Viewpoint:
Cell Stress, Vol. 4, No. 8, pp. 191 - 198; doi: 10.15698/cst2020.08.227
Exploiting the circuit breaker cancer evolution model in human clear cell renal cell carcinoma

1 Molecular Oncology, Department of Medicine, Washington University, St. Louis, MO 63110, USA.
2 Human Oncology and Pathogenesis Program, Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA.
3 Department of Pathology, Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA.
4 Department of Pathology and Laboratory Medicine, Weill Cornell Medical College, Cornell University, New York, NY 10065, USA.
Keywords: kidney cancer evolution, circuit breaker, VHL, HIF, PBRM1, MTOR.
Abbreviations:
CB – circuit breaker;
ccRCC – clear cell RCC;
HIF – hypoxia inducible factor;
OG – oncogene;
mccRCC – metastatic ccRCC;
RCC – renal cell carcinoma;
TSG – tumor suppressor gene;
VHL – Von-Hippel Lindau.
Received originally: 19/04/2020 Received in revised form: 16/06/2020
Accepted: 17/06/2020
Published: 25/06/2020
Correspondence:
James J. Hsieh, M.D., Ph.D, 660 S. Euclid Avenue, Box 8069, St. Louis, MO 63110, USA; Tel: 314-273-1688; Jhsieh@wustl.edu
Conflict of interest statement: JJH has received consulting fees from Eisai Inc., Japan; clinical trial funding from Merck & Co., Inc., USA, and AstraZeneca Pharmaceuticals, UK; and research funding from BostonGene, USA.
Please cite this article as: James J. Hsieh and Emily H. Cheng (2020). Exploiting the circuit breaker cancer evolution model in human clear cell renal cell carcinoma. Cell Stress 4(8): 191-198. doi: 10.15698/cst2020.08.227
Abstract
The incessant interactions between susceptible humans and their respective macro/microenvironments registered throughout their lifetime result in the ultimate manifestation of individual cancers. With the average lifespan exceeding 50 years of age in humans since the beginning of 20th century, aging – the “time” factor – has played an ever-increasing role alongside host and environmental factors in cancer incidences. Cancer is a genetic/epigenetic disease due to gain-of-function mutations in cancer-causing genes (oncogene; OG) and/or loss-of-function mutations in tumor-suppressing genes (tumor suppressor genes; TSG). In addition to their integral relationship with cancer, a timely deployment of specific OG and/or TSG is in fact needed for higher organisms like human to cope with respective physiological and pathological conditions. Over the past decade, extensive human kidney cancer genomics have been performed and novel mouse models recapitulating human kidney cancer pathobiology have been generated. With new genomic, genetic, mechanistic, clinical and therapeutic insights accumulated from studying clear cell renal cell carcinoma (ccRCC)–the most common type of kidney cancer, we conceived a cancer evolution model built upon the OG-TSG signaling pair analogous to the electrical circuit breaker (CB) that permits necessary signaling output and at the same time prevent detrimental signaling overdrive. Hence, this viewpoint aims at providing a step-by-step mechanistic explanation/illustration concerning how inherent OG-TSG CBs intricately operate in concert for the organism’s wellbeing; and how somatic mutations, the essential component for genetic adaptability, inadvertently triggers a sequential outage of specific sets of CBs that normally function to maintain and protect and individual tissue homeostasis.
INTRODUCTION
On average a human adult body encompasses fifty trillion cells (~5×1013) with a daily turnover of hundred billion cells (~1011) within which each contains two copies of ~three billion base-pair DNA haploid genome. Approximately, the routine maintenance of a healthy human body necessitates ~1021 DNA base pairs transaction every day. Remarkably, despite this astronomical chemical challenge genome integrity is inherently maintained by sophisticated DNA replication, proof-reading, and repair mechanisms evolved over billion years. However, the precision of DNA transaction in multi-cellular complex organisms can only be near-perfect to allow finite replication errors needed for genetic adaptation and thereby evolution. Nevertheless, individual cells are equipped with intrinsic cell death machineries to assure continuous genome integrity, which works alongside with immune system’s surveillance, killing, and removal of detected pre-cancer/cancer cells [1][2][3]. The human genome encodes ~20,000 protein coding sequences of which 2-10% are cancer-related genes such as oncogenes (OGs) and tumor suppressor genes (TSGs) [2][4]. Notably, these cancer-related genes normally participate in key biological processes such as embryonic development and tissue homeostasis such as MLL (a Trx-G gene) and BMI-1 (a Pc-G gene)[5]. In response to imminent or chronic tissue stresses such as ischemia tissue injury these “specialty genes” are called upon to act appropriately till resolution of respective insults [2][5][6]. Hence, we envisioned that specific oncogene and tumor suppressor gene pairs (OG-TSG) could function as regulatory modules similar to electrical circuit breakers (CBs) that mitigate power/signal overload [5]. Additionally, these OG-TSG CBs can interconnect to provide additional layers of regulation and safety that operate in a tissue context-dependent manner, which helps explain the tissue-type specific propensity of dysfunction in certain OG and TSG [7]. With these basic principles, hereafter we will begin to explore this hypothetic OG-TSG CB cancer evolution model inspired when we studied clear cell renal cell carcinoma (ccRCC) in human, mouse, and cell line models.
–
Kidney cancer
Kidney cancer accounts for ~2% of all cancer diagnoses (~74,000; ~300,000) and deaths (~15,000; ~134,000) annually at the United States and the World, respectively[8][9]. Renal cell carcinoma (RCC) encompasses a large heterogeneous group of cancers derived from renal tubular epithelial cells, which encompasses >10 molecular and histopathological subtypes [10]. RCC major subtypes (≥5%) include clear cell RCC (ccRCC) at ~75%, papillary RCC (pRCC) at ~15%, and chromophobe RCC (chRCC) at ~5% [11]. Accordingly, metastatic ccRCC (mccRCC) accounts for most of the kidney cancer mortality [12]. Although mccRCC is refractory to conventional chemotherapy, marked therapeutic advances have been made over the past 15 years, culminating in 15 FDA-approved agents for mccRCC [9]. These agents exploit seven molecular mechanisms: (1) IL-2 and (-Interferon are cytokines; (2) Sorafenib, Sunitinib, Pazopanib, Axitinib and Bevacizumab inhibit VEGF pathway; (3) Cabozantinib inhibits VEGFR2, cMET and AXL; (4) Lenvatinib inhibits VEGFR2 and FGFR; (5) Everolimus and Temsirolimus are inhibitors of mTORC1; (6) Nivolumab, Avelumab, and Pembrolizumab are PD-1/L1 blocking antibodies; and (7) Ipilimumab is an anti-CTLA-4 antibody [13]. However, mccRCC remains lethal and treatment response is highly heterogeneous among patients upon individual treatments [14], likely due to tumor genomic and host genetic heterogeneities.
–
The genomics of ccRCC
Modern multi-omics of human ccRCC consisting of genomics, transcriptomics, proteomics and metabolomics not only highlights its profound inter-/intra-tumor heterogeneity but also showcases underline cancer evolution constraints that could be exploit as therapeutic biomarkers [14][15][16][17][18][19][20][21][22][23][24][25]. The most conspicuous genomic event in ccRCC is the near universal (>90%) one copy loss of the short arm of Chromosome 3 [26]. The most striking genomic discovery in ccRCC is the extreme chromosomal proximity of the four most prevalently mutated kidney cancer TSGs—VHL (~80%), PBRM1 (29-46%), SETD2 (8-30%), and BAP1 (6-19%)—spanning chromosome 3p21-3p25 [6][27][28]. Hence, one genetic event incurred the 3p loss in renal epithelial cells simultaneously creates a haplo-insufficient state of four critical ccRCC TSGs. Remarkably, 3p loss represents the first somatic driver event in sporadic ccRCC, which takes place during adolescence that predates the most common second somatic event by ~20 years, i.e., the genetic/epigenetic inaction of the VHL gene, giving rise to the most recent common ancestor (MRCA) within a given ccRCC patient whose ccRCC is eventually diagnosed at 60 years of age [29]. Notably, unlike most cancers that are initiated by gain-of-function mutations in OGs ccRCC results from sequential losses of TSGs [6].
–
The shared genetic events of hereditary and sporadic in human ccRCC
The Von-Hippel Lindau (VHL) disease, an autosomal dominant hereditary cancer syndrome caused by the loss-of-function germline mutation in the VHL gene [30], is characterized by the development of hemangioblastoma of the central nervous system and retina, ccRCC, and pheochromocytoma [31][32]. It was later demonstrated that VHL is inactivated in ~90% of sporadic ccRCC through either genetic mutation or promoter methylation [9][33]. Biallelic inactivation of the TSG VHL is therefore established as an early event in both germline mutant VHL-associated and sporadic ccRCC [6].
DISCUSSION
The VHL-HIF-hypoxia-metabolism
Studies on oxygen sensing led to the discovery of Hypoxia-Inducible Factors (HIFs) [34]. VHL is a multipurpose adaptor protein and chiefly effects as the substrate recognition module of the VCB (VHL-Elongin C-Elongin B)-Cul2 E3 ligase which ubiquitinates HIF-1□ and HIF-2□ [35]. Under normal oxygen conditions, HIF1/2□ is prolyl hydroxylated by EGLN, ubiquitinated by VCB-Cul2-VHL, and rapidly degraded by the 26S Proteasome [36]; whereas under low oxygen conditions such as high altitude or ischemia, HIF□ is stabilized to initiate a myriad of hypoxia-specific transcriptional programs [34][36][37][38]. The pathologic loss of VHL in ccRCC results in persistently elevated HIFs accounting for the observed clear cell morphology and highly vascularity [11][12][38][39][40]. However, the long latency (>30 years) for VHL syndrome patients to develop ccRCC [31] and the insufficiency of VHL loss alone to induce ccRCC in mice [41] argue for the necessity of cooperative events [42].
–
ccRCC signifies prevalent loss-of-function mutations in TSGs at the renal epithelium
Unlike many cancers that originate from gain-of-function mutations in OGs such as EGFR and RAS, ccRCC manifests with prevalent loss-of-function mutations in TSGs, making the development of predictive biomarkers for individual targeted therapies and/or immunotherapies extremely challenging. Nevertheless, new therapeutic modalities, novel genetically engineered mouse models, clinically relevant patient-derived cell line/xenograft models, and outcome-based biomarker studies altogether have begun to shed light on how these seemingly distinct research areas are in fact exquisitely interconnected [43]. Here, we wish to update essential findings in ccRCC, and present a novel concept of “Interconnected OG-TSG Circuit Breaker Cancer Evolution Model in ccRCC”.
–
The first oncogenic driver event in ccRCC
In hereditary VHL-loss ccRCC, the first genetic event is the inheritance of a loss-of-function copy of the VHL gene; whereas in sporadic ccRCC, the first genetic event is the loss of chromosome 3p. The ensuing epi/genetic event in developing both kinds of ccRCC converges on the complete inactivation of VHL [6]. Hence, generally speaking ccRCC is a VHL-loss kidney cancer, and complete VHL inactivation is the quintessential first functional/genetic truncal event [7].
–
The exemplary interconnected VHL/HIF/PBRM1/TSC/MTORC1 CBs in ccRCC
Among the three newly identified 3p21 TSGs in ccRCC, PBRM1 is best studied in molecular mechanisms, mouse models, and human clinical outcomes [22][28][32][42][44][45][46][47][48]. Accordingly, we will further elaborate recent key research findings on PBRM1 and attempt to reconcile how and why PBRM1 loss in ccRCC impact the efficacy of select targeted therapies and immune checkpoint inhibitors.
–
PBRM1
–
The SWI/SNF are macromolecular protein complexes that utilize ATP to mobilize nucleosome, modulate chromatin structure, and thereby regulate central cellular, developmental and oncogenic processes [49][50]. They come with many flavors due to their interchangeable, dynamic compositions in nature [51]. Notably, mutations of individual SWI/SNF subunits are detected and exhibit preferential enrichment in ~20% human cancer of various types [52]. PBRM1 is the defining component of the PBAF complex and is most commonly mutated in ccRCC [53]. Remarkably, the in vivo tumor suppressor role of PBRM1 in ccRCC was confirmed and reported in 2017 by three independent laboratories using three different genetically engineered mouse models where combined losses of VHL and PBRM1 lead to multifocal ccRCC in mouse kidney, whereas individual losses did not [42][54][55]. How PBRM1 loss might have contributed to the ccRCC pathogenesis will be discussed hereafter.
–
The hypoxia scenario: the normal physiology
–
Upon tissue injury, insufficient blood supply incurs, thereby resulting in low oxygen tension, which in turn stabilizes HIF-1 to act accordingly (Fig. 1A), i.e. inhibition of mitochondria oxidative phosphorylation and promotion of revascularization. Once the tissue repair is complete, normal oxygen tension is re-established, HIF-1 is prolyl hydroxylated which is recognized by VHL, ubiquitinated by VCB-Cullin 2 E3 ligase, and degraded by 26S proteasome, and tissue returns to a normal homeostatic state [34][36].
–
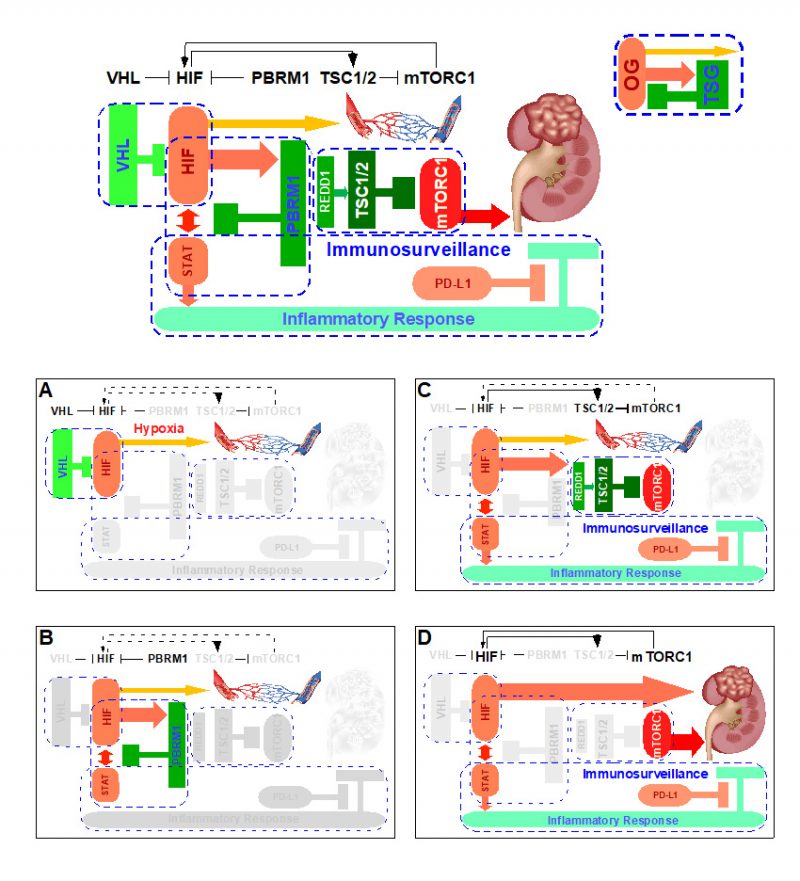 | FIGURE 1: The interconnected OG-TSG CBs operate in renal epithelium to control signaling output and prevent tumorigenesis. (A) Depicts the physiological employment of the inherent VHL-HIF CB to gauge tissue response to low oxygen tension, and (B-D) examine how the sequential losses of a pre-determined set of “OG-TSG CBs” once the first CB is inactivated result in ccRCC. |
–
The VHL loss scenario: inactivation of one CB
–
The complete pathologic loss of VHL due to chromosome 3p loss, mutations, and/or promoter methylation results in the aberrant abundance of HIF-1 protein, resulting in a hypoxia-like molecular response despite normal oxygen tension, i.e. pseudohypoxia. Under this scenario, the VHL loss inactivates the first ccRCC CB “VHL-HIF” (Fig. 1B) present in the renal cortex. In vivo mouse model studies demonstrated that the activation of pseudohypoxia program is evident when comparing the twelve week old normal appearing mouse renal cortex with kidney specific deletion of Vhl to that of wild-type control [42].
–
The VHL/PBRM1 loss scenario: inactivation of two CBs
–
The singular loss of VHL is insufficient in initiating ccRCC, which is recognized in both mouse and human VHL loss models [56]. The ensuing complete loss of PBRM1 inactivates the second CB “HIF-PBRM1-STAT” in the renal cortex (Fig. 1C), leading to the increased transcription output of HIF and STAT targets [42]. Of note, the singular loss of PBRM1 did not activate HIF or STAT targets [42]. It is known that HIF-1 and STAT3 cooperate to activate the expression of HIF-1 targets [57][58]. Consequently, the dysregulated interplay between HIF and STAT upon combined losses of VHL and PBRM1 creates a feed-forward amplification loop that maximizes downstream gene expression [42]. The role of PBRM1 in restricting HIF signaling output was independently reported using cell-based assays [59]. Under this scenario, the subsequent PBRM1 loss inactivates the second ccRCC CB “HIF-PBRM1-STAT”.
–
The VHL/PBRM1/TSC loss scenario: inactivation of three CBs
–
Despite the fact that HIF/STAT signaling overdrive was evident in the twelve week-old Vhl-/-;Pbrm1-/- mouse kidney, the long latency (ten months) and the incomplete penetrance (~50%) to the ultimate development of multifocal ccRCC in this model suggested that additional OG-TSG CBs could be at play [42]. To this end, gene expression and immunohistochemical analyses comparing twelve month-old ccRCC tumors to twelve week-old renal cortices of Vhl-/-;Pbrm1-/- mice detected hyperactive mTORC1 signaling in tumors in addition to the demonstrated pre-existing activation of HIF/STAT and suppression of mitochondrial pathways [42]. Hence, the prevention of aberrant activation of mTORC1 pathway probably constitutes the third ccRCC CB. Of note, mTORC1 serves as the central nutrition state integrator of the cell and its main control is conferred by the TSG TSC1/2 complex [60]. Remarkably, loss-of-function mutations in TSC1 or TSC2 (~10%), and gain-of-function mutations in MTOR (~6%) are common in ccRCC and correlated with rapalog response in therapeutic outlier studies; and Tsc1 and Tsc2 expression levels are down-regulated in Vhl-/-;Pbrm1-/- mouse ccRCC tumors [17][22][42][61][62][63]. Accordingly, the third CB in place to prevent ccRCC pathogenesis after losses of VHL and PBRM1 is “TSC-mTORC1” (Fig. 1D). Of note, the inactivation of the “PTEN/PI3K” CB which functions upstream of the “TSC-mTORC1” CB is observed in 7% of ccRCC [22]. Mechanistically, the inactivation of the first “VHL-HIF” and the second “HIF-PBRM1-STAT” CBs and the resulting HIF output overdrive apparently activates Redd1, a known HIF1 target and an activator of TSC2 [42][64], which positions “TSC-mTORC1′ as the preferred third CB after the losses of VHL and PBRM1 [56].
–
A hypothetical immunosurveillance CB in ccRCC
–
As STATs are key transcription factors in cancer inflammation and immunity [65], the activation of STAT pathway due to the combined loss of VHL and PBRM1 could render the resulting tumors prone to immune regulation [42]. Recent approvals of single agent Nivolumab (anti-PD-1 antibody) as second line and the combination of Ipilimumab (anti-CTLA-4 antibody) and Nivolumab as first line treatment options for ccRCC have dramatically altered the therapeutic landscape of metastatic kidney cancer [66][67]. Intriguingly, a recent paper identified PBRM1 loss as a potential genomic biomarker for the treatment response to these immune checkpoint inhibitors [68][69] and others suggested otherwise [70][71], which needs further validation facing the daunting intratumor heterogeneity of ccRCC. Nevertheless, these mechanistic, mouse, and human ccRCC studies support a working hypothesis in which the disarmed “STAT-PD-1” immunosurveillance CB can be reactivated through biological means such as anti-PD-1/L1 antibodies for therapeutic exploitation (Fig. 1D). This might be one of the important rationales of why immunotherapy has activity in ccRCC, a tumor generally associated with low tumor mutation burden and a lack of microsatellite instability.
FUTURE DIRECTION
Integrated applied pathology to exploit functional pathology in advancing precision cancer therapeutics
The holy grail of contemporary cancer research is to be able to predict not only how an individual patient may benefit from currently available front-line therapies, but also how an individual tumor’s molecular identity could potentially inform resistance mechanisms and thereby help implement a novel, tailored combination therapeutic strategy to greatly improve clinical outcome. One of the most challenging issues concerning metastatic ccRCC care is the known conspicuous intra-tumor and inter-tumor heterogeneity, which probably contributes to clinical outcomes. Nevertheless, like all individual human subjects develop from the same two copies of genome, all cancer cells evolve from the same set of genetic materials carried in the host cell. Hence, we hypothesize that intrinsic programming principles are in place to guide embryonic development, maintain tissue homeostasis, and restrict tumorigenesis [7][14]. To visualize this third-generation hypothesis, we propose a “OG-TSG CB Constrained Braided Cancer River” model by integrating our first-generation “Braided River Model” and second-generation “CB Model” to further expound on this carcinogenic principle inspired from studying ccRCC, which might be applicable to additional cancer types.
–
The OG-TSG CB constrained braided cancer river model
–
Despite conspicuous tumor heterogeneity, long-term clinical benefits on single agent targeted therapy are not uncommonly observed with metastatic ccRCC patients, implicating underlying cancer evolutionary constraints that force nonrandom sequences of parallel gene/pathway/function/phenotype convergences (Fig. 2). Indeed, our multi-region genomics study performed on ccRCC therapeutic outliers treated with single agent mTORC1 inhibitors rendered invaluable insights concerning this hypothesis [62]. We first proposed a braided cancer river model to help depict individual cancer evolution and advise therapeutic options.[7] The “Braided River” model highlights parallel and convergent events occurring throughout individual ccRCC tumorigenesis. It illustrates the stepwise, ordered accumulation of different driver mutations in kidney cancer development to acquire cancer hallmarks. A late chaotic evolutionary time point was incorporated to explain the limited effectiveness of combined targeted therapies in highly aggressive cancers, when “speedy” driver mutations quickly accumulate to enable extreme subclonal evolution. With the new concept of interconnected OG-TSG CBs, we propose a “CB-Constrained Braided Cancer River” model in which inherent context-dependent OG-TSG CBs are positioned at the gene/pathway/function/phenotype convergent points (Fig. 2). In other words, CBs function like dams to limit flow and prevent flooding. This model stipulates that each individual cancer river initiates with a truncal driver event, and once that occurs the ensuing branching events could be predicted. Accordingly, restoring a pre-determined, preferred set of CBs at once could offer effective measures and guide trial designs.
–
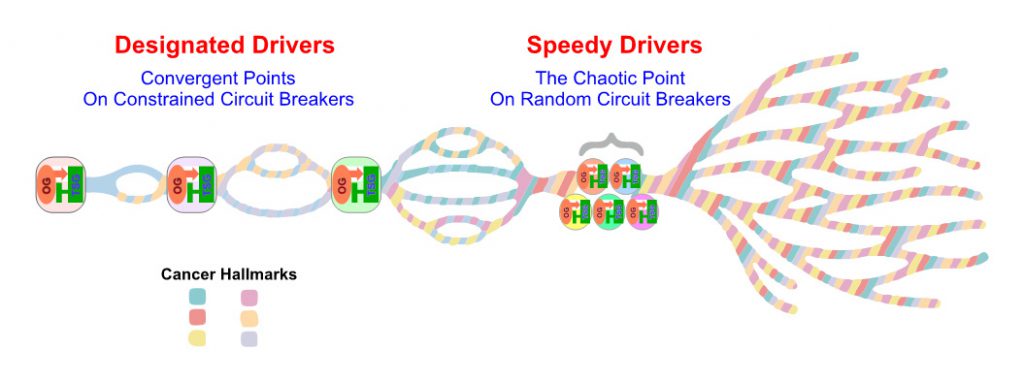 | FIGURE 2. The diagram depicts the “OG-TSG CB Constrained Braided Cancer River” model to explain the non-randomness of cancer evolution and devise personalized cancer treatment strategy. |
REFERENCES
- Nowell PC (1976). The clonal evolution of tumor cell populations. Science 194(4260): 23-8. 10.1126/science.959840
- Hanahan D, Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144(5):646-74. 10.1016/j.cell.2011.02.013
- Dunn GP, Old LJ, Schreiber RD (2004). The immunobiology of cancer immunosurveillance and immunoediting. Immunity 21(2):137-48. 10.1016/j.immuni.2004.07.017
- Stratton MR, Campbell PJ, Futreal PA (2009). The cancer genome. Nature 458(7239):719-24. 10.1038/nature07943
- Liu H, Takeda S, Cheng EH, Hsieh JJ (2008). Biphasic MLL takes helm at cell cycle control: implications in human mixed lineage leukemia. Cell Cycle 7(4):428-35. 10.4161/cc.7.4.5426
- Hsieh JJ, Le VH, Oyama T, Ricketts CJ, Ho TH, Cheng EH (2018). Chromosome 3p Loss-Orchestrated VHL, HIF, and Epigenetic Deregulation in Clear Cell Renal Cell Carcinoma. J Clin Oncol 2018:JCO2018792549. 10.1200/JCO.2018.79.2549
- Wei EY, Hsieh JJ (2015). A river model to map convergent cancer evolution and guide therapy in RCC. Nat Rev Urol 12(12):706-12. 10.1038/nrurol.2015.260
- Siegel RL, Miller KD, Jemal A (2020). Cancer statistics, 2020. CA Cancer J Clin 70(1):7-30. 10.3322/caac.21590
- Huang JJ, Hsieh JJ (2020). The Therapeutic Landscape of Renal Cell Carcinoma: From the Dark Age to the Golden Age. Semin Nephrol 40(1):28-41. 10.1016/j.semnephrol.2019.12.004
- Moch H, Humphrey PA, Ulbright TM, Reuter VE (2016). Who classification of tumours of the urinaryt system and male genital organs. 4th Edition, Volume 8. WHO/IARC Classification of Tumours, Lyon, France.
- Hsieh JJ, Le V, Cao D, Cheng EH, Creighton CJ (2018). Genomic classifications of renal cell carcinoma: a critical step towards the future application of personalized kidney cancer care with pan-omics precision. J Pathol 244(5):525-37. 10.1002/path.5022
- Hsieh JJ, Purdue MP, Signoretti S, Swanton C, Albiges L, Schmidinger M, Heng DY, Larkin J, Ficarra V (2017). Renal cell carcinoma. Nat Rev Dis Primers 3:17009. 10.1038/nrdp.2017.9
- McKay RR, Bosse D, Choueiri TK (2018). Evolving Systemic Treatment Landscape for Patients With Advanced Renal Cell Carcinoma. J Clin Oncol 2018:JCO2018790253. 10.1200/JCO.2018.79.0253
- Hsieh JJ, Manley BJ, Khan N, Gao J, Carlo MI, Cheng EH (2017). Overcome tumor heterogeneity-imposed therapeutic barriers through convergent genomic biomarker discovery: A braided cancer river model of kidney cancer. Semin Cell Dev Biol 64:98-106. 10.1016/j.semcdb.2016.09.002
- Gerlinger M, Horswell S, Larkin J, Rowan AJ, Salm MP, Varela I, Fisher R, McGranahan N, Matthews N, Santos CR, Martinez P, Phillimore B, Begum S, Rabinowitz A, Spencer-Dene B, Gulati S, Bates PA, Stamp G, Pickering L, Gore M, Nicol DL, Hazell S, Futreal PA, Stewart A, Swanton C (2014). Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet 46(3):225-33. 10.1038/ng.2891
- Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, McDonald NQ, Butler A, Jones D, Raine K, Latimer C, Santos CR, Nohadani M, Eklund AC, Spencer-Dene B, Clark G, Pickering L, Stamp G, Gore M, Szallasi Z, Downward J, Futreal PA, Swanton C (2012). Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 366(10):883-92. 10.1056/NEJMoa1113205
- Hsieh JJ, Chen D, Wang PI, Marker M, Redzematovic A, Chen YB, Selcuklu SD, Weinhold N, Bouvier N, Huberman KH, Bhanot U, Chevinsky MS, Patel P, Pinciroli P, Won HH, You D, Viale A, Lee W, Hakimi AA, Berger MF, Socci ND, Cheng EH, Knox J, Voss MH, Voi M, Motzer RJ (2017). Genomic Biomarkers of a Randomized Trial Comparing First-line Everolimus and Sunitinib in Patients with Metastatic Renal Cell Carcinoma. Eur Urol 71(3):405-14. 10.1016/j.eururo.2016.10.007
- Pena-Llopis S, Vega-Rubin-de-Celis S, Liao A, Leng N, Pavia-Jimenez A, Wang S, Yamasaki T, Zhrebker L, Sivanand S, Spence P, Kinch L, Hambuch T, Jain S, Lotan Y, Margulis V, Sagalowsky AI, Summerour PB, Kabbani W, Wong SW, Grishin N, Laurent M, Xie XJ, Haudenschild CD, Ross MT, Bentley DR, Kapur P, Brugarolas J (2012). BAP1 loss defines a new class of renal cell carcinoma. Nat Genet 44(7):751-9. 10.1038/ng.2323
- Durinck S, Stawiski EW, Pavia-Jimenez A, Modrusan Z, Kapur P, Jaiswal BS, Zhang N, Toffessi-Tcheuyap V, Nguyen TT, Pahuja KB, Chen YJ, Saleem S, Chaudhuri S, Heldens S, Jackson M, Pena-Llopis S, Guillory J, Toy K, Ha C, Harris CJ, Holloman E, Hill HM, Stinson J, Rivers CS, Janakiraman V, Wang W, Kinch LN, Grishin NV, Haverty PM, Chow B, Gehring JS, Reeder J, Pau G, Wu TD, Margulis V, Lotan Y, Sagalowsky A, Pedrosa I, de Sauvage FJ, Brugarolas J, Seshagiri S (2015). Spectrum of diverse genomic alterations define non-clear cell renal carcinoma subtypes. Nat Genet 47(1):13-21. 10.1038/ng.3146
- Sato Y, Yoshizato T, Shiraishi Y, Maekawa S, Okuno Y, Kamura T, Shimamura T, Sato-Otsubo A, Nagae G, Suzuki H, Nagata Y, Yoshida K, Kon A, Suzuki Y, Chiba K, Tanaka H, Niida A, Fujimoto A, Tsunoda T, Morikawa T, Maeda D, Kume H, Sugano S, Fukayama M, Aburatani H, Sanada M, Miyano S, Homma Y, Ogawa S (2013). Integrated molecular analysis of clear-cell renal cell carcinoma. Nat Genet 45(8):860-7. 10.1038/ng.2699
- Casuscelli J, Weinhold N, Gundem G, Wang L, Zabor EC, Drill E, Wang PI, Nanjangud GJ, Redzematovic A, Nargund AM, Manley BJ, Arcila ME, Donin NM, Cheville JC, Thompson RH, Pantuck AJ, Russo P, Cheng EH, Lee W, Tickoo SK, Ostrovnaya I, Creighton CJ, Papaemmanuil E, Seshan VE, Hakimi AA, Hsieh JJ (2017). Genomic landscape and evolution of metastatic chromophobe renal cell carcinoma. JCI insight 2(12): e92688. 10.1172/jci.insight.92688
- Cancer Genome Atlas Research Network (2013). Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 499(7456):43-9. 10.1038/nature12222
- Cancer Genome Atlas Research Network, et al. (2016). Comprehensive Molecular Characterization of Papillary Renal-Cell Carcinoma. N Engl J Med 374(2):135-45. 10.1056/NEJMoa1505917
- Davis CF, Ricketts CJ, Wang M, Yang L, Cherniack AD, Shen H, Buhay C, Kang H, Kim SC, Fahey CC, Hacker KE, Bhanot G, Gordenin DA, Chu A, Gunaratne PH, Biehl M, Seth S, Kaipparettu BA, Bristow CA, Donehower LA, Wallen EM, Smith AB, Tickoo SK, Tamboli P, Reuter V, Schmidt LS, Hsieh JJ, Choueiri TK, Hakimi AA, Cancer Genome Atlas Research N, Chin L, Meyerson M, Kucherlapati R, Park WY, Robertson AG, Laird PW, Henske EP, Kwiatkowski DJ, Park PJ, Morgan M, Shuch B, Muzny D, Wheeler DA, Linehan WM, Gibbs RA, Rathmell WK, Creighton CJ (2014). The somatic genomic landscape of chromophobe renal cell carcinoma. Cancer Cell 26(3):319-30. 10.1016/j.ccr.2014.07.014
- Sankin A, Hakimi AA, Mikkilineni N, Ostrovnaya I, Silk MT, Liang YP, Mano R, Chevinsky M, Motzer RJ, Solomon SB, Cheng EH, Durack JC, Coleman JA, Russo P, Hsieh JJ (2014). The impact of genetic heterogeneity on biomarker development in kidney cancer assessed by multiregional sampling. Cancer Med-Us 3(6):1485-92. 10.1002/cam4.293
- Zbar B, Brauch H, Talmadge C, Linehan M (1987). Loss of alleles of loci on the short arm of chromosome 3 in renal cell carcinoma. Nature 327(6124):721-4. 10.1038/327721a0
- Hakimi AA, Pham CG, Hsieh JJ (2013). A clear picture of renal cell carcinoma. Nat Genet 45(8):849-50. 10.1038/ng.2708
- Casuscelli J, Vano Y-A, Fridman WH, Hsieh JJ (2017). Molecular Classification of Renal Cell Carcinoma and Its Implication in Future Clinical Practice. Kidney Cancer 1(1):3-13. 10.3233/KCA-170008
- Mitchell TJ, et al. (2018). Timing the Landmark Events in the Evolution of Clear Cell Renal Cell Cancer: TRACERx Renal. Cell 173(3):611-23 e17. 10.1016/j.cell.2018.02.020
- Linehan WM, Lerman MI, Zbar B (1995). Identification of the von Hippel-Lindau (VHL) gene. Its role in renal cancer. JAMA 273(7):564-70. 10.1001/jama.1995.03520310062031
- Lonser RR, Glenn GM, Walther M, Chew EY, Libutti SK, Linehan WM, Oldfield EH (2003). von Hippel-Lindau disease. Lancet. 361(9374):2059-67. 10.1016/S0140-6736(03)13643-4
- Fisher R, Horswell S, Rowan A, Salm MP, de Bruin EC, Gulati S, McGranahan N, Stares M, Gerlinger M, Varela I, Crockford A, Favero F, Quidville V, Andre F, Navas C, Gronroos E, Nicol D, Hazell S, Hrouda D, O’Brien T, Matthews N, Phillimore B, Begum S, Rabinowitz A, Biggs J, Bates PA, McDonald NQ, Stamp G, Spencer-Dene B, Hsieh JJ, Xu J, Pickering L, Gore M, Larkin J, Swanton C (2014). Development of synchronous VHL syndrome tumors reveals contingencies and constraints to tumor evolution. Genome Biol 15(8):433. 10.1186/s13059-014-0433-z.
- Linehan WM, Srinivasan R, Schmidt LS (2010). The genetic basis of kidney cancer: a metabolic disease. Nat Rev Urol 7(5):277-85. 10.1038/nrurol.2010.47
- Semenza GL (2013). HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest 123(9):3664-71. 10.1172/JCI67230
- Frew IJ, Krek W (2008). pVHL: a multipurpose adaptor protein. Sci Signal 1(24):pe30. 10.1126/scisignal.124pe30
- Ivan M, Kaelin WG Jr (2017). The EGLN-HIF O2-Sensing System: Multiple Inputs and Feedbacks. Mol Cell 66(6):772-9. 10.1016/j.molcel.2017.06.002
- Masson N, Ratcliffe PJ (2014). Hypoxia signaling pathways in cancer metabolism: the importance of co-selecting interconnected physiological pathways. Cancer Metab 2(1):3. 10.1186/2049-3002-2-3
- Nakazawa MS, Keith B, Simon MC (2016). Oxygen availability and metabolic adaptations. Nat Rev Cancer 16(10):663-73. 10.1038/nrc.2016.84
- Schito L, Semenza GL (2016). Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer 2(12):758-70. 10.1016/j.trecan.2016.10.016
- Hakimi AA, Reznik E, Lee CH, Creighton CJ, Brannon AR, Luna A, Aksoy BA, Liu EM, Shen RL, Lee W, Chen Y, Stirdivant SM, Russo P, Chen YB, Tickoo SK, Reuter VE, Cheng EH, Sander C, Hsieh JJ (2019). An Integrated Metabolic Atlas of Clear Cell Renal Cell Carcinoma. Cancer Cell 29(1):104-16. 10.1016/j.ccell.2015.12.004
- Kapitsinou PP, Haase VH (2008). The VHL tumor suppressor and HIF: insights from genetic studies in mice. Cell Death Differ 15(4):650-9. 10.1038/sj.cdd.4402313
- Nargund AM, Pham CG, Dong Y, Wang PI, Osmangeyoglu HU, Xie Y, Aras O, Han S, Oyama T, Takeda S, Ray CE, Dong Z, Berge M, Hakimi AA, Monette S, Lekaye CL, Koutcher JA, Leslie CS, Creighton CJ, Weinhold N, Lee W, Tickoo SK, Wang Z, Cheng EH, Hsieh JJ (2017). The SWI/SNF Protein PBRM1 Restrains VHL-Loss-Driven Clear Cell Renal Cell Carcinoma. Cell Rep 18(12):2893-906. 10.1016/j.celrep.2017.02.074
- Wolf MM, Kimryn Rathmell W, Beckermann KE (2020). Modeling clear cell renal cell carcinoma and therapeutic implications. Oncogene 39(17):3413-3426. 10.1038/s41388-020-1234-3
- Lee CH, Pham CG, Hsieh JJ (2015). PBRM1: A Critical Subunit of the SWI/SNF Chromatin Redmodeling Complex. Renal Cell Carcinoma. In: Bukowski RM, Motzer RJ, Figlin RA, editors. Molecular Targets and Clinical Applications. Springer, New York; pp 111-51. 10.1007/978-1-4939-1622-1_5
- Becerra MF, Manley BJ, Casuscelli J, Redzematovic A, Tennenbaum DM, Motzer RJ, Feldman DR, Voss MH, Arcila M, Russo P, Coleman JA, Hsieh JJ, Hakimi AA (2016). Genomic Lanscape of Clear Cell Renal Cell Carcinoma Metastasis. J Urology 195(4):E919-E20.
- Chen FJ,et al. (2016). Multilevel Genomics-Based Taxonomy of Renal Cell Carcinoma. Cell Rep 14(10):2476-89. 10.1016/j.celrep.2016.02.024
- Hakimi AA, Ostrovnaya I, Reva B, Schultz N, Chen YB, Gonen M, Liu H, Takeda S, Voss MH, Tickoo SK, Reuter VE, Russo P, Cheng EH, Sander C, Motzer RJ, Hsieh JJ (2013). Adverse Outcomes in Clear Cell Renal Cell Carcinoma with Mutations of 3p21 Epigenetic Regulators BAP1 and SETD2: A Report by MSKCC and the KIRC TCGA Research Network. Clin Cancer Res 19(12):3259-67. 10.1158/1078-0432.CCR-12-3886
- Hakimi AA, Chen YB, Wren J, Gonen M, Abdel-Wahab O, Heguy A, Liu H, Takeda S, Tickoo SK, Reuter VE, Voss MH, Motzer RJ, Coleman JA, Cheng EH, Russo P, Hsieh JJ (2013). Clinical and pathologic impact of select chromatin-modulating tumor suppressors in clear cell renal cell carcinoma. Eur Urol 63(5):848-54. 10.1016/j.eururo.2012.09.005
- Clapier CR, Cairns BR (2009). The biology of chromatin remodeling complexes. Annu Rev Biochem 78:273-304. 10.1146/annurev.biochem.77.062706.153223
- Biegel JA, Busse TM, Weissman BE (2014). SWI/SNF chromatin remodeling complexes and cancer. Am J Med Genet C Semin Med Genet 166C(3):350-66. 10.1002/ajmg.c.31410
- Hodges C, Kirkland JG, Crabtree GR (2016). The Many Roles of BAF (mSWI/SNF) and PBAF Complexes in Cancer. Cold Spring Harb Perspect Med 6(8): a026930. 10.1101/cshperspect.a026930
- Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, Crabtree GR (2013). Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet 45(6):592-601. 10.1038/ng.2628
- Varela I, et al. (2011). Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature 469(7331):539-42. 10.1038/nature09639
- Gu YF, Cohn S, Christie A, McKenzie T, Wolff NC, Do QN, Madhuranthakam A, Pedrosa I, Wang T, Dey A, Busslinger M, Xie XJ, Hammer RE, McKay RM, Kapur P, Brugarolas J (2017). Modeling Renal Cell Carcinoma in Mice: Bap1 and Pbrm1 Inactivation Drive Tumor Grade. Cancer Discov 7(8):900-917. 10.1158/2159-8290.CD-17-0292
- Espana-Agusti J, Warren A, Chew SK, Adams DJ, Matakidou A (2017). Loss of PBRM1 rescues VHL dependent replication stress to promote renal carcinogenesis. Nat Commun 8(1):2026. 10.1038/s41467-017-02245-1
- Nargund AM, Osmanbeyoglu HU, Cheng EH, Hsieh JJ (2017). SWI/SNF tumor suppressor gene PBRM1/BAF180 in human clear cell kidney cancer. Mol Cell Oncol 4(4):e1342747. 10.1080/23723556.2017.1342747
- Luo W, Semenza GL (2012). Emerging roles of PKM2 in cell metabolism and cancer progression. Trends Endocrinol Metab 23(11):560-6. 10.1016/j.tem.2012.06.010
- Jung JE, Lee HG, Cho IH, Chung DH, Yoon SH, Yang YM, Lee JW, Choi S, Park JW, Ye SK, Chung MH (2005). STAT3 is a potential modulator of HIF-1-mediated VEGF expression in human renal carcinoma cells. FASEB J 19(10):1296-8. 10.1096/fj.04-3099fje
- Gao W, Li W, Xiao T, Liu XS, Kaelin WG Jr (2017). Inactivation of the PBRM1 tumor suppressor gene amplifies the HIF-response in VHL-/-clear cell renal carcinoma. Proc Natl Acad Sci U S A 114(5):1027-32. 10.1073/pnas.1619726114
- Saxton RA, Sabatini DM (2017). mTOR Signaling in Growth, Metabolism, and Disease. Cell 169(2):361-71. 10.1016/j.cell.2017.03.035
- Xu J, Pham CG, Albanese SK, Dong Y, Oyama T, Lee CH, Rodrik-Outmezguine V, Yao Z, Han S, Chen D, Parton DL, Chodera JD, Rosen N, Cheng EH, Hsieh JJ (2016). Mechanistically distinct cancer-associated mTOR activation clusters predict sensitivity to rapamycin. J Clin Invest 126(9):3526-40. 10.1172/JCI86120
- Voss MH, Hakimi AA, Pham CG, Brannon AR, Chen YB, Cunha LF, Akin O, Liu H, Takeda S, Scott SN, Socci ND, Viale A, Schultz N, Sander C, Reuter VE, Russo P, Cheng EH, Motzer RJ, Berger MF, Hsieh JJ (2014). Tumor genetic analyses of patients with metastatic renal cell carcinoma and extended benefit from mTOR inhibitor therapy. Clin Cancer Res 20(7):1955-64. 10.1158/1078-0432.CCR-13-2345
- Kwiatkowski DJ, Choueiri TK, Fay AP, Rini BI, Thorner AR, De Velasco G, Tyburczy M, Hamieh L, Albiges L, Agarwal N, Ho TH, Song J, Pignon JC, Barrios PM, Michaelson MD, Van Allen EM, Krajewski KM, Porta C, Pal SK, Bellmunt J, McDermott DF, Heng DY, Gray KP, Signoretti S (2016). Mutations in TSC1, TSC2, and MTOR are associated with response to rapalogs in patients with metastatic Renal Cell Carcinoma. Clin Cancer Res 22(10):2445-52. 10.1158/1078-0432.CCR-15-2631
- Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, Hafen E, Witters LA, Ellisen LW, Kaelin WG Jr (2004). Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev 18(23):2893-904. 10.1101/gad.1256804
- Yu H, Pardoll D, Jove R (2009). STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer 9(11):798-809. 10.1038/nrc2734
- Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, Tykodi SS, Sosman JA, Procopio G, Plimack ER, Castellano D, Choueiri TK, Gurney H, Donskov F, Bono P, Wagstaff J, Gauler TC, Ueda T, Tomita Y, Schutz FA, Kollmannsberger C, Larkin J, Ravaud A, Simon JS, Xu LA, Waxman IM, Sharma P, CheckMate I (2015). Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med 373(19):1803-13. 10.1056/NEJMoa1510665
- Motzer RJ, Tannir NM, McDermott DF, Aren Frontera O, Melichar B, Choueiri TK, Plimack ER, Barthelemy P, Porta C, George S, Powles T, Donskov F, Neiman V, Kollmannsberger CK, Salman P, Gurney H, Hawkins R, Ravaud A, Grimm MO, Bracarda S, Barrios CH, Tomita Y, Castellano D, Rini BI, Chen AC, Mekan S, McHenry MB, Wind-Rotolo M, Doan J, Sharma P, Hammers HJ, Escudier B, CheckMate I (2018). Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N Engl J Med 378(14):1277-1290. 10.1056/NEJMoa1712126
- Miao D, Margolis CA, Gao W, Voss MH, Li W, Martini DJ, Norton C, Bosse D, Wankowicz SM, Cullen D, Horak C, Wind-Rotolo M, Tracy A, Giannakis M, Hodi FS, Drake CG, Ball MW, Allaf ME, Snyder A, Hellmann MD, Ho T, Motzer RJ, Signoretti S, Kaelin WG, Jr., Choueiri TK, Van Allen EM (2018). Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science 359(6377):801-806. 10.1126/science.aan5951
- Braun DA, Ishii Y, Walsh AM, Van Allen EM, Wu CJ, Shukla SA, Choueiri TK (2019). Clinical Validation of PBRM1 Alterations as a Marker of Immune Checkpoint Inhibitor Response in Renal Cell Carcinoma. JAMA Oncol 5(11):1631-1633. 10.1001/jamaoncol.2019.3158
- Liu XD, Kong W, Peterson CB, McGrail DJ, Hoang A, Zhang X, Lam T, Pilie PG, Zhu H, Beckermann KE, Haake SM, Isgandrova S, Martinez-Moczygemba M, Sahni N, Tannir NM, Lin SY, Rathmell WK, Jonasch E (2020). PBRM1 loss defines a nonimmunogenic tumor phenotype associated with checkpoint inhibitor resistance in renal carcinoma. Nat Commun 11(1):2135. 10.1038/s41467-020-15959-6
- McDermott DF, Huseni MA, Atkins MB, Motzer RJ, Rini BI, Escudier B, Fong L, Joseph RW, Pal SK, Reeves JA, Sznol M, Hainsworth J, Rathmell WK, Stadler WM, Hutson T, Gore ME, Ravaud A, Bracarda S, Suarez C, Danielli R, Gruenwald V, Choueiri TK, Nickles D, Jhunjhunwala S, Piault-Louis E, Thobhani A, Qiu J, Chen DS, Hegde PS, Schiff C, Fine GD, Powles T (2018). Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat Med 24(6):749-57. 10.1038/s41591-018-0053-3
–
ACKNOWLEDGMENTS
JJH and EHC are supported by NIH R01 CA223231.
COPYRIGHT
© 2020

Exploiting the circuit breaker cancer evolution model in human clear cell renal cell carcinoma by Hsieh and Cheng is licensed under a Creative Commons Attribution 4.0 International License.


