Reviews:
Cell Stress, Vol. 1, No. 2, pp. 79 - 89; doi: 10.15698/cst2017.11.110
Emerging roles of mitochondria and autophagy in liver injury during sepsis

1 Department of Forensic Medicine, Graduate School of Medical and Dental Sciences, Tokyo Medical and Dental University, 1-5-45, Yushima, Bunkyo-ku, Tokyo, 113-8519, Japan.
Keywords: autophagy, mitophagy, non-canonical autophagy, sepsis, mitochondrial dysfunction, liver, immune response, secretion.
Abbreviations:
ALT – alanine transaminase,
ATG – autophagy related,
ATP – adenosine triphosphate,
CLP – cecal ligation and puncture,
CPS-1 – carbamoylphosphat synthase,
GSDM – gasdermin,
IL – interleukin,
KO – knock out,
LPS – lipopolysaccharide,
NAAP – nicotic acid adenine dinucleotide phosphate,
PI3K – phosphatidylinositol-3-kinase,
ROS – reactive oxygen species,
TLR – toll-like receptor,
TNF – tumor necrosis factor,
ULK – Unc-51-like kinase,
WT – wild type.
Received originally: 15/06/2017 Received in revised form: 17/09/2017
Accepted: 18/09/2017
Published: 02/10/2017
Correspondence:
Toshihiko Aki, PhD, Department of Forensic Medicine, Graduate School of Medical and Dental Sciences, Tokyo Medical and Dental University, 1-5-45, Yushima, Bunkyo-ku, Tokyo, 113-8519, Japan; Tel: +81-3-5803-5978; Fax: +81-3-5803-0128 aki.legm@tmd.ac.jp
Conflict of interest statement: The authors declare that there is no conflict of interest to disclose.
Please cite this article as: Toshihiko Aki, Kana Unuma and Koichi Uemura (2017). Emerging roles of mitochondria and autophagy in liver injury during sepsis. Cell Stress 1(2): 79-89. doi: 10.15698/cst2017.11.110
Abstract
Recent research indicates crucial roles of autophagy during sepsis. In animal models of sepsis induced by cecal ligation and puncture (CLP) or the systemic administration of lipopolysaccharides (LPS), autophagy is implicated in the activation and/or damage of various cells/organs, such as immune cells, heart, lung, kidney, and liver. Since sepsis is associated with an increased production of pro- as well as anti-inflammatory cytokines, it has long been considered that hypercytokinemia is a fetal immune response leading to multiple organ failure (MOF) and mortality of humans during sepsis. However, a recent paradigm illuminates the crucial roles of mitochondrial dysfunction as well as the perturbation of autophagy in the pathogenesis of sepsis. In the livers of animal models of sepsis, autophagy is involved in the elimination of damaged mitochondria to prevent the generation of mitochondrial ROS and the initiation of the mitochondrial apoptotic pathway. In addition, many reports now indicate that the role of autophagy is not restricted to the elimination of hazardous malfunctioning mitochondria within the cells; autophagy has been shown to be involved in the regulation of inflammasome activation and the release of cytokines as well as other inflammatory substances. In this review, we summarize recent literature describing the versatile role of autophagy and its possible implications in the pathogenesis of sepsis in the liver.
INTRODUCTION
Autophagy, derived from the Greek (auto” meaning “self” and “phagy” meaning “eat”), is a cellular system to degrade intracellular unnecessary materials, including proteins and organelles, for recycling [1]. Until now, three types of autophagy have been reported; macroautophagy, microautophagy, and chaperone-mediated autophagy. However, molecular mechanisms and physiological implications of the latter two types of autophagy (microautophagy, and chaperone-mediated autophagy) are still obscure compared to macroautophagy. In this review, therefore, we feature only macroautophagy and hereafter refer to macroautophagy as autophagy. During autophagy, cytoplasmic materials are engulfed by newly synthesized vesicles, which are called isolation membranes or phagophores [2][3]. Closed phagophores in which cytoplasmic constituents are included become to be double membrane structures called autophagosomes. Outer membrane of autophagosomes fuses with lysosomal membrane and resultant fusion structures are called autolysosomes. Cytoplasmic constituents within the inner membrane of autophagosomes are delivered into the luminal space of lysosomes, where hydrolytic enzymes such as cathepsins degrade them.
–
Autophagy is executed through a set of genes called atg (autophagy related) genes [4]. Unc-51-like kinase-1 and -2 (ULK1 and ULK2), mammalian homologues of yeast Atg1, form a complex with Atg13, FAK family kinase-interacting protein of 200 kDa (FIP200), and Atg101 [3]. In healthy cells, this ULK complex resides in the cytosol and is inactivated by mammalian target of rapamycin complex (mTORC). Upon starvation, mTORC is inactivated and the ULK complex becomes activated to initiate the process of autophagy. One of the targets of ULK complex is class III phosphatidylinositol 3-kinase (PI3K) complex, which includes beclin-1 (Atg6) and Atg14. Class III PI3K complex facilitates the production of phosphatidylinositol 3-phosphate (PI3P) for autophagosome formation. Ubiqutin-like protein conjugation system is also involved in the process of autophagy [5]. For example, after conjugation with the ubiquitin-like Atg12 protein, the Atg5 protein works as an E3 ubiquitin ligase to conjugate phosphatidylethanolamine (PE) to Atg8 [5][6]. Atg7 is an E1 ubiquitin-activating enzyme that activates both Atg8 and Atg12 [5][6]. These two ubiqutin-like conjugation systems characterize the process of autophagy.
–
By generating liver-specific atg5 or atg7 gene knock out (KO) animals, it has been demonstrated that basal autophagy is required to maintain liver homeostasis. Komatsu, et al. showed that in liver-specific conditional atg7 KO mice, starvation-induced autophagy is severely impaired [7]. Conditional atg7 KO mice also develop a variety of degenerative syndromes, such as hepatomegaly in response to antibacterial drugs, steatosis, and the accumulation of protein aggregates resembling Mallory bodies [7]. Development of multiple liver tumors has also been reported in mice with partial deletion of atg5 [8]. Anyway, autophagy protects hepatocytes and maintains homeostasis in both healthy and degenerating liver.
–
Sepsis caused by infection, trauma, or endotoxins leads to an excessive and uncontrolled immune cell response [9][10]. Sepsis has an extremely high mortality rate with approximately 50% of patients delivered to the ICU (intensive care unit) with suspected sepsis eventually dying [11]. During sepsis, the acute inflammatory phase leads to serious shock, which finally causes multiple organ failure (MOF) and subsequent mortality. Despite the self-explanatory role of inflammation in the pathogenesis of sepsis, there is also evidence against a critical role of immune reactions in the development of sepsis; whether or not there is a correlation between plasma levels of circulating cytokines and severity of outcome in patients suffering from sepsis remains a subject of debate [12][13]. In addition, elevated blood cytokine levels usually resolve during the progression of sepsis.
–
Concurrent with the rising suspicion that uncontrolled immune responses might not play a central role in the pathogenesis of sepsis, mitochondrial dysfunction has come into the spotlight. Examinations of skeletal muscle biopsy samples from critically ill patients suffering from sepsis have shown that mitochondrial damage is more severe in patients who subsequently die than in those who eventually survive [14]. Moreover, in contrast to the transient elevation in circulating cytokines, mitochondrial dysfunction persists even after cytokine levels return to basal levels [15]. Given these indications, although cytokine production and mitochondrial dysfunction should be mutually connected, at least during the acute phase of the inflammatory response, the high mortality rate of sepsis might be attributable not only to the cytokine storm that eventually quiets down, but also to mitochondrial dysfunction that persists throughout the duration of the disease.
MITOCHONDRIAL REPROGRAMMING IN MACROPHAGES DURING SEPSIS
The molecular mechanisms underlying mitochondrial dysfunction caused by LPS (lipopolysaccharide)stimulation in the liver are poorly understood. On the other hand, a possible mechanism for mitochondrial reprogramming in immune cells has recently been proposed. Macrophages undergo metabolic reprogramming upon activation by immunostimulation such as LPS stimulation [16]. Along with this change, “rested” macrophages become “activated” cells, in which cytokine production is activated. During this cellular transition from an anti-inflammatory to a pro-inflammatory phenotype, macrophages become more dependent on glycolysis rather than mitochondrial OXPHOS (oxidative phosphorylation) for survival [16][17]. Hypoxia-inducible factor1α (HIF1α) is the inducible subunit of HIF1. HIF is a transcriptional activating complex that activates glycolytic genes, vascular endothelial growth factor (VEGF), and erythropoietin (EPO) in response to hypoxia. During this transition, the mitochondrial metabolite succinate accumulates in cells and stabilizes HIF1α by suppressing prolyl hydroxylase, which ordinarily leads HIF1α to proteasomal degradation in healthy cells [18]. The stabilization of HIF1α in LPS-stimulated macrophages not only results in a cellular transition of metabolism, but also contributes to cytokine production by binding directly to the IL-1β promoter [18]. Furthermore, increased succinate levels are thought to cause reverse electron transport at Complex I, which leads to ROS (reactive oxygen species) generation from the complex [19]. ROS have been shown to be required for inflammasome activation and the subsequent production of inflammatory cytokines [20]. Furthermore, increased succinate levels in LPS-stimulated macrophages contribute to inflammatory cytokine production by activating HIF1α as well as inflammasomes. Nevertheless, it remains to be examined whether this mechanism is also responsible for the generation of ROS in other cells. Furthermore, molecules that pave the pathway from LPS ligation to mitochondrial reprogramming are also poorly understood to date.
AUTOPHAGY IN THE LIVER
Direct evidence of autophagy in liver injury during sepsis has been reported by Watanabe, et al. [21]. They surveyed liver samples from patients suffering from observable sepsis, as well as from CLP (cecal ligation and puncture)-induced sepsis model mice, and showed a massive accumulation of autophagic vacuoles in hepatocytes [21]. Later, it was shown that autophagy plays a crucial role in protecting hepatocytes from septic insults. For example, in atg7 KO mice, liver injuries caused by LPS/D-galactosamine (GalN), a potent hepatotoxin [22], are more severe than in wild type (WT) mice [23]. Likewise, it has been demonstrated that the systemic administration of LPS causes more severe damage in atg7-deficient liver than in WT liver [24].
–
Although its role in protecting hepatocytes against septic insult has been established, the cellular degradation activity of autophagy might not be fully activated in the liver during sepsis: several reports have pointed out perturbations in autophagy. There is a report indicating that in the hearts of sepsis model mice, autophagic flux is increased during the initial phase of sepsis, but declines at a later phase [25]. The same research group has also reported similar results in the liver during sepsis [26]. These observations coincide with the damage of lysosomes in LPS-stimulated cells. Nevertheless, other reports indicate the completion of the entire autophagy process in the liver of CLP-sepsis mice [27].
INFLAMMATORY CYTOKINES AND SEPTIC LIVER INJURY
Several studies have proposed TNFα as a crucial mediator of liver toxicity during sepsis [23][24][28]. Lower levels of TNFα secretion are observed in WT Kupffer cells, liver-residential macrophages, than in atg7-deficient Kupffer cells, suggesting that autophagy suppresses TNFα secretion from the cells [29]. Therefore, this should represent one of the mechanisms by which autophagy ameliorates liver injury. Stellate cells are also responsible for the secretion of pro-inflammatory cytokines in response to LPS stimulation in the liver [30]. Since hepatic stellate cells reside mainly along the sinusoids, these cells should participate in the earliest immune responses in the liver during sepsis. Hepatocytes can also secrete TNFα, at least in response to IL-1β stimulation [31]. In addition, hepatocytes themselves express the LPS receptor, Toll-like receptor 4 (TLR4). Thus, TNFα secreted from Kupffer and stellate cells, as well as from hepatocytes themselves, should be responsible for hepatocyte injury (Fig. 1).
–
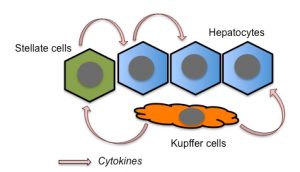 | FIGURE 1: Release of inflammatory cytokines from hepatocytes, Kupffer cells, and stellate cells during liver inflammation caused by LPS. In the liver of mice systemically administered LPS, residential liver macrophages, Kupffer cells are activated to secrete inflammatory cytokines such as TNFα and IL-1β. These cytokines in turn activate stellate cells as well as hepatocytes. Both stellate cells and hepatocytes themselves are activated by LPS and subsequently release cytokines. |
–
LPS binds to its cognate receptor, TLR4, and promotes the synthesis of cytokines, chemokines, and type-I interferons [32]. TLR4 signaling is mediated by a number of adapter molecules, such as MyD88, TIRAP, TRIF, and TRAM. TLR4 signaling could be divided into MyD88-dependent and -independent pathways. For example, cytokine and interferon productions in response to LPS stimulation are mediated by MyD88-dependent and -independent pathways, respectively. There are at least four types of inflammasome, NLRP1, NLRP3, NLRC4, and AIM2 [32]. Activation of inflammasomes, such as the NLRP3 inflammasomes, for which the mechanism of activation is best characterized, occurs in at least two steps (Fig. 2). During the first step, NF-kB is activated to induce the expression of NLRP3 as well as cytokines. Then, in the second step, NLPR3 forms a complex, the so-called “inflammasome”, with other molecules including caspase-1 (also called IL-1β-converting enzyme or ICE) and ASC. Indeed, it has been shown that LPS stimulation rapidly induces the expression of NLRP3 and other inflammasome components in hepatocytes [33][34]. This induction is followed by the expression of TNFα as well as IL-1β, suggesting that hepatocytes produce inflammatory cytokines in response to LPS stimulation through the formation of inflammasomes [33][34]. In parallel with inflammasome activation, LPS damages mitochondria as well as lysosomes. ROS from damaged mitochondria as well as cathepsins from lysosomes have been shown to be essential for inflammasome activation [35][36]. Indeed, autophagy negatively affects inflammasome activation by eliminating ROS from mitochondria [37].
–
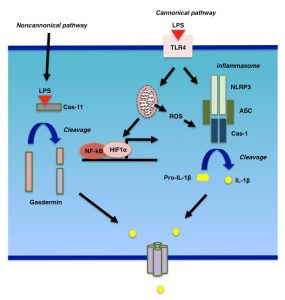 | FIGURE 2: Cytokine secretion in response to LPS stimulation. There are two pathways for the LPS-initiated secretion of cytokines. In the conventional pathway, LPS binds to its cognate cell surface receptor, TLR4. The activation of TLR4 leads to the activation of NF-kB as well as HIF1α, the latter of which is mediated by mitochondrial dysfunction. Both NF-kB and HIF1α are involved in the expression cytokines as well as inflammasome components. Inflammasomes, comprising NLRP3, ASC, and Cas-1 for example, are involved in the activation of Cas-1. In an unconventional pathway, LPS binds directly to cytoplasmic Cas-11, which cleaves gasdermin to form a pore in the plasma membrane. |
–
In addition to the canonical pathway for intracellular LPS signaling, which is initiated by the ligation of LPS to TLR4, LPS also binds directly to and activates cytosolic caspase-11 in mice (caspase-4 and -5 are the human homologues of murine caspase-11) [38][39]. This pathway is a non-cannonical pathway that leads macrophages to a certain form of cell death called pyroptosis. During pyroptosis, caspase-1 cleaves gasdermin D (GSDMD) [40][41], and the cleaved GSDMD assembles on the plasma membrane to form a pore through which IL-1β or other inflammatory modulators, such as HMGB1, are secreted [42]. Recently, it has been shown that gasdermin E (GSDME), also known as DFNA5, fulfills a similar role to GSDMD in non-immune cells [43][44]. To obtain pore-forming status, GSDME is cleaved by caspase-3 rather than caspase-1 [43][44]. The role of gasdermins in LPS-stimulated liver injury is a subject awaiting examination.
CARBAMOYLPHOSPHATE SYNTHASE-1
Carbamoylphosphate synthase1 (CPS-1) is a urea cycle enzyme that catalyzes the generation of carbamoyl phosphate from ammonia and bicarbonate. CPS-1 expression is restricted to the liver and to some extent the intestine. In an experiment to find liver proteins whose expressions are dysregulated during sepsis, Struck et al., using a comparative proteomics approach, found CPS-1 [15]. They also showed that plasma CPS-1 levels in baboons increase gradually over time following the injection of LPS. The kinetics of plasma CPS-1 levels differs sharply from that of TNFα: CPS-1 levels begin to increase several hours after LPS injection and continue increasing for the duration of the experiment [15]. On the other hand, TNFα levels increase rapidly within several minutes of the injection and return to basal levels within several hours [15]. Thus, when the transient increase in TNFα in circulation has resolved, CPS-1 levels in turn begin to increase. The increase in CPS-1 in circulation during sepsis should not derive from the enzymatic nature of this protein, but rather from its cellular localization: CPS-1 localizes to the mitochondrial matrix space. Indeed, Crouser et al. also confirmed the release of CPS-1 into circulation in CLP-sepsis model mice [45]. They showed severe mitochondrial degeneration, such as decreased respiration and increased carbonylation of mitochondrial proteins [45]. In their experiments, the increase of CPS-1 in the plasma took place much faster than that of alanine transaminase (ALT) during CLP-sepsis: peak CPS-1 levels were observed after 24 hours while ALT levels peaked after 48 hours of CLP injection [45]. Thus, the occurrence of plasma CPS-1 should result from the release of liver mitochondria into circulation, not from hepatocyte necrosis. They also supposed the lysosomal clearance of damaged mitochondria through autophagy, so-called mitophagy [45].
–
CPS-1 release into the circulation prior to ALT during sepsis has also been confirmed in the LPS model [46]. Furthermore, we have also shown that CPS-1 release from LPS-stimulated hepatocytes is associated with the release of mitochondrial, autophagosomal, and lysosomal proteins, confirming the lysosomal exocytosis of autophagosomes that include mitochondria [47]. Lysosomal exocytosis also occurs in LPS-stimulated MEF cells, but not in LPS-stimulated atg5-KO MEF cells, further confirming the essential role of autophagy in this process [47].
MODULATION OF LIVER INJURY DURING SEPSIS THROUGH AUTOPHAGY-MODULATING COMPOUNDS
In accordance with the supposed perturbation of autophagy at least in the later stages of sepsis [25][26], a panel of autophagy-inducing compounds has been shown to ameliorate septic liver injury (Table 1). Carbamazepine (CBZ), which is an anti-epileptic drug that was recently identified as an autophagy enhancer, can alleviate liver injury in mice with CLP-sepsis [26]. In CLP-mice, the bacterial loads to the liver and circulation are significantly suppressed by CBZ, suggesting the role of autophagy in bacterial clearance [26]. Another report points to the importance of Ca2+-signaling in LPS-induced autophagy in the liver. Nicotinic acid adenine dinucleotide phosphate (NAADP) is a Ca2+-mobilizing molecule that is different from cyclic ADP-ribose (cADPR) and myo-inositol 1,4,5-triphosphate (IP3) [48]. NAADP mobilizes Ca2+ from different intracellular storage sites, including ER and lysosomes [48], and intracellular levels of NAADP are regulated by CD38, an ADP-ribosyl cyclase. In the liver of LPS-induced sepsis model mice, it has been shown that CD38 KO results in decreased autophagy in response to LPS stimulation [49]. LPS/GalN-induced liver injuries such as apoptosis and hemorrhage are also decreased in CD38-KO mice as compared to WT mice [49]. An aglycone, genipin, which has been shown to reduce LPS/GalN-induced liver injury [50], was recently demonstrated to provide liver protection by up-regulating autophagy [51].
–
| Table 1. Chemicals that affect liver injury during sepsis by modulating autophagy. |
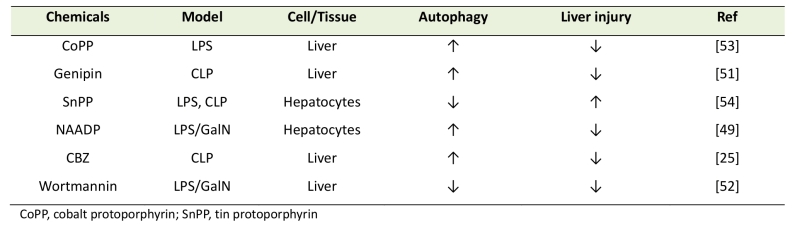 |
| [25][49][51][52][53][54] |
–
On the other hand, there are several reports demonstrating autophagy as an exacerbating factor in sepsis (Table 2). Wortmannin is a PI3K inhibitor that is frequently used as an inhibitor of autophagy [55] because PI3K is required in the early stages of autophagy [56]. Liver injuries caused by LPS/GalN in mice have been shown to be alleviated by pretreatment with wortmannin [52]: wortmannin alleviates not only LPS/GalN-induced increases in autophagy, but also cytokine production, ERK and JNK activation, and the death of hepatocytes. Deteriorated LPS-induced liver injury has also been reported in mice deficient in Kir6.2, an ATP-sensitive K+-channel [57]. In this case, however, increased autophagy in response to LPS was observed in KO mice as compared to WT mice, suggesting that autophagy might be involved in liver injury rather than liver protection.
–
| Table 2. Release of intracellular contents from cells. |
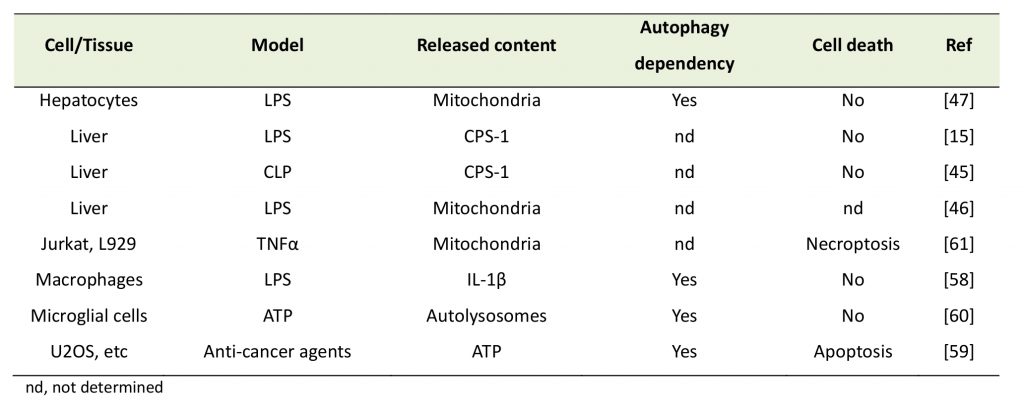 |
| [15][45][46][47][58][59][60][61] |
–
AUTOPHAGY-DEPENDENT AND –INDEPENDENT CASES OF INTRACELLULAR CONTENT SECRETION DURING IMMUNE RESPONSES
Although autophagy is considered to be an intracellular degradation mechanism, recent research progress has shown it plays roles in other processes than the intracellular degradation of excess and/or unnecessary materials (Fig. 3). Unconventional functions of autophagy include a broad range of cellular activities, among which is the secretion of intracellular contents. Recent research suggests that autophagy plays important roles in septic liver failure, not only through intracellular degradation, but also via the secretion of cell contents.
–
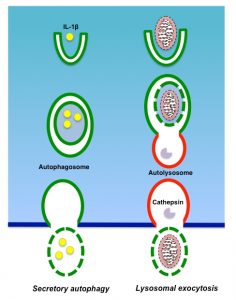 | FIGURE 3: Secretion through autophagy machinery: secretory autophagy and lysosomal exocyto-sis. During secretory autophagy, autophagosomes do not fuse with lysosomes, but are delivered to the plasma membrane. During lysosomal exocytosis, autolysosomes fuse with the plasma membrane to extrude the contents. |
–
TFEB, which has been identified as a transcriptional activator of lysosomal biogenesis [62], has also been found to be an inducer of autophagy [63] and lysosomal exocytosis [64]. In healthy and/or nutrient-rich cells, TFEB is phosphorylated by the mTORC1 at the lysosomal membrane and retained in the cytoplasm [65]. The cytoplasmic chaperone 14-3-3 is involved in this cytoplasmic retention by binding to phosphorylated TFEB [66]. In unhealthy and/or starved cells, TFEB is activated through the release of calcium ions from lysosomes into the cytoplasm, a process that is mediated by the lysosomal membrane calcium ion channel, MCOLN1 [64]. A cytoplasmic calcium-dependent phosphatase, calcineurin, is responsible for the dephosphorylation of TFEB [67]. It should be noted that MCOLN1 has also been suggested to be an NAADP-dependent calcium channel [49], although other channels have also been proposed as receptors for NAADP. It has also been shown that the CD38-catalized generation of NAADP and the resultant calcium mobilization lead to TFEB activation [49].
–
Autophagy-dependent secretion of IL-1β
The secretion of IL-1β is one of the most important responses of immune cells during sepsis. Since IL-1β is a leaderless protein and is not secreted via a conventional secretary pathway, the mechanism of its secretion remains to be elucidated. IL-1β has been shown to be secreted by an autophagy-dependent mechanism under some circumstances [58]. Further studies by the same group have identified TRIM16 as a receptor recognizing IL-1β-containing cargo as well as sec22b, syntaxin 3/4, and SNAP23/29 as components of the SNARE complex that mediates the docking of autophagosomes to the plasma membrane [68].
–
Mast cell degranulation
Using bone-marrow-derived mast cells (BMMCs), Ushio et al. have shown that the ligation of the Fc receptor, which triggers the secretion of mediators of allergic responses such as histamine and leukotriene, is an autophagy-dependent process [69]. They showed that although bone marrow cells derived from atg7-KO mice differentiate mast cells as WT cells do, mast cells derived from atg7-KO mice do not secrete granules in response to Fc receptor ligation, confirming autophagy-dependent degranulation [69]. They also showed the co-localization of LC3-II and CD63, a marker of secretory lysosomes. Zhang et al. have also shown that mast cells stimulated by IgE secrete mitochondrial contents including mtDNA and ATP [70]. They have shown that the addition of mitochondrial contents to mast cells in turn causes cell degranulation during which not only histamines and prostaglandins, but also IL-1β, TNFα, and IL-18 are secreted [70]. Although they did not examine whether this process is dependent on autophagy or not, the secretion of mitochondrial components should occur during the degranulation of mast cells and be involved in allergic responses.
–
Mitochondrial extrusion via an autophagy-independent pathway
Nakajima and colleagues showed that MEFs from c-Flip KO mice stimulated by TNFα extrude mitochondria from the cells [71]. Prior to their release into the extracellular milieu, the mitochondria undergo fragmentation and cytoplasmic vacuolization is associated with the secretion [71]. The cytoplasmic vacuoles seem to be derived from the plasma membrane since the membranes of the vacuoles were stained by the FM1-43 dye, which is incorporated into the plasma membrane. It has also been shown that z-VAD, cycloheximide, cytochalasin B, and pacilitaxel can all suppress the mitochondrial extrusion, suggesting that caspase activity, ongoing protein synthesis, actin and the tubulin cytoskeleton are all required for this phenomenon [71]. Interestingly, 3MA, an inhibitor of autophagy, scarcely affects mitochondrial extrusion, suggesting that autophagy should not be involved in the process.
–
ATP release from cells through autophagy-dependent lysosomal exocytosis
ATP is often released from damaged cells and acts in DAMPs (damage-associated molecular patterns) to activate immune cells. Tumor cells often show immunological responses, such as the release of ATP, during chemotherapy-induced cell death [72]. Martins et al. showed that the secretion of ATP is executed through the exocytosis of lysosomes in which ATP is stored [59]. Autophagy is involved in this ATP secretion since it has been shown that the inhibition of autophagy does not interfere with lysosomal exocytosis but does block ATP secretion [59].
–
Autolysosome release from microglial cells
Takenouchi et al. have provided evidence for the release of autolysosomal contents from cells [60]. In microglial cells stimulated by ATP through the purinergic receptor P2X7, they found that LC3-II levels increase over time [60]. This increase cannot be attributed to the induction of autophagy because 3MA does not suppress the increase in LC3-II. Rather, they found that lysosomal activity is impaired. Lysosomal dysfunction might be attributed, at least in part, to the release of lysosomal contents from the cells. The content released from the cells consists not only of lysosomal luminal proteins such as cathepsins, but also autophagosomal proteins such as LC3-II. Thus, after fusion with lysosomes, autophagosomes are digested within lysosomes as well as secreted into the extracellular space.
–
Release of mitochondria from TNFα-stimulated cells
Necroptosis is a form of programmed cell death that is executed by defined molecules such as receptor-interacting serine/threonine-protein (RIP) 1 and 3, and MLKL (mixed lineage kinase domain like pseudokinase), but morphologically resembles necrosis rather than apoptosis [73]. Necroptosis occurs when cells do not undergo apoptosis for some reason, such as a lack of caspases. During TNFα-induced necroptosis, Maeda and Fadeel found that mitochondria are released from the cells into the extracellular space [61]. Importantly, they showed that the released mitochondria are intact, as they contain mtDNA and stain positively with mitochondrial membrane potential-dependent dyes such as MitoTracker. The release of mitochondria is efficiently inhibited by the necroptosis inhibitor nec-1s, suggesting the involvement of necroptosis machinery in the secretion. They also showed that the exposure of monocyte-derived macrophages and dendritic cells to purified mitochondria results in the production of inflammatory cytokines including TNFα, IL-6, IL-8, and IL-10, confirming that the released mitochondria can act in DAMPs to activate immune cells.
CONCLUDING REMARKS
Many literatures have shown that autophagy is essential to maintain liver homeostasis under both healthy and disease conditions. Autophagy is consisted from multiple steps and the completion of the whole processes of autophagy is the prerequisite for its cytoprotective role. Although mitochondrial dysfunction and subsequent induction of autophagy to eliminate damaged mitochondria should be important to protect liver and other organs/tissues from septic injuries, the fate of damaged mitochondria might be dependent on lysosomes. There seems to be at least two destinations of mitochondria during sepsis: digestion within the cells and secretion into extracellular space. Factors determining the fate of mitochondria should be examined in the future studies. In addition, chaperone-mediated autophagy has also been shown to be necessary for liver homeostasis [74]. Although the involvement of autophagy (macroautophagy) has been proved by use of conditional atg5- or atg7-KO mice in many studies, atg5/atg7-independent macroautophagy (alternative or non-canonical macroautophagy) has also been proposed [75]. Thus, possible involvement of these types of autophagy in the septic injury in the liver should be examined in the future.
–
The extrusion of intracellular contents/substances from immunologically activated cells raises the question as to why cells actively secrete these contents. One explanation is that the intracellular capacity to degrade dysfunctional materials reaches its limit. Indeed, several studies have pointed out that the flux of autophagy is prone to stagnate in the liver during sepsis [26]. An alternative explanation is that the cells extrude intracellular contents for specific purposes. For example, a number of studies have shown that autophagy-dependent secretion of intracellular materials, such as mitochondria, resulted in the activation of immune cells. Mitochondria, which have prokaryotic features such as CpG-methylated DNA as well as formylmethionine, act as DAMPs and have been shown to be involved in inflammation during sterile trauma [76]. Indeed, several studies, including ours, have shown that purified mitochondria have the capacity to activate immune cells [47][61][76]. Thus, the release of mitochondria from immunologically activated cells might be involved in the propagation of inflammation, which might be good or bad for human health depending on the situation. Further studies are needed to elucidate the exact roles of autophagy and mitochondrial release in liver injury during sepsis.
References
- D.J. Klionsky, "Autophagy revisited: A conversation with Christian de Duve", Autophagy, vol. 4, pp. 740-743, 2008. http://dx.doi.org/10.4161/auto.6398
- A. Orsi, H.E. Polson, and S.A. Tooze, "Membrane trafficking events that partake in autophagy", Current Opinion in Cell Biology, vol. 22, pp. 150-156, 2010. http://dx.doi.org/10.1016/j.ceb.2009.11.013
- N. Mizushima, T. Yoshimori, and Y. Ohsumi, "The Role of Atg Proteins in Autophagosome Formation", Annual Review of Cell and Developmental Biology, vol. 27, pp. 107-132, 2011. http://dx.doi.org/10.1146/annurev-cellbio-092910-154005
- C. He, and D.J. Klionsky, "Regulation Mechanisms and Signaling Pathways of Autophagy", Annual Review of Genetics, vol. 43, pp. 67-93, 2009. http://dx.doi.org/10.1146/annurev-genet-102808-114910
- Y. Ohsumi, "Molecular dissection of autophagy: two ubiquitin-like systems", Nature Reviews Molecular Cell Biology, vol. 2, pp. 211-216, 2001. http://dx.doi.org/10.1038/35056522
- Y. Ohsumi, and N. Mizushima, "Two ubiquitin-like conjugation systems essential for autophagy", Seminars in Cell & Developmental Biology, vol. 15, pp. 231-236, 2004. http://dx.doi.org/10.1016/j.semcdb.2003.12.004
- M. Komatsu, S. Waguri, T. Ueno, J. Iwata, S. Murata, I. Tanida, J. Ezaki, N. Mizushima, Y. Ohsumi, Y. Uchiyama, E. Kominami, K. Tanaka, and T. Chiba, "Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice", The Journal of Cell Biology, vol. 169, pp. 425-434, 2005. http://dx.doi.org/10.1083/jcb.200412022
- A. Takamura, M. Komatsu, T. Hara, A. Sakamoto, C. Kishi, S. Waguri, Y. Eishi, O. Hino, K. Tanaka, and N. Mizushima, "Autophagy-deficient mice develop multiple liver tumors", Genes & Development, vol. 25, pp. 795-800, 2011. http://dx.doi.org/10.1101/gad.2016211
- R.S. Hotchkiss, and I.E. Karl, "The Pathophysiology and Treatment of Sepsis", New England Journal of Medicine, vol. 348, pp. 138-150, 2003. http://dx.doi.org/10.1056/NEJMra021333
- D. Rittirsch, M.A. Flierl, and P.A. Ward, "Harmful molecular mechanisms in sepsis", Nature Reviews Immunology, vol. 8, pp. 776-787, 2008. http://dx.doi.org/10.1038/nri2402
- G. Kumar, N. Kumar, A. Taneja, T. Kaleekal, S. Tarima, E. McGinley, E. Jimenez, A. Mohan, R.A. Khan, J. Whittle, E. Jacobs, and R. Nanchal, "Nationwide Trends of Severe Sepsis in the 21st Century (2000–2007)", Chest, vol. 140, pp. 1223-1231, 2011. http://dx.doi.org/10.1378/chest.11-0352
- G. Buyse, C. de Greef, L. Raeymaekers, G. Droogmans, B. Nilius, and J. Eggermont, "The ubiquitously expressed pICln protein forms homomeric complexes in vitro.", Biochemical and biophysical research communications, 1996. http://www.ncbi.nlm.nih.gov/pubmed/8579598
- J.F. Frencken, L.A. van Vught, L.M. Peelen, D.S.Y. Ong, P.M.C. Klein Klouwenberg, J. Horn, M.J.M. Bonten, T. van der Poll, and O.L. Cremer, "An Unbalanced Inflammatory Cytokine Response Is Not Associated With Mortality Following Sepsis: A Prospective Cohort Study", Critical Care Medicine, vol. 45, pp. e493-e499, 2017. http://dx.doi.org/10.1097/CCM.0000000000002292
- D. Brealey, M. Brand, I. Hargreaves, S. Heales, J. Land, R. Smolenski, N.A. Davies, C.E. Cooper, and M. Singer, "Association between mitochondrial dysfunction and severity and outcome of septic shock", The Lancet, vol. 360, pp. 219-223, 2002. http://dx.doi.org/10.1016/S0140-6736(02)09459-X
- J. Struck, M. Uhlein, N.G. Morgenthaler, W. Fürst, C. Höflich, S. Bahrami, A. Bergmann, H. Volk, and H. Redl, "Release of the mitochondrial enzyme carbamoyl phosphate synthase under septic conditions.", Shock (Augusta, Ga.), 2005. http://www.ncbi.nlm.nih.gov/pubmed/15897806
- E.L. Mills, and L.A. O'Neill, "Reprogramming mitochondrial metabolism in macrophages as an anti‐inflammatory signal", European Journal of Immunology, vol. 46, pp. 13-21, 2016. http://dx.doi.org/10.1002/eji.201445427
- E.M. Palsson‐McDermott, and L.A.J. O'Neill, "The Warburg effect then and now: From cancer to inflammatory diseases", BioEssays, vol. 35, pp. 965-973, 2013. http://dx.doi.org/10.1002/bies.201300084
- G.M. Tannahill, A.M. Curtis, J. Adamik, E.M. Palsson-McDermott, A.F. McGettrick, G. Goel, C. Frezza, N.J. Bernard, B. Kelly, N.H. Foley, L. Zheng, A. Gardet, Z. Tong, S.S. Jany, S.C. Corr, M. Haneklaus, B.E. Caffrey, K. Pierce, S. Walmsley, F.C. Beasley, E. Cummins, V. Nizet, M. Whyte, C.T. Taylor, H. Lin, S.L. Masters, E. Gottlieb, V.P. Kelly, C. Clish, P.E. Auron, R.J. Xavier, and L.A.J. O’Neill, "Succinate is an inflammatory signal that induces IL-1β through HIF-1α", Nature, vol. 496, pp. 238-242, 2013. http://dx.doi.org/10.1038/nature11986
- E.L. Mills, B. Kelly, A. Logan, A.S. Costa, M. Varma, C.E. Bryant, P. Tourlomousis, J.H.M. Däbritz, E. Gottlieb, I. Latorre, S.C. Corr, G. McManus, D. Ryan, H.T. Jacobs, M. Szibor, R.J. Xavier, T. Braun, C. Frezza, M.P. Murphy, and L.A. O’Neill, "Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages", Cell, vol. 167, pp. 457-470.e13, 2016. http://dx.doi.org/10.1016/j.cell.2016.08.064
- R. Zhou, A.S. Yazdi, P. Menu, and J. Tschopp, "A role for mitochondria in NLRP3 inflammasome activation", Nature, vol. 469, pp. 221-225, 2010. http://dx.doi.org/10.1038/nature09663
- E. Watanabe, J.T. Muenzer, W.G. Hawkins, C.G. Davis, D.J. Dixon, J.E. McDunn, D.J. Brackett, M.R. Lerner, P.E. Swanson, and R.S. Hotchkiss, "Sepsis induces extensive autophagic vacuolization in hepatocytes: a clinical and laboratory-based study", Laboratory Investigation, vol. 89, pp. 549-561, 2009. http://dx.doi.org/10.1038/labinvest.2009.8
- C. Galanos, M.A. Freudenberg, and W. Reutter, "Galactosamine-induced sensitization to the lethal effects of endotoxin.", Proceedings of the National Academy of Sciences of the United States of America, 1979. http://www.ncbi.nlm.nih.gov/pubmed/293694
- M. Amir, E. Zhao, L. Fontana, H. Rosenberg, K. Tanaka, G. Gao, and M.J. Czaja, "Inhibition of hepatocyte autophagy increases tumor necrosis factor-dependent liver injury by promoting caspase-8 activation", Cell Death & Differentiation, vol. 20, pp. 878-887, 2013. http://dx.doi.org/10.1038/cdd.2013.21
- G. Lalazar, G. Ilyas, S.A. Malik, K. Liu, E. Zhao, M. Amir, Y. Lin, K.E. Tanaka, and M.J. Czaja, "Autophagy confers resistance to lipopolysaccharide-induced mouse hepatocyte injury", American Journal of Physiology-Gastrointestinal and Liver Physiology, vol. 311, pp. G377-G386, 2016. http://dx.doi.org/10.1152/ajpgi.00124.2016
- C. Hsieh, P. Pai, H. Hsueh, S. Yuan, and Y. Hsieh, "Complete Induction of Autophagy Is Essential for Cardioprotection in Sepsis", Annals of Surgery, vol. 253, pp. 1190-1200, 2011. http://dx.doi.org/10.1097/SLA.0b013e318214b67e
- C. Lin, S. Lo, D. Perng, D.B. Wu, P. Lee, Y. Chang, P. Kuo, M. Yu, S.F. Yuan, and Y. Hsieh, "Complete Activation of Autophagic Process Attenuates Liver Injury and Improves Survival in Septic Mice", Shock, vol. 41, pp. 241-249, 2014. http://dx.doi.org/10.1097/SHK.0000000000000111
- W. Takahashi, E. Watanabe, L. Fujimura, H. Watanabe-Takano, H. Yoshidome, P.E. Swanson, T. Tokuhisa, S. Oda, and M. Hatano, "Kinetics and protective role of autophagy in a mouse cecal ligation and puncture-induced sepsis", Critical Care, vol. 17, pp. R160, 2013. http://dx.doi.org/10.1186/cc12839
- M. Nagaki, and H. Moriwaki, "Implication of cytokines: Roles of tumor necrosis factor‐α in liver injury", Hepatology Research, vol. 38, 2008. http://dx.doi.org/10.1111/j.1872-034X.2008.00422.x
- H. Fukada, S. Yamashina, K. Izumi, M. Komatsu, K. Tanaka, K. Ikejima, and S. Watanabe, "Suppression of autophagy sensitizes Kupffer cells to endotoxin", Hepatology Research, vol. 42, pp. 1112-1118, 2012. http://dx.doi.org/10.1111/j.1872-034X.2012.01024.x
- A. Dangi, C. Huang, A. Tandon, D. Stolz, T. Wu, and C.R. Gandhi, "Endotoxin‐stimulated Rat Hepatic Stellate Cells Induce Autophagy in Hepatocytes as a Survival Mechanism", Journal of Cellular Physiology, vol. 231, pp. 94-105, 2015. http://dx.doi.org/10.1002/jcp.25055
- E. Yoshigai, T. Hara, H. Inaba, I. Hashimoto, Y. Tanaka, M. Kaibori, T. Kimura, T. Okumura, A. Kwon, and M. Nishizawa, "Interleukin‐1β induces tumor necrosis factor‐α secretion from rat hepatocytes", Hepatology Research, vol. 44, pp. 571-583, 2013. http://dx.doi.org/10.1111/hepr.12157
- H. Guo, J.B. Callaway, and J.P. Ting, "Inflammasomes: mechanism of action, role in disease, and therapeutics", Nature Medicine, vol. 21, pp. 677-687, 2015. http://dx.doi.org/10.1038/nm.3893
- S.G. Boaru, E. Borkham-Kamphorst, L. Tihaa, U. Haas, and R. Weiskirchen, "Expression analysis of inflammasomes in experimental models of inflammatory and fibrotic liver disease", Journal of Inflammation, vol. 9, 2012. http://dx.doi.org/10.1186/1476-9255-9-49
- S.G. Boaru, E. Borkham-Kamphorst, E. Van de Leur, E. Lehnen, C. Liedtke, and R. Weiskirchen, "NLRP3 inflammasome expression is driven by NF-κB in cultured hepatocytes", Biochemical and Biophysical Research Communications, vol. 458, pp. 700-706, 2015. http://dx.doi.org/10.1016/j.bbrc.2015.02.029
- V. Hornung, F. Bauernfeind, A. Halle, E.O. Samstad, H. Kono, K.L. Rock, K.A. Fitzgerald, and E. Latz, "Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization", Nature Immunology, vol. 9, pp. 847-856, 2008. http://dx.doi.org/10.1038/ni.1631
- K. Nakahira, J.A. Haspel, V.A.K. Rathinam, S. Lee, T. Dolinay, H.C. Lam, J.A. Englert, M. Rabinovitch, M. Cernadas, H.P. Kim, K.A. Fitzgerald, S.W. Ryter, and A.M.K. Choi, "Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome", Nature Immunology, vol. 12, pp. 222-230, 2010. http://dx.doi.org/10.1038/ni.1980
- C. Shi, K. Shenderov, N. Huang, J. Kabat, M. Abu-Asab, K.A. Fitzgerald, A. Sher, and J.H. Kehrl, "Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction", Nature Immunology, vol. 13, pp. 255-263, 2012. http://dx.doi.org/10.1038/ni.2215
- J.A. Hagar, D.A. Powell, Y. Aachoui, R.K. Ernst, and E.A. Miao, "Cytoplasmic LPS Activates Caspase-11: Implications in TLR4-Independent Endotoxic Shock", Science, vol. 341, pp. 1250-1253, 2013. http://dx.doi.org/10.1126/science.1240988
- N. Kayagaki, M.T. Wong, I.B. Stowe, S.R. Ramani, L.C. Gonzalez, S. Akashi-Takamura, K. Miyake, J. Zhang, W.P. Lee, A. Muszyński, L.S. Forsberg, R.W. Carlson, and V.M. Dixit, "Noncanonical Inflammasome Activation by Intracellular LPS Independent of TLR4", Science, vol. 341, pp. 1246-1249, 2013. http://dx.doi.org/10.1126/science.1240248
- N. Kayagaki, I.B. Stowe, B.L. Lee, K. O’Rourke, K. Anderson, S. Warming, T. Cuellar, B. Haley, M. Roose-Girma, Q.T. Phung, P.S. Liu, J.R. Lill, H. Li, J. Wu, S. Kummerfeld, J. Zhang, W.P. Lee, S.J. Snipas, G.S. Salvesen, L.X. Morris, L. Fitzgerald, Y. Zhang, E.M. Bertram, C.C. Goodnow, and V.M. Dixit, "Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling", Nature, vol. 526, pp. 666-671, 2015. http://dx.doi.org/10.1038/nature15541
- J. Shi, Y. Zhao, K. Wang, X. Shi, Y. Wang, H. Huang, Y. Zhuang, T. Cai, F. Wang, and F. Shao, "Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death", Nature, vol. 526, pp. 660-665, 2015. http://dx.doi.org/10.1038/nature15514
- R.A. Aglietti, A. Estevez, A. Gupta, M.G. Ramirez, P.S. Liu, N. Kayagaki, C. Ciferri, V.M. Dixit, and E.C. Dueber, "GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes", Proceedings of the National Academy of Sciences, vol. 113, pp. 7858-7863, 2016. http://dx.doi.org/10.1073/pnas.1607769113
- C. Rogers, T. Fernandes-Alnemri, L. Mayes, D. Alnemri, G. Cingolani, and E.S. Alnemri, "Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death", Nature Communications, vol. 8, 2017. http://dx.doi.org/10.1038/ncomms14128
- Y. Wang, W. Gao, X. Shi, J. Ding, W. Liu, H. He, K. Wang, and F. Shao, "Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin", Nature, vol. 547, pp. 99-103, 2017. http://dx.doi.org/10.1038/nature22393
- E.D. Crouser, M.W. Julian, J.E. Huff, J. Struck, and C.H. Cook, "Carbamoyl phosphate synthase-1: A marker of mitochondrial damage and depletion in the liver during sepsis", Critical Care Medicine, vol. 34, pp. 2439-2446, 2006. http://dx.doi.org/10.1097/01.CCM.0000230240.02216.21
- K. Unuma, T. Aki, S. Matsuda, T. Funakoshi, K. Yoshida, and K. Uemura, "Elimination and active extrusion of liver mitochondrial proteins during lipopolysaccharide administration in rat", Hepatology Research, vol. 43, pp. 526-534, 2012. http://dx.doi.org/10.1111/j.1872-034X.2012.01084.x
- K. Unuma, T. Aki, T. Funakoshi, K. Hashimoto, and K. Uemura, "Extrusion of mitochondrial contents from lipopolysaccharide-stimulated cells: Involvement of autophagy", Autophagy, vol. 11, pp. 1520-1536, 2015. http://dx.doi.org/10.1080/15548627.2015.1063765
- A. Galione, and M. Ruas, "NAADP receptors", Cell Calcium, vol. 38, pp. 273-280, 2005. http://dx.doi.org/10.1016/j.ceca.2005.06.031
- S. Rah, Y. Lee, and U. Kim, "NAADP‐mediated Ca 2+ signaling promotes autophagy and protects against LPS‐induced liver injury", The FASEB Journal, vol. 31, pp. 3126-3137, 2017. http://dx.doi.org/10.1096/fj.201601290R
- S. TAKEUCHI, T. GOTO, K. MIKAMI, K. MIURA, S. OHSHIMA, K. YONEYAMA, M. SATO, T. SHIBUYA, D. WATANABE, and E. KATAOKA, "Genipin prevents fulminant hepatic failure resulting in reduction of lethality through the suppression of TNF-α production", Hepatology Research, vol. 33, pp. 298-305, 2005. http://dx.doi.org/10.1016/j.hepres.2005.08.009
- H. Cho, S. Kim, J. Choi, and S. Lee, "Genipin alleviates sepsis‐induced liver injury by restoring autophagy", British Journal of Pharmacology, vol. 173, pp. 980-991, 2016. http://dx.doi.org/10.1111/bph.13397
- Y. Li, X. Wang, Z. Wei, H. Mao, M. Gao, Y. Liu, Y. Ma, X. Liu, C. Guo, L. Zhang, and X. Wang, "Pretreatment with wortmannin alleviates lipopolysaccharide/d-galactosamine-induced acute liver injury", Biochemical and Biophysical Research Communications, vol. 455, pp. 234-240, 2014. http://dx.doi.org/10.1016/j.bbrc.2014.10.152
- K. Unuma, T. Aki, S. Matsuda, T. Funakoshi, K. Yoshida, and K. Uemura, "Inducer of heme oxygenase‐1 cobalt protoporphyrin accelerates autophagy and suppresses oxidative damages during lipopolysaccharide treatment in rat liver", Hepatology Research, vol. 43, pp. 91-96, 2012. http://dx.doi.org/10.1111/j.1872-034X.2012.01049.x
- E.H. Carchman, J. Rao, P.A. Loughran, M.R. Rosengart, and B.S. Zuckerbraun, "Heme oxygenase‐1–mediated autophagy protects against hepatocyte cell death and hepatic injury from infection/sepsis in mice", Hepatology, vol. 53, pp. 2053-2062, 2011. http://dx.doi.org/10.1002/hep.24324
- E.F. Blommaart, U. Krause, J.P. Schellens, H. Vreeling-Sindelárová, and A.J. Meijer, "The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes.", European journal of biochemistry, 1997. http://www.ncbi.nlm.nih.gov/pubmed/9030745
- A. Kihara, Y. Kabeya, Y. Ohsumi, and T. Yoshimori, "Beclin–phosphatidylinositol 3‐kinase complex functions at the trans ‐Golgi network", EMBO reports, vol. 2, pp. 330-335, 2001. http://dx.doi.org/10.1093/embo-reports/kve061
- R. Du, J. Tan, N. Yan, L. Wang, C. Qiao, J. Ding, M. Lu, and G. Hu, "Kir6.2 knockout aggravates lipopolysaccharide-induced mouse liver injury via enhancing NLRP3 inflammasome activation", Journal of Gastroenterology, vol. 49, pp. 727-736, 2013. http://dx.doi.org/10.1007/s00535-013-0823-0
- N. Dupont, S. Jiang, M. Pilli, W. Ornatowski, D. Bhattacharya, and V. Deretic, "Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β", The EMBO Journal, vol. 30, pp. 4701-4711, 2011. http://dx.doi.org/10.1038/emboj.2011.398
- I. Martins, Y. Wang, M. Michaud, Y. Ma, A.Q. Sukkurwala, S. Shen, O. Kepp, D. Métivier, L. Galluzzi, J. Perfettini, L. Zitvogel, and G. Kroemer, "Molecular mechanisms of ATP secretion during immunogenic cell death", Cell Death & Differentiation, vol. 21, pp. 79-91, 2013. http://dx.doi.org/10.1038/cdd.2013.75
- T. Takenouchi, M. Nakai, Y. Iwamaru, S. Sugama, M. Tsukimoto, M. Fujita, J. Wei, A. Sekigawa, M. Sato, S. Kojima, H. Kitani, and M. Hashimoto, "The Activation of P2X7 Receptor Impairs Lysosomal Functions and Stimulates the Release of Autophagolysosomes in Microglial Cells", The Journal of Immunology, vol. 182, pp. 2051-2062, 2009. http://dx.doi.org/10.4049/jimmunol.0802577
- A. Maeda, and B. Fadeel, "Mitochondria released by cells undergoing TNF-α-induced necroptosis act as danger signals", Cell Death & Disease, vol. 5, pp. e1312-e1312, 2014. http://dx.doi.org/10.1038/cddis.2014.277
- M. Sardiello, M. Palmieri, A. di Ronza, D.L. Medina, M. Valenza, V.A. Gennarino, C. Di Malta, F. Donaudy, V. Embrione, R.S. Polishchuk, S. Banfi, G. Parenti, E. Cattaneo, and A. Ballabio, "A Gene Network Regulating Lysosomal Biogenesis and Function", Science, vol. 325, pp. 473-477, 2009. http://dx.doi.org/10.1126/science.1174447
- C. Settembre, C. Di Malta, V.A. Polito, M.G. Arencibia, F. Vetrini, S. Erdin, S.U. Erdin, T. Huynh, D. Medina, P. Colella, M. Sardiello, D.C. Rubinsztein, and A. Ballabio, "TFEB Links Autophagy to Lysosomal Biogenesis", Science, vol. 332, pp. 1429-1433, 2011. http://dx.doi.org/10.1126/science.1204592
- D. Medina, A. Fraldi, V. Bouche, F. Annunziata, G. Mansueto, C. Spampanato, C. Puri, A. Pignata, J. Martina, M. Sardiello, M. Palmieri, R. Polishchuk, R. Puertollano, and A. Ballabio, "Transcriptional Activation of Lysosomal Exocytosis Promotes Cellular Clearance", Developmental Cell, vol. 21, pp. 421-430, 2011. http://dx.doi.org/10.1016/j.devcel.2011.07.016
- C. Settembre, R. Zoncu, D.L. Medina, F. Vetrini, S. Erdin, S. Erdin, T. Huynh, M. Ferron, G. Karsenty, M.C. Vellard, V. Facchinetti, D.M. Sabatini, and A. Ballabio, "A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB", The EMBO Journal, vol. 31, pp. 1095-1108, 2012. http://dx.doi.org/10.1038/emboj.2012.32
- A. Roczniak-Ferguson, C.S. Petit, F. Froehlich, S. Qian, J. Ky, B. Angarola, T.C. Walther, and S.M. Ferguson, "The Transcription Factor TFEB Links mTORC1 Signaling to Transcriptional Control of Lysosome Homeostasis", Science Signaling, vol. 5, 2012. http://dx.doi.org/10.1126/scisignal.2002790
- D.L. Medina, S. Di Paola, I. Peluso, A. Armani, D. De Stefani, R. Venditti, S. Montefusco, A. Scotto-Rosato, C. Prezioso, A. Forrester, C. Settembre, W. Wang, Q. Gao, H. Xu, M. Sandri, R. Rizzuto, M.A. De Matteis, and A. Ballabio, "Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB", Nature Cell Biology, vol. 17, pp. 288-299, 2015. http://dx.doi.org/10.1038/ncb3114
-
T. Kimura, J. Jia, S. Kumar, S.W. Choi, Y. Gu, M. Mudd, N. Dupont, S. Jiang, R. Peters, F. Farzam, A. Jain, K.A. Lidke, C.M. Adams, T. Johansen, and V. Deretic, "Dedicated
SNARE s and specializedTRIM cargo receptors mediate secretory autophagy", The EMBO Journal, vol. 36, pp. 42-60, 2016. http://dx.doi.org/10.15252/embj.201695081 - H. Ushio, T. Ueno, Y. Kojima, M. Komatsu, S. Tanaka, A. Yamamoto, Y. Ichimura, J. Ezaki, K. Nishida, S. Komazawa-Sakon, F. Niyonsaba, T. Ishii, T. Yanagawa, E. Kominami, H. Ogawa, K. Okumura, and H. Nakano, "Crucial role for autophagy in degranulation of mast cells", Journal of Allergy and Clinical Immunology, vol. 127, pp. 1267-1276.e6, 2011. http://dx.doi.org/10.1016/j.jaci.2010.12.1078
- B. Zhang, S. Asadi, Z. Weng, N. Sismanopoulos, and T.C. Theoharides, "Stimulated Human Mast Cells Secrete Mitochondrial Components That Have Autocrine and Paracrine Inflammatory Actions", PLoS ONE, vol. 7, pp. e49767, 2012. http://dx.doi.org/10.1371/journal.pone.0049767
- A. Nakajima, H. Kurihara, H. Yagita, K. Okumura, and H. Nakano, "Mitochondrial Extrusion through the Cytoplasmic Vacuoles during Cell Death", Journal of Biological Chemistry, vol. 283, pp. 24128-24135, 2008. http://dx.doi.org/10.1074/jbc.M802996200
- L. Galluzzi, L. Senovilla, L. Zitvogel, and G. Kroemer, "The secret ally: immunostimulation by anticancer drugs", Nature Reviews Drug Discovery, vol. 11, pp. 215-233, 2012. http://dx.doi.org/10.1038/nrd3626
- L. Galluzzi, and G. Kroemer, "Necroptosis: A Specialized Pathway of Programmed Necrosis", Cell, vol. 135, pp. 1161-1163, 2008. http://dx.doi.org/10.1016/j.cell.2008.12.004
- J. Schneider, Y. Suh, and A. Cuervo, "Deficient Chaperone-Mediated Autophagy in Liver Leads to Metabolic Dysregulation", Cell Metabolism, vol. 20, pp. 417-432, 2014. http://dx.doi.org/10.1016/j.cmet.2014.06.009
- Y. Nishida, S. Arakawa, K. Fujitani, H. Yamaguchi, T. Mizuta, T. Kanaseki, M. Komatsu, K. Otsu, Y. Tsujimoto, and S. Shimizu, "Discovery of Atg5/Atg7-independent alternative macroautophagy", Nature, vol. 461, pp. 654-658, 2009. http://dx.doi.org/10.1038/nature08455
- Q. Zhang, M. Raoof, Y. Chen, Y. Sumi, T. Sursal, W. Junger, K. Brohi, K. Itagaki, and C.J. Hauser, "Circulating mitochondrial DAMPs cause inflammatory responses to injury", Nature, vol. 464, pp. 104-107, 2010. http://dx.doi.org/10.1038/nature08780
COPYRIGHT
© 2017

Emerging roles of mitochondria and autophagy in liver injury during sepsis by Aki et al. is licensed under a Creative Commons Attribution 4.0 International License.


