tripathi RP, Gupta JP, Mathew TL, Ravindranath T, Jain V (2005). Optimizing cancer radiotherapy with 2-deoxy-d-glucose dose escalation studies in patients with glioblastoma multiforme. Strahlenther Onkol 181(8): 507-514. 10.1007/s00066-005-1320-z
- Venkataramanaa NK, Venkatesh PK, Dwarakanath BS, Vani S (2013). Protective effect on normal brain tissue during a combinational therapy of 2-deoxy-d-glucose and hypofractionated irradiation in malignant gliomas. Asian J Neurosurg 8(1): 9-14. 10.4103/1793-5482.110274
- Ko YH, Pedersen PL, Geschwind JF (2001). Glucose catabolism in the rabbit VX2 tumor model for liver cancer: characterization and targeting hexokinase. Cancer Lett 173(1): 83-91. 10.1016/s0304-3835(01)00667-x
- Ganapathy-Kanniappan S, Geschwind JF, Kunjithapatham R, Buijs M, Vossen JA, Tchernyshyov I, Cole RN, Syed LH, Rao PP, Ota S, Vali M (2009). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is pyruvylated during 3-bromopyruvate mediated cancer cell death. Anticancer Res 29(12): 4909-4918. doi
- Lis P, Dylag M, Niedzwiecka K, Ko YH, Pedersen PL, Goffeau A, Ulaszewski S (2016). The HK2 dependent “Warburg Effect” and mitochondrial oxidative phosphorylation in cancer: targets for effective therapy with 3-bromopyruvate. Molecules 21(12). 10.3390/molecules21121730
- Ko YH, Verhoeven HA, Lee MJ, Corbin DJ, Vogl TJ, Pedersen PL (2012). A translational study “case report” on the small molecule “energy blocker” 3-bromopyruvate (3BP) as a potent anticancer agent: from bench side to bedside. J Bioenerg Biomembr 44(1): 163-170. 10.1007/s10863-012-9417-4
- El Sayed SM, Mohamed WG, Seddik MA, Ahmed AS, Mahmoud AG, Amer WH, Helmy Nabo MM, Hamed AR, Ahmed NS, Abd-Allah AA (2014). Safety and outcome of treatment of metastatic melanoma using 3-bromopyruvate: a concise literature review and case study. Chin J Cancer 33(7): 356-364. 10.5732/cjc.013.10111
- El Sayed SM (2018). Enhancing anticancer effects, decreasing risks and solving practical problems facing 3-bromopyruvate in clinical oncology: 10 years of research experience. Int J Nanomedicine 13: 4699-4709. 10.2147/IJN.S170564
- Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, Kang Y, Fleming JB, Bardeesy N, Asara JM, Haigis MC, DePinho RA, Cantley LC, Kimmelman AC (2013). Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496(7443): 101-105. 10.1038/nature12040
- Biancur DE, Paulo JA, Malachowska B, Quiles Del Rey M, Sousa CM, Wang X, Sohn ASW, Chu GC, Gygi SP, Harper JW, Fendler W, Mancias JD, Kimmelman AC (2017). Compensatory metabolic networks in pancreatic cancers upon perturbation of glutamine metabolism. Nature Commun 8: 15965. 10.1038/ncomms15965
- Lampa M, Arlt H, He T, Ospina B, Reeves J, Zhang B, Murtie J, Deng G, Barberis C, Hoffmann D, Cheng H, Pollard J, Winter C, Richon V, Garcia-Escheverria C, Adrian F, Wiederschain D, Srinivasan L (2017). Glutaminase is essential for the growth of triple-negative breast cancer cells with a deregulated glutamine metabolism pathway and its suppression synergizes with mTOR inhibition. PLoS One 12(9): e0185092. 10.1371/journal.pone.0185092
- Casals N, Zammit V, Herrero L, Fado R, Rodriguez-Rodriguez R, Serra D (2016). Carnitine palmitoyltransferase 1C: from cognition to cancer. Prog Lipid Res 61: 134-148. 10.1016/j.plipres.2015.11.004
- Dheeraj A, Agarwal C, Schlaepfer IR, Raben D, Singh R, Agarwal R, Deep G (2018). A novel approach to target hypoxic cancer cells via combining beta-oxidation inhibitor etomoxir with radiation. Hypoxia 6: 23-33. 10.2147/HP.S163115
- Fukawa T, Yan-Jiang BC, Min-Wen JC, Jun-Hao ET, Huang D, Qian CN, Ong P, Li Z, Chen S, Mak SY, Lim WJ, Kanayama HO, Mohan RE, Wang RR, Lai JH, Chua C, Ong HS, Tan KK, Ho YS, Tan IB, Teh BT, Shyh-Chang N (2016). Excessive fatty acid oxidation induces muscle atrophy in cancer cachexia. Nat Med 22(6): 666-671. 10.1038/nm.4093
- Hubinger A, Weikert G, Wolf HP, Gries FA (1992). The effect of etomoxir on insulin sensitivity in type 2 diabetic patients. Horm Metab Res 24(3): 115-118. 10.1055/s-2007-1003271
- Schmidt-Schweda S, Holubarsch C (2000). First clinical trial with etomoxir in patients with chronic congestive heart failure. Clin Sci (Lond) 99(1): 27-35. 10887055
- Ashton TM, McKenna WG, Kunz-Schughart LA, Higgins GS (2018). Oxidative phosphorylation as an emerging target in cancer therapy. Clin Cancer Res 24(11): 2482-2490. 10.1158/1078-0432.Ccr-17-3070
- Ju R, Guo L, Li J, Zhu L, Yu X, Chen C, Chen W, Ye C, Zhang D (2016). Carboxyamidotriazole inhibits oxidative phosphorylation in cancer cells and exerts synergistic anti-cancer effect with glycolysis inhibition. Cancer Lett 370(2): 232-241. 10.1016/j.canlet.2015.10.025
- Wilk A, Wyczechowska D, Zapata A, Dean M, Mullinax J, Marrero L, Parsons C, Peruzzi F, Culicchia F, Ochoa A, Grabacka M, Reiss K (2015). Molecular mechanisms of fenofibrate-induced metabolic catastrophe and glioblastoma cell death. Mol Cell Biol 35(1): 182-198. 10.1128/mcb.00562-14
- Suissa S, Azoulay L (2014). Metformin and Cancer: Mounting Evidence Against an Association. Diabetes Care 37(7): 1786. 10.2337/dc14-0500
- Ben Sahra I, Laurent K, Loubat A, Giorgetti-Peraldi S, Colosetti P, Auberger P, Tanti JF, Le Marchand-Brustel Y, Bost F (2008). The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene 27(25): 3576-3586. 10.1038/sj.onc.1211024
- Zakikhani M, Dowling R, Fantus IG, Sonenberg N, Pollak M (2006). Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res 66(21): 10269-10273. 10.1158/0008-5472.Can-06-1500
- Dirat B, Ader I, Golzio M, Massa F, Mettouchi A, Laurent K, Larbret F, Malavaud B, Cormont M, Lemichez E, Cuvillier O, Tanti JF, Bost F (2015). Inhibition of the GTPase Rac1 mediates the antimigratory effects of metformin in prostate cancer cells. Mol Cancer Ther 14(2): 586-596. 10.1158/1535-7163.Mct-14-0102
- Anisimov VN, Berstein LM, Egormin PA, Piskunova TS, Popovich IG, Zabezhinski MA, Kovalenko IG, Poroshina TE, Semenchenko AV, Provinciali M, Re F, Franceschi C (2005). Effect of metformin on life span and on the development of spontaneous mammary tumors in HER-2/neu transgenic mice. Exp Gerontol 40(8-9): 685-693. 10.1016/j.exger.2005.07.007
- Huang X, Wullschleger S, Shpiro N, McGuire VA, Sakamoto K, Woods YL, McBurnie W, Fleming S, Alessi DR (2008). Important role of the LKB1-AMPK pathway in suppressing tumorigenesis in PTEN-deficient mice. Biochem J 412(2): 211-221. 10.1042/bj20080557
- Cerezo M, Tichet M, Abbe P, Ohanna M, Lehraiki A, Rouaud F, Allegra M, Giacchero D, Bahadoran P, Bertolotto C, Tartare-Deckert S, Ballotti R, Rocchi S (2013). Metformin blocks melanoma invasion and metastasis development in AMPK/p53-dependent manner. Mol Cancer Ther 12(8): 1605-1615. 10.1158/1535-7163.Mct-12-1226-t
- Buzzai M, Jones RG, Amaravadi RK, Lum JJ, DeBerardinis RJ, Zhao F, Viollet B, Thompson CB (2007). Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res 67(14): 6745-6752. 10.1158/0008-5472.Can-06-4447
- El-Mir MY, Nogueira V, Fontaine E, Averet N, Rigoulet M, Leverve X (2000). Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem 275(1): 223-228. 10.1074/jbc.275.1.223
- Owen MR, Doran E, Halestrap AP (2000). Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J 348(Pt 3): 607-614. 10.1042/0264-6021:3480607
- Bridges HR, Jones AJY, Pollak MN, Hirst J (2014). Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem J 462(3): 475-487. 10.1042/BJ20140620
- Saraei P, Asadi I, Kakar MA, Moradi-Kor N (2019). The beneficial effects of metformin on cancer prevention and therapy: a comprehensive review of recent advances. Cancer Manag Res 11: 3295-3313. 10.2147/CMAR.S200059
- Coyle C, Cafferty FH, Vale C, Langley RE (2016). Metformin as an adjuvant treatment for cancer: a systematic review and meta-analysis. Ann Oncol 27(12): 2184-2195. 10.1093/annonc/mdw410
- Chappie TA, Humphrey JM, Allen MP, Estep KG, Fox CB, Lebel LA, Liras S, Marr ES, Menniti FS, Pandit J, Schmidt CJ, Tu M, Williams RD, Yang FV (2007). Discovery of a Series of 6,7-Dimethoxy-4-pyrrolidylquinazoline PDE10A Inhibitors. J Med Chem 50(2): 182-185. 10.1021/jm060653b
- Benej M, Hong X, Vibhute S, Scott S, Wu J, Graves E, Le QT, Koong AC, Giaccia AJ, Yu B, Chen CS, Papandreou I, Denko NC (2018). Papaverine and its derivatives radiosensitize solid tumors by inhibiting mitochondrial metabolism. Proc Natl Acad Sci U S A 115(42): 10756-10761. 10.1073/pnas.1808945115
- Shimizu T, Ohta Y, Ozawa H, Matsushima H, Takeda K (2000). Papaverine combined with prostaglandin E2 synergistically induces neuron-like morphological changes and decrease of malignancy in human prostatic cancer LNCaP cells. Anticancer Res 20(2a): 761-767. 10810351
- Sajadian S, Vatankhah M, Majdzadeh M, Kouhsari SM, Ghahremani MH, Ostad SN (2015). Cell cycle arrest and apoptogenic properties of opium alkaloids noscapine and papaverine on breast cancer stem cells. Toxicol Mech Methods 25(5): 388-395. 10.3109/15376516.2015.1045656
- Noureini SK, Wink M (2014). Antiproliferative effect of the isoquinoline alkaloid papaverine in hepatocarcinoma HepG-2 cells–inhibition of telomerase and induction of senescence. Molecules 19(8): 11846-11859. 10.3390/molecules190811846
- Inada M, Shindo M, Kobayashi K, Sato A, Yamamoto Y, Akasaki Y, Ichimura K, Tanuma S-i (2019). Anticancer effects of a non-narcotic opium alkaloid medicine, papaverine, in human glioblastoma cells. PLOS ONE 14(5): e0216358. 10.1371/journal.pone.0216358
- Martinez-Outschoorn UE, Peiris-Pages M, Pestell RG, Sotgia F, Lisanti MP (2017). Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol 14(1): 11-31. 10.1038/nrclinonc.2016.60
- Guo L, Shestov AA, Worth AJ, Nath K, Nelson DS, Leeper DB, Glickson JD, Blair IA (2016). Inhibition of mitochondrial complex II by the anticancer agent lonidamine. J Biol Chem 291(1): 42-57. 10.1074/jbc.M115.697516
- Nixon GL, Moss DM, Shone AE, Lalloo DG, Fisher N, O’Neill PM, Ward SA, Biagini GA (2013). Antimalarial pharmacology and therapeutics of atovaquone. J Antimicrob Chemother 68(5): 977-985. 10.1093/jac/dks504
- Ashton TM, Fokas E, Kunz-Schughart LA, Folkes LK, Anbalagan S, Huether M, Kelly CJ, Pirovano G, Buffa FM, Hammond EM, Stratford M, Muschel RJ, Higgins GS, McKenna WG (2016). The anti-malarial atovaquone increases radiosensitivity by alleviating tumour hypoxia. Nat Commun 7: 12308. 10.1038/ncomms12308
- Birth D, Kao WC, Hunte C (2014). Structural analysis of atovaquone-inhibited cytochrome bc1 complex reveals the molecular basis of antimalarial drug action. Nat Commun 5: 4029. 10.1038/ncomms5029
- Dixon R, Pozniak AL, Watt HM, Rolan P, Posner J (1996). Single-dose and steady-state pharmacokinetics of a novel microfluidized suspension of atovaquone in human immunodeficiency virus-seropositive patients. Antimicrob Agents Chemother 40(3): 556-560. 10.1128/aac.40.3.556
- Fiorillo M, Lamb R, Tanowitz HB, Mutti L, Krstic-Demonacos M, Cappello AR, Martinez-Outschoorn UE, Sotgia F, Lisanti MP (2016). Repurposing atovaquone: targeting mitochondrial complex III and OXPHOS to eradicate cancer stem cells. Oncotarget 7(23): 34084-34099. 10.18632/oncotarget.9122
- Falloon J, Sargent S, Piscitelli SC, Bechtel C, LaFon SW, Sadler B, Walker RE, Kovacs JA, Polis MA, Davey RT, Jr., Lane HC, Masur H (1999). Atovaquone suspension in HIV-infected volunteers: pharmacokinetics, pharmacodynamics, and TMP-SMX interaction study. Pharmacotherapy 19(9): 1050-1056. 10.1592/phco.19.13.1050.31598
- Diepart C, Karroum O, Magat J, Feron O, Verrax J, Calderon PB, Gregoire V, Leveque P, Stockis J, Dauguet N, Jordan BF, Gallez B (2012). Arsenic trioxide treatment decreases the oxygen consumption rate of tumor cells and radiosensitizes solid tumors. Cancer Res 72(2): 482-490. 10.1158/0008-5472.Can-11-1755
- Jordan BF, Sonveaux P (2012). Targeting tumor perfusion and oxygenation to improve the outcome of anticancer therapy. Front Pharmacol 3: 94. 10.3389/fphar.2012.00094
- Crokart N, Radermacher K, Jordan BF, Baudelet C, Cron GO, Gregoire V, Beghein N, Bouzin C, Feron O, Gallez B (2005). Tumor radiosensitization by antiinflammatory drugs: evidence for a new mechanism involving the oxygen effect. Cancer Res 65(17): 7911-7916. 10.1158/0008-5472.Can-05-1288
- Ni R, Cao T, Xiong S, Ma J, Fan GC, Lacefield JC, Lu Y, Le Tissier S, Peng T (2016). Therapeutic inhibition of mitochondrial reactive oxygen species with mito-TEMPO reduces diabetic cardiomyopathy. Free Radic Biol Med 90: 12-23. 10.1016/j.freeradbiomed.2015.11.013
- Murphy MP, Smith RAJ (2007). Targeting antioxidants to mitochondria by conjugation to lipophilic cations. 47(1): 629-656. 10.1146/annurev.pharmtox.47.120505.105110
- James AM, Sharpley MS, Manas AR, Frerman FE, Hirst J, Smith RA, Murphy MP (2007). Interaction of the mitochondria-targeted antioxidant MitoQ with phospholipid bilayers and ubiquinone oxidoreductases. J Biol Chem 282(20): 14708-14718. 10.1074/jbc.M611463200
- Liou G-Y, Döppler H, DelGiorno Kathleen E, Zhang L, Leitges M, Crawford Howard C, Murphy Michael P, Storz P (2016). Mutant KRas-induced mitochondrial oxidative stress in acinar cells upregulates EGFR signaling to drive formation of pancreatic precancerous lesions. Cell Rep 14(10): 2325-2336. 10.1016/j.celrep.2016.02.029
- Chiu SM, Oleinick NL (2001). Dissociation of mitochondrial depolarization from cytochrome c release during apoptosis induced by photodynamic therapy. Br J Cancer 84(8): 1099-1106. 10.1054/bjoc.2000.1714
- Vantieghem A, Xu Y, Declercq W, Vandenabeele P, Denecker G, Vandenheede JR, Merlevede W, de Witte PA, Agostinis P (2001). Different pathways mediate cytochrome c release after photodynamic therapy with hypericin. Photochem Photobiol 74(2): 133-142. 10.1562/0031-8655(2001)074<0133:dpmccr>2.0.co;2
- Varnes ME, Chiu SM, Xue LY, Oleinick NL (1999). Photodynamic therapy-induced apoptosis in lymphoma cells: translocation of cytochrome c causes inhibition of respiration as well as caspase activation. Biochem Biophys Res Commun 255(3): 673-679. 10.1006/bbrc.1999.0261
- Baskaran R, Lee J, Yang SG (2018). Clinical development of photodynamic agents and therapeutic applications. Biomater Res 22: 25. 10.1186/s40824-018-0140-z
- Sareen D, Darjatmoko SR, Albert DM, Polans AS (2007). Mitochondria, Calcium, and Calpain are Key Mediators of Resveratrol-Induced Apoptosis in Breast Cancer. Mol Pharmacol 72(6): 1466. 10.1124/mol.107.039040
- Ma X, Tian X, Huang X, Yan F, Qiao D (2007). Resveratrol-induced mitochondrial dysfunction and apoptosis are associated with Ca2+ and mCICR-mediated MPT activation in HepG2 cells. Mol Cell Biochem 302(1-2): 99-109. 10.1007/s11010-007-9431-8
- Yallapu MM, Nagesh PK, Jaggi M, Chauhan SC (2015). Therapeutic applications of curcumin nanoformulations. AAPS j 17(6): 1341-1356. 10.1208/s12248-015-9811-z
- Levitsky DO, Dembitsky VM (2014). Anti-breast cancer agents derived from plants. Nat Prod Bioprospect. 10.1007/s13659-014-0048-9
- Zhou Z, Zhu C, Cai Z, Zhao F, He L, Lou X, Qi X (2018). Betulin induces cytochrome c release and apoptosis in colon cancer cells via NOXA. Oncol Lett 15(5): 7319-7327. 10.3892/ol.2018.8183
- Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR, Nunnari J (2008). Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell 14(2): 193-204. 10.1016/j.devcel.2007.11.019
- Wang J, Hansen K, Edwards R, Van Houten B, Qian W (2015). Mitochondrial division inhibitor 1 (mdivi-1) enhances death receptor-mediated apoptosis in human ovarian cancer cells. Biochem Biophys Res Commun 456(1): 7-12. 10.1016/j.bbrc.2014.11.010
- Kim B, Kim JS, Yoon Y, Santiago MC, Brown MD, Park JY (2013). Inhibition of Drp1-dependent mitochondrial division impairs myogenic differentiation. Am J Physiol Regul Integr Comp Physiol 305(8): R927-938. 10.1152/ajpregu.00502.2012
- Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T (2006). Dynasore, a cell-permeable inhibitor of dynamin. Dev Cell 10(6): 839-850. 10.1016/j.devcel.2006.04.002
- Shen F, Gai J, Xing J, Guan J, Fu L, Li Q (2018). Dynasore suppresses proliferation and induces apoptosis of the non-small-cell lung cancer cell line A549. Biochem Biophys Res Commun 495(1): 1158-1166. 10.1016/j.bbrc.2017.11.109
- Zhang Y, Nolan M, Yamada H, Watanabe M, Nasu Y, Takei K, Takeda T (2016). Dynamin2 GTPase contributes to invadopodia formation in invasive bladder cancer cells. Biochem Biophys Res Commun 480(3): 409-414. 10.1016/j.bbrc.2016.10.063
- Yamada H, Abe T, Li SA, Masuoka Y, Isoda M, Watanabe M, Nasu Y, Kumon H, Asai A, Takei K (2009). Dynasore, a dynamin inhibitor, suppresses lamellipodia formation and cancer cell invasion by destabilizing actin filaments. Biochem Biophys Res Commun 390(4): 1142-1148. 10.1016/j.bbrc.2009.10.105
- Zhong B, Shi D, Wu F, Wang S, Hu H, Cheng C, Qing X, Huang X, Luo X, Zhang Z, Shao Z (2019). Dynasore suppresses cell proliferation, migration, and invasion and enhances the antitumor capacity of cisplatin via STAT3 pathway in osteosarcoma. Cell Death Dis 10(10): 687. 10.1038/s41419-019-1917-2
- Yue W, Chen Z, Liu H, Yan C, Chen M, Feng D, Yan C, Wu H, Du L, Wang Y, Liu J, Huang X, Xia L, Liu L, Wang X, Jin H, Wang J, Song Z, Hao X, Chen Q (2014). A small natural molecule promotes mitochondrial fusion through inhibition of the deubiquitinase USP30. Cell Res 24(4): 482-496. 10.1038/cr.2014.20
- Yazbeck VY, Buglio D, Georgakis GV, Li Y, Iwado E, Romaguera JE, Kondo S, Younes A (2008). Temsirolimus downregulates p21 without altering cyclin D1 expression and induces autophagy and synergizes with vorinostat in mantle cell lymphoma. Exp Hematol 36(4): 443-450. 10.1016/j.exphem.2007.12.008
- Liu W, Huang S, Chen Z, Wang H, Wu H, Zhang D (2014). Temsirolimus, the mTOR inhibitor, induces autophagy in adenoid cystic carcinoma: in vitro and in vivo. Pathol Res Pract 210(11): 764-769. 10.1016/j.prp.2014.03.008
- Van Haaften RI, Haenen GR, van Bladeren PJ, Bogaards JJ, Evelo CT, Bast A (2003). Inhibition of various glutathione S-transferase isoenzymes by RRR-alpha-tocopherol. Toxicol In Vitro 17(3): 245-251. 10.1016/s0887-2333(03)00038-9
- Sampayo-Reyes A, Zakharyan RA (2006). Tocopherol esters inhibit human glutathione S-transferase omega. Acta Biochim Pol 53(3): 547-552. 10.18388/abp.2006_3326
- Angulo-Molina A, Reyes-Leyva J, Lopez-Malo A, Hernandez J (2014). The role of alpha tocopheryl succinate (alpha-TOS) as a potential anticancer agent. Nutr Cancer 66(2): 167-176. 10.1080/01635581.2014.863367
- dos Santos GA, Abreu e Lima RS, Pestana CR, Lima AS, Scheucher PS, Thome CH, Gimenes-Teixeira HL, Santana-Lemos BA, Lucena-Araujo AR, Rodrigues FP, Nasr R, Uyemura SA, Falcao RP, de The H, Pandolfi PP, Curti C, Rego EM (2012). (+)alpha-Tocopheryl succinate inhibits the mitochondrial respiratory chain complex I and is as effective as arsenic trioxide or ATRA against acute promyelocytic leukemia in vivo. Leukemia 26(3): 451-460. 10.1038/leu.2011.216
- Dong LF, Low P, Dyason JC, Wang XF, Prochazka L, Witting PK, Freeman R, Swettenham E, Valis K, Liu J, Zobalova R, Turanek J, Spitz DR, Domann FE, Scheffler IE, Ralph SJ, Neuzil J (2008). Alpha-tocopheryl succinate induces apoptosis by targeting ubiquinone-binding sites in mitochondrial respiratory complex II. Oncogene 27(31): 4324-4335. 10.1038/onc.2008.69
- Dourado DF, Fernandes PA, Ramos MJ, Mannervik B (2013). Mechanism of glutathione transferase P1-1-catalyzed activation of the prodrug canfosfamide (TLK286, TELCYTA). Biochemistry 52(45): 8069-8078. 10.1021/bi4005705
- Vergote I, Finkler NJ, Hall JB, Melnyk O, Edwards RP, Jones M, Keck JG, Meng L, Brown GL, Rankin EM, Burke JJ, Boccia RV, Runowicz CD, Rose PG (2010). Randomized phase III study of canfosfamide in combination with pegylated liposomal doxorubicin compared with pegylated liposomal doxorubicin alone in platinum-resistant ovarian cancer. Int J Gynecol Cancer 20(5): 772-780. 10.1111/igc.0b013e3181daaf59
- Rosen LS, Laxa B, Boulos L, Wiggins L, Keck JG, Jameson AJ, Parra R, Patel K, Brown GL (2004). Phase 1 study of TLK286 (Telcyta) administered weekly in advanced malignancies. Clin Cancer Res 10(11): 3689-3698. 10.1158/1078-0432.Ccr-03-0687
- Kavanagh JJ, Gershenson DM, Choi H, Lewis L, Patel K, Brown GL, Garcia A, Spriggs DR (2005). Multi-institutional phase 2 study of TLK286 (TELCYTA, a glutathione S-transferase P1-1 activated glutathione analog prodrug) in patients with platinum and paclitaxel refractory or resistant ovarian cancer. Int J Gynecol Cancer 15(4): 593-600. 10.1111/j.1525-1438.2005.00114.x
- Lorusso D, Mainenti S, Pietragalla A, Ferrandina G, Foco G, Masciullo V, Scambia G (2009). Brostallicin (PNU-166196), a new minor groove DNA binder: preclinical and clinical activity. Expert Opin Investig Drugs 18(12): 1939-1946. 10.1517/13543780903401284
- Pezzola S, Antonini G, Geroni C, Beria I, Colombo M, Broggini M, Marchini S, Mongelli N, Leboffe L, MacArthur R, Mozzi AF, Federici G, Caccuri AM (2010). Role of glutathione transferases in the mechanism of brostallicin activation. Biochemistry 49(1): 226-235. 10.1021/bi901689s
- Hattinger CM, Pasello M, Ferrari S, Picci P, Serra M (2010). Emerging drugs for high-grade osteosarcoma. Expert Opin Emerg Drugs 15(4): 615-634. 10.1517/14728214.2010.505603
- Weber DD, Aminzadeh-Gohari S, Tulipan J, Catalano L, Feichtinger RG, Kofler B (2020). Ketogenic diet in the treatment of cancer – Where do we stand? Mol Metabol 33: 102-121. 10.1016/j.molmet.2019.06.026
- De Giorgis V, Veggiotti P (2013). GLUT1 deficiency syndrome 2013: current state of the art. Seizure 22(10): 803-811. 10.1016/j.seizure.2013.07.003
- Wexler ID, Hemalatha SG, McConnell J, Buist NR, Dahl HH, Berry SA, Cederbaum SD, Patel MS, Kerr DS (1997). Outcome of pyruvate dehydrogenase deficiency treated with ketogenic diets. Studies in patients with identical mutations. Neurology 49(6): 1655-1661. 10.1212/wnl.49.6.1655
- Abdelwahab MG, Fenton KE, Preul MC, Rho JM, Lynch A, Stafford P, Scheck AC (2012). The ketogenic diet is an effective adjuvant to radiation therapy for the treatment of malignant glioma. PLOS ONE 7(5): e36197. 10.1371/journal.pone.0036197
- Maurer GD, Brucker DP, Bahr O, Harter PN, Hattingen E, Walenta S, Mueller-Klieser W, Steinbach JP, Rieger J (2011). Differential utilization of ketone bodies by neurons and glioma cell lines: a rationale for ketogenic diet as experimental glioma therapy. BMC Cancer 11: 315. 10.1186/1471-2407-11-315
- Zuccoli G, Marcello N, Pisanello A, Servadei F, Vaccaro S, Mukherjee P, Seyfried TN (2010). Metabolic management of glioblastoma multiforme using standard therapy together with a restricted ketogenic diet: Case Report. Nutr Metab 7: 33. 10.1186/1743-7075-7-33
- Elsakka AMA, Bary MA, Abdelzaher E, Elnaggar M, Kalamian M, Mukherjee P, Seyfried TN (2018). Management of glioblastoma multiforme in a patient treated with ketogenic metabolic therapy and modified standard of care: a 24-month follow-up. Front Nutr 5: 20. 10.3389/fnut.2018.00020
- Seyfried TN, Shelton L, Arismendi-Morillo G, Kalamian M, Elsakka A, Maroon J, Mukherjee P (2019). Provocative question: should ketogenic metabolic therapy become the standard of care for glioblastoma? Neurochem Res 44(10): 2392-2404. 10.1007/s11064-019-02795-4
- Strickaert A, Saiselet M, Dom G, De Deken X, Dumont JE, Feron O, Sonveaux P, Maenhaut C (2017). Cancer heterogeneity is not compatible with one unique cancer cell metabolic map. Oncogene 36(19): 2637-2642. 10.1038/onc.2016.411
- Danhier P, Banski P, Payen VL, Grasso D, Ippolito L, Sonveaux P, Porporato PE (2017). Cancer metabolism in space and time: beyond the Warburg effect. Biochim Biophys Acta Bioenerg 1858(8): 556-572. 10.1016/j.bbabio.2017.02.001
- Sonveaux P, Vegran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, De Saedeleer CJ, Kennedy KM, Diepart C, Jordan BF, Kelley MJ, Gallez B, Wahl ML, Feron O, Dewhirst MW (2008). Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest 118(12): 3930-3942. 10.1172/JCI36843
- Payen VL, Mina E, Van Hee VF, Porporato PE, Sonveaux P (2020). Monocarboxylate transporters in cancer. Mol Metab 33: 48-66. 10.1016/j.molmet.2019.07.006
- Yong C, Stewart GD, Frezza C (2020). Oncometabolites in renal cancer. Nat Rev Nephrol 16(3): 156-172. 10.1038/s41581-019-0210-z
- Payen VL, Brisson L, Dewhirst MW, Sonveaux P (2015). Common responses of tumors and wounds to hypoxia. Cancer J 21(2): 75-87. 10.1097/PPO.0000000000000098
- Wu H, Chen Q (2015). Hypoxia activation of mitophagy and its role in disease pathogenesis. Antioxid Redox Signal 22(12): 1032-1046. 10.1089/ars.2014.6204
- Mazurek S (2011). Pyruvate kinase type M2: a key regulator of the metabolic budget system in tumor cells. Int J Biochem Cell Biol 43(7): 969-980. 10.1016/j.biocel.2010.02.005
- Ju YS, Alexandrov LB, Gerstung M, Martincorena I, Nik-Zainal S, Ramakrishna M, Davies HR, Papaemmanuil E, Gundem G, Shlien A, Bolli N, Behjati S, Tarpey PS, Nangalia J, Massie CE, Butler AP, Teague JW, Vassiliou GS, Green AR, Du MQ, Unnikrishnan A, Pimanda JE, Teh BT, Munshi N, Greaves M, Vyas P, El-Naggar AK, Santarius T, Collins VP, Grundy R, et al. (2014). Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. Elife 3: e02935. 10.7554/eLife.02935
- Gopal RK, Calvo SE, Shih AR, Chaves FL, McGuone D, Mick E, Pierce KA, Li Y, Garofalo A, Van Allen EM, Clish CB, Oliva E, Mootha VK (2018). Early loss of mitochondrial complex I and rewiring of glutathione metabolism in renal oncocytoma. Proc Natl Acad Sci U S A 115(27): E6283-E6290. 10.1073/pnas.1711888115
- Hardie RA, van Dam E, Cowley M, Han TL, Balaban S, Pajic M, Pinese M, Iconomou M, Shearer RF, McKenna J, Miller D, Waddell N, Pearson JV, Grimmond SM, Australian Pancreatic Cancer Genome I, Sazanov L, Biankin AV, Villas-Boas S, Hoy AJ, Turner N, Saunders DN (2017). Mitochondrial mutations and metabolic adaptation in pancreatic cancer. Cancer Metab 5: 2. 10.1186/s40170-017-0164-1
- Rai PK, Craven L, Hoogewijs K, Russell OM, Lightowlers RN (2018). Advances in methods for reducing mitochondrial DNA disease by replacing or manipulating the mitochondrial genome. Essays Biochem 62(3): 455-465. 10.1042/EBC20170113
–
ACKNOWLEDGMENTS
Works at authors’ lab are supported by European Union’s Horizon 2020 research innovation pro-gram under the Marie Skłodowska-Curie grant agree-ments No 642623 RADIATE, No 722605 TRANSMIT and No 860245THERADNET, the Belgian Fonds National de la Re-cherche Scientifique (F.R.S.-FNRS), the Fondation belge contre le cancer (Fundamental Research Grant. FAF-F/2018/1282), the Belgian Télévie and the Louvain Foun-dation. DG is a PhD Fellow of Marie Skłodowska-Curie grant No 642623 RADIATE and a PhD Fellow of the Télévie. She further received a Bourse du Patrimoine from UCLou-vain. LXZ is a PhD Fellow of Marie Skłodowska-Curie grant No 722605 TRANSMIT. JAV is an UCLouvain PhD Research Fellow. PS is a F.R.S.-FNRS Senior Research Associate.
COPYRIGHT
© 2020

Mitochondria in cancer by Grasso et al. is licensed under a Creative Commons Attribution 4.0 International License.


