Reviews:
Cell Stress, Vol. 1, No. 1, pp. 11 - 36; doi: 10.15698/cst2017.10.104
VDAC1 at the crossroads of cell metabolism, apoptosis and cell stress
1 Department of Life Sciences and the National Institute for Biotechnology in the Negev, Ben-Gurion University of the Negev, Beer-Sheva, 84105, Israel.
2 Department of Drug Discovery & Biomedical Sciences, Medical University of South Carolina, Charleston, SC, USA.
Keywords: apoptosis, cancer, metabolism, mitochondria, VDAC1.
Abbreviations:
Aβ – Amyloid beta,
AD – Alzheimer’s disease,
ALS – amyotrophic lateral sclerosis,
AIF - apoptosis-inducing factor,
ANT - adenine nucleotide translocase,
Bcl-2 - B-cell lymphoma 2,
CSC – cancer stem cell,
CVD – cardiovascular disease,
Cyto c - cytochrome c,
DIDS - 4,4-diisothiocyanostilbene-2,2-disulfonic acid,
HK – hexokinase,
IMM - inner mitochondrial membrane,
IMS – intermembrane space,
NSCLC - non-small cell lung cancer,
miRNA – micro RNA
OMM - outer mitochondrial membrane,
OXPHOS – oxidative phosphorylation,
PTP - permeability transition pore,
RNAi - RNA interference,
ROS - reactive oxygen species,
TSPO - translocator protein,
T2D – type 2 diabetes,
VDAC - voltage-dependent anion channel.
Received originally: 05/06/2017 Received in revised form: 21/09/2017
Accepted: 21/09/2017
Published: 01/10/2017
Correspondence:
Prof. Varda Shoshan-Barmatz, Department of Life Sciences, Ben-Gurion University of the Negev, Beer-Sheva 84105, Israel; Fax : 972-8-647 2992 vardasb@bgu.ac.il
Conflict of interest statement: The authors declare no conflict of interest.
Please cite this article as: Varda Shoshan-Barmatz, Eduardo N. Maldonado and Yakov Krelin (2017). VDAC1 at the crossroads of cell metabolism, apoptosis and cell stress. Cell Stress 1(1): 11-36. doi: 10.15698/cst2017.10.104
Abstract
This review presents current knowledge related to VDAC1 as a multi-functional mitochondrial protein acting on both sides of the coin, regulating cell life and death, and highlighting these functions in relation to disease. It is now recognized that VDAC1 plays a crucial role in regulating the metabolic and energetic functions of mitochondria. The location of VDAC1 at the outer mitochondrial membrane (OMM) allows the control of metabolic cross-talk between mitochondria and the rest of the cell and also enables interaction of VDAC1 with proteins involved in metabolic and survival pathways. Along with regulating cellular energy production and metabolism, VDAC1 is also involved in the process of mitochondria-mediated apoptosis by mediating the release of apoptotic proteins and interacting with anti-apoptotic proteins. VDAC1 functions in the release of apoptotic proteins located in the mitochondrial intermembrane space via oligomerization to form a large channel that allows passage of cytochrome c and AIF and their release to the cytosol, subsequently resulting in apoptotic cell death. VDAC1 also regulates apoptosis via interactions with apoptosis regulatory proteins, such as hexokinase, Bcl2 and Bcl-xL, some of which are also highly expressed in many cancers. This review also provides insight into VDAC1 function in Ca2+ homeostasis, oxidative stress, and presents VDAC1 as a hub protein interacting with over 100 proteins. Such interactions enable VDAC1 to mediate and regulate the integration of mitochondrial functions with cellular activities. VDAC1 can thus be considered as standing at the crossroads between mitochondrial metabolite transport and apoptosis and hence represents an emerging cancer drug target.
VDAC ISOFORMS, STRUCTURE, AND CHANNEL ACTIVITY
VDAC isoforms and cellular localization
Three different VDAC isoforms, VDAC1, VDAC2 and VDAC3, sharing ~70% identity and structural and some functional properties [1][2], are expressed in mammalian mitochondria, with VDAC1 being the major protein expressed. However, significantly differences in the functions of the three isoforms were found [1][3][4], suggesting they assume different physiological roles [1][5]. The three isoforms are expressed in most tissue types, with VDAC1 expression being higher than that of VDAC2 and VDAC3 in most but not all tissues [1][2].
–
Both VDAC1- and VDAC3-deficient mice are viable. However, VDAC1−/− mice (inbred C57BL/6 background) were born in less than expected numbers according to the Mendelian ratio, suggesting partial embryonic lethality. Studies using VDAC1−/− mice confirmed the importance of this protein as a carrier of metabolites across the OMM [6]. In mice, deletion of VDAC1 and VDAC2 reduces respiratory capacity [7], and the absence of VDAC3 causes male sterility, while a lack of both VDAC1 and VDAC3 causes growth retardation [8] and is associated with deficits in learning behavior and synaptic plasticity [9]. In this review, the focus will be on the VDAC1 isoform.
–
Using various approaches, VDAC was detected not only in the mitochondria but also in other cell compartments [3], such as the plasma membrane [3][10], including the caveolae and caveolae-like domains [11], the sarcoplasmic reticulum (SR) of skeletal muscles [12], and the ER of rat cerebellum [13][14]. A possible mechanism for targeting VDAC protein to the plasma membrane proposes that this version of the protein contains an N-terminal signal peptide responsible for targeting to the cell membrane [15][16]. The exact function of extra-mitochondrial VDAC is unknown, although several possible roles have been proposed (reviewed in [17]).
–
VDAC1 structure, channel conductance, properties and regulation
The three-dimensional structure of VDAC isoform 1 was determined at atomic resolution, revealing that VDAC1 is composed of 19 transmembrane β-strands connected by flexible loops to form a β-barrel, with strands β1 and β19 being in parallel conformation along with a 25-residue-long N-terminal region that lies inside the pore [18][19][20] (Fig.1A). The N-terminal region is proposed to move in the open space [21] and translocate from the internal pore to the channel surface [22] (Fig.1B). This segment is ideally positioned to regulate the conductance of ions and metabolites passing through the VDAC1 pore [20][18].
–
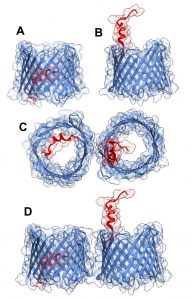 | FIGURE 1: Three-dimensional structure of VDAC1. VDAC1 monomer and dimer structures. (A) Side-view of the crystal structure of VDAC1 (PDB code: 3EMN). The β-barrel is formed by 19 β strands and the N-terminal domain (colored red) is folded into the pore interior. (B) A proposed model for the conformation of VDAC1 with its N-terminal on the outside of the VDAC1 pore. (C) Top-view of VDAC1 dimer with the N-terminal helix nested inside the VDAC1 pore in one monomer and outside of the pore in the other. (D) Side-view of proposed dimer of VDAC1. Figures were prepared using PyMOL software. |
–
The pore diameter of the channel has been estimated to be between 3 and 3.8 nm [18], and is decreased to about 1.5 nm when the N-terminal α-helix is located within the pore [18][19][20]. The stretch of multiple glycine residues (21GlyTyrGlyPheGly25) [1][5] connecting the N-terminal domain to β-strand 1 of the barrel is thought to provide the flexibility required for N-terminal region translocation out of the internal pore of the channel [22]. The reported results suggest that the N-terminal region mobility is involved in channel gating, interaction with anti-apoptotic proteins, and VDAC1 dimer formation [22], as well as serving the interaction site of apoptosis-regulating proteins of the Bcl-2 family (i.e., Bax, Bcl-2, and Bcl-xL) [22][23][24][25][26] and hexokinase (HK) [23][27].
–
Purified and membrane-embedded VDAC1 is able to assemble into dimers, trimers, tetramers, hexamers, and higher-order moieties [1][28][29][30][31][32][33][34][35][36]. The contact sites between VDAC1 molecules in dimers and higher oligomers were identified [37]. Under physiological conditions, VDAC1 is present as a monomer and dimer, with a contact site involving β-strands 1, 2, and 19. However, upon apoptosis induction, VDAC1 dimers undergo conformational changes to assemble into higher oligomeric states with contact sites also involving β-strands 8 and 16 [37]. VDAC1 oligomerization has been proposed to play important physiological roles in the regulation of VDAC1 function, including contributing to stabilizing the protein [38], serving as a platform for other proteins that oligomerize, such as HK [36] and creatine kinase [39], and finally, in mediating Cytochrome c release and the binding of apoptosis-regulating proteins [23][28][36] (see below).
–
VDAC1 has been purified from mitochondria isolated from liver, brain, and other tissues [40], and its channel properties were characterized following reconstitution into a planar lipid bilayer (PLB). Such bilayer-reconstituted VDAC1 assumes a variety of voltage-dependent conducting states, with different selectivities and permeabilities. VDAC1 shows symmetrical bell-shaped voltage-dependent conductance. At low voltages (-20 to +20 mV), VDAC1 exists in a high conductive state (~4 nS at 1 M KCl), and shows a preference for transporting anions over cations, while at high positive or negative potentials (> 40 mV), VDAC1 switches to lower conductance states permeable to small ions [41][42]. VDAC1 is permeable to small ions (e.g. Cl–, K+, Na+), and also to large anions, such as glutamate [41] and ATP [43], and to large cations, such as acetylcholine and dopamine [41].
–
The interactions of VDAC1 with Ca2+, ATP, glutamate, NADH, and different proteins were suggested to modulate its activity [44][45][46][47]. VDAC1 has been shown to be phosphorylated by protein kinase A (PKA) [48] and protein kinase C (PKC)ε [49], and VDAC1 and VDAC2 were found to be phosphorylated at a particular Tyr residue under hypoxic conditions [50].
VDAC1, A MULTI-FUNCTIONAL CHANNEL CONTROLLING CELL ENERGY AND METABOLISM
The OMM, the interface between the cytosol and a mitochondrion, is also a limiting boundary for modulating cell bioenergetics, mediated via VDAC1. The metabolites and ions that reach the matrix must first cross the OMM to reach the mitochondrial intermembrane space (IMS), from where they are then transported by about 53 secondary transport proteins called mitochondrial carriers (MCs). The mitochondrial carrier proteins of the family SLC25 (solute carrier family 25, located in the inner mitochondrial membrane (IMM)) are substrate-specific and mediate electrochemical, chemical and membrane potential gradient-dependent transport. The SLC25 family includes carriers for Pi (PiC), ADP/ATP (ANT) and aspartate/glutamate, pyruvate, acyl carnitine, oxoglutarate, and citrate, among others [51]. On the other hand, VDAC1 is the sole channel mediating the flux of ions, nucleotides and other metabolites up to ~5,000 Da (e.g. pyruvate, malate, succinate, NADH/NAD+) across the OMM, as well as hemes and cholesterol [1][52]. Thus, at the OMM, VDAC1 is perfectly positioned to function as gatekeeper for the entry and exit of substrates and products into and out of the mitochondria, and to interact with proteins that mediate and regulate the integration of mitochondrial functions with other cellular activities [1][29][30][31][42][53][54] (Fig. 2).
–
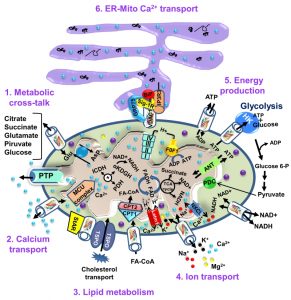 | FIGURE 2: VDAC1 as a multi-functional channel involved in metabolite, cholesterol and Ca2+ transport, energy production and in ER-mitochondria structural and functional association. The functions of VDAC1 in cell life include control of the metabolic cross-talk between the mitochondria and the rest of the cell energy production, regulation of glycolysis via binding of HK, Ca2+ signaling and cholesterol transport. The various functions of VDAC1 in the cell and mitochondria functions are presented. These include: 1. Control of the metabolic cross-talk between mitochondria and the rest of the cell; 2. Transport of Ca2+ to and from the IMS and acting in Ca2+ signaling; 3. Lipid metabolism; 4. Transport of ions, such as Mg2+, Zn+, Na+ and K+; 5. Mediating cellular energy production by transporting ATP/ADP and NAD+/NADH and acyl-CoA (FA-CoA) from the cytosol to and from the IMS, and regulating glycolysis via association with HK; 6. Structurally and functionally contributing to ER-mitochondria contacts, mediating Ca2+ transport from the ER to mitochondria. Ca2+ influx and efflux systems in the IMM are shown. The mitochondrial Ca2+ uniporter (MCU), in association with a calcium-sensing accessory subunit (MCU1), mediates Ca2+ transport from the IMS into the matrix. The ryanodine receptor (RyR) in the IMM mediates Ca2+ influx. NCLX, a Na+/Ca2+ exchanger, mediates Ca2+ efflux from the matrix to the IMS. High levels of matrix Ca2+ trigger the opening of the PTP, a fast Ca2+ release channel. Molecular fluxes are indicated by arrows. The function of Ca2+ in regulating energy production is mediated via activation of the TCA cycle enzymes pyruvate dehydrogenase (PDH), isocitrate dehydrogenase (ICDH) and α-ketoglutarate dehydrogenase (α-KGDH), leading to enhanced activity of the TCA cycle. The electron transport chain (ETC) and the ATP synthase (FoF1) are also presented. VDAC1 mediates the transfer of acyl-CoAs across the OMM to the IMS, where they are converted into acylcarnitine by CPT1a for further processing by β-oxidation. VDAC1 is involved in cholesterol transport by being a constituent of a multi-protein complex, the transduceosome, containing StAR/TSPO/VDAC1.The ER-mitochondria association is presented with key proteins indicated. These include the inositol 3 phosphate receptor type 3 (IP3R3), the sigma 1 receptor (Sig1R) (a reticular chaperone), binding immunoglobulin protein (BiP), the ER HSP70 chaperone, and glucose-regulated protein 75 (GRP75). IP3 activates IP3R in the ER to release Ca2+ that is directly transferred to the mitochondria via VDAC1. |
–
VDAC1 allows the shuttling of ATP/ADP and NAD+/NADH, with mitochondria-generated ATP being transported to the cytosol in exchange for ADP, which is utilized in oxidative phosphorylation (OXPHOS) to generate ATP. As such, VDAC1 controls the electron transport chain [1] (Fig. 2), as well as the normal flow of metabolites [55]. The importance of VDAC1 in channeling ATP from the mitochondria to kinases has been presented in several studies. These showed that VDAC1 interacts with HK and creatine kinase (CrK) to convert newly generated ATP into high-energy storage forms, like glucose-6-phosphate (G-6-P) and creatine phosphate in brain and muscle, respectively. The interaction of VDAC1 with HK mediates coupling between OXPHOS and glycolysis, while at the contact sites between the IMM and OMM, VDAC1 forms a complex with the adenine nucleotide translocase (ANT), and CrK [56]. Dimeric αβ-tubulin was proposed as a regulator of VDAC1 permeability to ATP, with heterodimers of αβ-tubulin decreasing the passage of ATP through the channel [57]. The importance of VDAC1 in cell energy and metabolism homeostasis is reflected in the findings that closure of VDAC [55] or down-regulation of VDAC1 expression decreased metabolite exchange between mitochondria and the cytosol and inhibited cell growth [58][59]. Moreover, VDAC1 is overexpressed in many cancer cells [32], as discussed below.
–
Cholesterol is another metabolite transported across the OMM [60] (Fig. 2), with VDAC1 being considered as a necessary component of a multi-protein complex, the transduceosome, involved in the process. In addition to VDAC1, the transduceosome also includes the OMM high-affinity cholesterol-binding protein translocator protein (TSPO) and the steroidogenic acute regulatory protein (StAR) [61] (Fig. 2). Cholesterol synthesis is highly elevated in various cancer cells, with hepatocellular carcinoma cells containing 2-10-fold more mitochondrial cholesterol (mainly in the OMM) than found in liver mitochondria [62]. The increased binding of HK to the mitochondria may increase synthesis and uptake of cholesterol into the mitochondria of cancer cells [63]. At high levels, cholesterol can reduce the activity of membrane-associated proteins and thus inhibit the metabolic functions of VDAC1 [64]. As such, VDAC1 is involved in cholesterol synthesis and transport, as well as being subject to cholesterol-mediated regulation.
–
Finally, it appears that VDAC1 is also part of a complex mediating the transport of fatty acids through the OMM in rat liver mitochondria [65]. In this case, it is hypothesized that VDAC1 acts as an anchor, linking the long-chain acyl-CoA synthetases (ACSLs) at the OMM to carnitine palmitoyltransferase 1a (CPT1a), which faces the IMS. According to the proposed model, upon activation by ACSL, VDAC1 transfers acyl-CoAs across the OMM to the IMS, where they are converted into acylcarnitine by CPT1a. Moreover, recently it was shown that fatty acid accumulation in hepatocytes leads to a lack of phosphorylation by GSK-3β, indicating interplay between lipids and VDAC function [66]. Furthermore, it was recently proposed that VDAC behaves as a lipid sensor [67].
CANCER, METABOLISM, MITOCHONDRIA, AND VDAC1
One of the main functions of the telomere is to prevent the metabolic reprogramming in cancer cells that require plasticity of the metabolic machinery, regardless of cellular or tissue origin, a critical process that promotes cell proliferation with alterations seen in the metabolism of several substrates, including glucose and glutamine [68][69]. In the 1920’s , Otto Warburg demonstrated increased lactic acid production resulting from high glycolysis in tumors, as compared to non-proliferating cells. The Warburg effect described a metabolic phenotype characterized by enhanced glycolysis and suppression of mitochondrial metabolism at any level of oxygen. However, although enhanced glycolysis is a prominent feature of most tumor cells, the mitochondria of cancer cells maintain a membrane potential, oxidize respiratory substrates, and generate NADH and ATP, among other functional parameters [70][71][72][73]. The view of cancer as a metabolic disease that originated with the experiments of Otto Warburg was gradually displaced by the concept of cancer as a genetic disease. Recently, evidence supporting a general hypothesis that genomic instability and essentially all hallmarks of cancer, including aerobic glycolysis, can be linked to impaired mitochondrial function and energy metabolism, has been reviewed [74][75]. Interestingly, no specific gene mutations or chromosomal abnormalities are common to all cancers [76], while nearly all cancers display aerobic glycolysis, regardless of their tissue or cellular origin. The view of cancer as primarily being a metabolic disease will impact approaches to cancer management and prevention.
–
Malignant cancer cells typically display high rates of glycolysis, even when fully oxygenated (aerobic glycolysis), and an altered redox balance [77][78][79]. To increase glycolysis, cancer cells up-regulate the transcription of genes involved in the glycolytic pathway (i.e., glucose transporters, glycolytic enzymes, etc.). Cancer cells in fact use both glycolysis and OXPHOS, with the ratio depending on the prevalent normoxic or hypoxic environmental conditions and their capacity to express adequate levels of oncogenes and tumor suppressor gene products for cell growth [80]. By regulating the metabolic and energetic functions of mitochondria, VDAC1 can, therefore, control the fate of cancer cells. Mitochondrial-bound HK, considered the rate-limiting enzyme of glycolysis, is over-expressed in cancer [1][81][82]. The association of HK with VDAC1 offers several advantages to cancer cells [1][32], such as direct access to mitochondrial sources of ATP, assumption of the role of an anti-apoptotic protein, reducing intracellular levels of reactive oxygen species (ROS) and increasing synthesis and uptake of cholesterol. The HK-VDAC1 complex formation is regulated by Akt [83] and glycogen synthase kinase 3 beta (GSK3β), while the HK-VDAC complex is disrupted by VDAC phosphorylation [84].
–
In recent years, cumulative evidence indicates that free tubulin in cancers cells interacts with VDAC [70][85]. Dimeric αβ-tubulin decreases the conductance of bilayer-reconstituted VDAC1 and VDAC2 and also decreases respiration in cardiac myocytes and isolated brain mitochondria [86][87]. In cancer cells, microtubule destabilization induced by colchicine or microtubule stabilization by paclitaxel increased and decreased free tubulin, leading to decreased and increased ΔΨm, respectively [70]. The dynamic regulation of ΔΨm by free tubulin appears to occur only in cancer cells. It has been proposed that the dynamic changes of ΔΨm brought about by free tubulin in tumor cells are related to αβ-tubulin heterodimers modulating VDAC conductance (Fig. 3) [70].
–
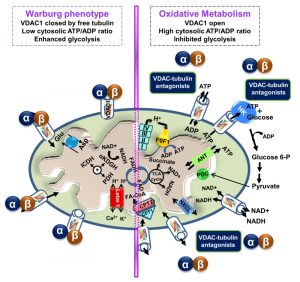 | FIGURE 3: VDAC1-tubulin interaction: a metabolic switch to modulate mitochondrial metabolism in cancer cells. In cancer cells, high levels of free tubulin close VDAC1, decreasing the flux of metabolites, ATP and ADP through the OMM. VDAC1 closing leads to low generation of mitochondrial ATP and subsequently, to a low cytosolic ATP/ADP ratio that favors glycolysis in the Warburg phenotype. Erastin, a VDAC-tubulin antagonist, opens VDAC1 by blocking the inhibitory effect of free tubulin. VDAC1 opening leads to increased mitochondrial metabolism and to a high cytosolic ATP/ADP ratio that inhibits glycolysis and reverts the Warburg phenotype. αβ indicates tubulin heterodimers. |
–
VDAC1 and VDAC2 isolated after VDAC2/3 or VDAC1/3 double knockdown in cancer cells were shown to be sensitive to tubulin inhibition. Even more, VDAC1 knockdown in tumor cells decreased ΔΨm, indicating that VDAC1 is critical for the maintenance of ΔΨm and is regulated by endogenous free tubulin [85]. Inhibition of VDAC1 conductance by free tubulin is a main contributor to the suppression of mitochondrial metabolism in the Warburg phenotype. Recently, the VDAC-tubulin interaction was proposed to serve as a metabolic switch to increase or decrease mitochondrial metabolism, ATP generation and cytosolic ATP/ADP ratios [88]. High and low cytosolic ATP/ADP ratios inhibit or favor aerobic glycolysis, respectively. Thus, blockage of the inhibitory effect of tubulin on VDAC by VDAC-tubulin antagonists promotes mitochondrial metabolism and reverses the Warburg phenotype (Fig. 3). The VDAC-tubulin interaction represents a new pharmacological target for the development of novel anti-cancer agents [88].
–
Silencing VDAC1 expression reduces cell energy homeostasis, inhibiting cells and tumor growth
As cellular metabolic and energy reprogramming are cancer hallmarks essential for tumor progression, and VDAC1 is a key regulator of these processes [1][30][32][47][52][89], down-regulation of VDAC1 expression is expected to impact cancer cell growth. VDAC1 down-regulation results in reduced metabolite exchange between the mitochondria and the cytosol, leading to inhibited cell growth. Indeed, silencing VDAC1 expression reduced cellular ATP levels and cell growth, with tight correlation between cell growth and cellular ATP levels being seen [58]. shRNA directed against hVDAC1 inhibited the development of a HeLa cervical tumor [90]. Nano-molar concentrations of a single siRNA specific to human VDAC1 silenced VDAC1 expression and inhibited the growth of various cancer cell types. In fact, such treatment inhibited solid tumor development and growth in lung cancers (over 90%) both in vitro and in vivo [59].
–
Recently, a global change in tumor hallmarks upon silencing VDAC1 expression was demonstrated in glioblastoma multiform (GBM) [91]. Using a sub-cutaneous or an intracranial-orthotopic GBM model, we demonstrated that si-VDAC1 inhibited tumor growth, with the residual tumor showing reversed oncogenic properties, such as reprogramed metabolism, angiogenesis, epithelial-mesenchymal transition (EMT), invasiveness and stemness, leading to differentiation into neuron- and astrocyte-like cells [91] (Fig. 4). These VDAC1 depletion-mediated effects involved alterations in transcription factors (TFs) that regulate signaling pathways associated with cancer hallmarks, allowing for attacks on the interplay between metabolism and oncogenic signaling networks (to be explored here), leading to cancer stem cell (CSC) differentiation into neuronal-like cells [91].
–
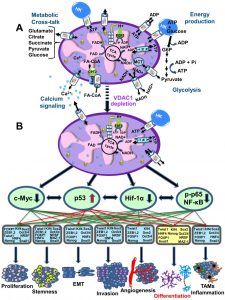 | FIGURE 4: VDAC1-depletion and metabolic reprogramming leading to alterations in key transcription factor levels and biological processes: a reversal of oncogenic properties and cell differentiation. (A) A schematic presentation of mitochondria in a cancer cell before treatment with hVDAC1 siRNA. Here, cancer cells maintain homeostatic energy and metabolic states, with HK bound to VDAC1 accelerating glycolysis and mitochondrial function to allow sufficient ATP and metabolite precursor levels to support cell growth and survival. (B) VDAC1 depletion leads to dramatic decreases in energy and metabolite generation. This leads to changes in master metabolism regulator (p53, HIF1-α, c-Myc and NF-kb, P65) expression levels, which alters the expression of transcription factors associated with stemness, EMT, cell proliferation, invasion, TAMs and angiogenesis, while leading to differentiation into astrocyte- or neuron-like cells. |
–
microRNA-mediated regulation of VDAC1
A number of microRNAs (miRNAs) targeting VDAC1 were reported and found to be modified under pathological conditions. miR-29a [92] and miR-320a [93] have been shown to reduce VDAC1 expression levels. Another miRNA species, miR-7, was shown to inhibit VDAC1 expression, proliferation and metastasis in hepatocellular carcinoma [94], possibly by affecting the permeability transition pore (PTP) [95]. Recently, lncRNA-H19/miR-675 was reported to regulate high glucose-induced apoptosis by targeting VDAC1, and thus provides a novel therapeutic strategy for the treatment of diabetic cardiomyopathy [96].
–
The therapeutic potential of a number of miRNAs able to regulate VDAC1 expression levels is clear in view of the observation that VDAC1 over-expression is associated with a variety of pathological conditions, including Alzheimer’s disease (AD) [97][98][99], and cardiovascular diseases (CVDs) [100]. In addition, hyperglycemia has been shown to increase VDAC1 expression in β-cells [101] and in the kidney [102].
VDAC1 AS A PLAYER IN MITOCHONDRIA-MEDIATED APOPTOSIS
Programmed cell death, or apoptosis, is the biological process by which a cell rapidly proceeds towards death upon receiving specific stimuli. The apoptotic machinery in humans consists of a molecular network comprising a large number of proteins that regulate a cascade of events leading to apoptosis through multiple parallel pathways. It is well accepted that mitochondria serve as integrators and amplifiers of apoptosis by mediating and regulating the release of pro-apoptotic proteins and/or disrupting cellular energy metabolism [103]. Upon transfer of an apoptotic signal into the cell, mitochondrial membrane permeability changes so as to facilitate the release of apoptogenic proteins, such as cytochrome c (Cyto c), apoptosis-inducing factor (AIF), and SMAC/Diablo, from the IMS into the cytosol [54][103]. These proteins participate in complex processes, resulting in the activation of proteases and nucleases, leading to protein and DNA degradation and cell death. However, it remains unclear how these apoptotic initiators cross the OMM and are thus released into the cytosol. Several hypotheses regarding the mechanism of mitochondria-mediated apoptosis have been proposed (Fig. 5) (for reviews, see [1][30][104]). The major models include OMM rupture and non-specific release of IMS proteins into the cytosol [55][105][106], opening of the PTP in response to over-production of ROS or Ca2+ overload [107], a large channel formed by Bax and/or Bak oligomers [108][109], a channel formed by hetero-oligomers of VDAC1 and Bax [110][111] or VDAC1 oligomers (Fig. 5) [23][28][29][30][31][36][104][112][113].
–
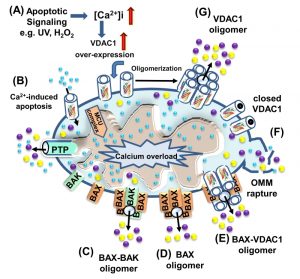 | FIGURE 5: VDAC1 function in cell death, with apoptosis inducers enhancing VDAC1 expression levels and oligomerization. A schematic representation of VDAC1 function in cell death - Different models for the release of apoptogenic proteins, such as Cyto c (purple) and AIF (yellow), are shown. (A) Proposed model suggesting that apoptotic stimuli or conditions cause enhanced VDAC1 expression via increases in [Ca2+]i levels or transcription factors, leading to activation of the VDAC1 promoter. The increase in VDAC1 expression shifts the equilibrium towards the VDAC1 oligomeric state, forming a hydrophilic protein-conducting channel capable of mediating the release of apoptogenic proteins (e.g., Cyto c and AIF) from the mitochondrial IMS to the cytosol. (B) Mitochondrial Ca2+ overload induces apoptosis. Ca2+ transport across the OMM, as mediated by VDAC1, and then across the IMM, as mediated by the MCU, leads to Ca2+ overload in the matrix. This, in turn, causes dissipation of the membrane potential, mitochondrial swelling, PTP opening, Cyto c/AIF release and the triggering of apoptotic cell death. (C) Bax/Bak oligomerization and activation, forming a route for Cyto c/AIF release. (D) Bax activation leads to its association with the OMM, followed by its oligomerization as a large oligomer/complex, forming a Cyto c/AIF-conducting channel. (E) The interaction of the pro-apoptotic protein Bax with VDAC1 forms hetro-oligomers that mediate Cyto c/AIF release. (F) Prolonged VDAC1 closure leads to mitochondrial matrix swelling and OMM rupture, resulting in the appearance of a non-specific release pathway for apoptogenic proteins. |
–
All of the apoptotic proteins known to translocate to the cytoplasm following an apoptotic stimulus reside in IMS. Thus, only the permeability of the OMM needs to be modified for their release [114][115][116][117]. Hence, VDAC1, as an OMM channel, could mediate Cyto c release. Indeed, VDAC1 is now accepted as a key player in mitochondria-mediated apoptosis, with VDAC1 silencing or over-expression affecting apoptosis induction [1][23][33][118][119][120][121][122]. Exogenous over-expression of VDAC from various sources was found to induce apoptotic cell death regardless of cell type [113][118][119][120][121][122]. VDAC1 over-expression-induced cell death was prevented by RuR [122][123], Bcl2, DIDS [120] or by over-expression of HK-I [118][122][124], with all these agents directly interacting with VDAC1. Finally, reducing VDAC1 expression by siRNA efficiently prevented cisplatin-induced apoptosis and Bax activation in non-small cell lung cancer (NSCLC) cells [125], and inhibited selenite-induced PTP opening in HeLa cells [126]. VDAC1-siRNA also attenuated endostatin-induced apoptosis [127]. In addition to the evidence above, release of Cyto c via purified VDAC reconstituted into Cyto c-encapsulating liposomes has been demonstrated [36][128][129]. It is thus proposed that VDAC1 oligomerization is a key step in the release of the pro-apoptotic proteins from the IMS to the cytosol [23][28][29][30][31][36][104][112][113].
–
A VDAC1 oligomeric structure as a Cyto c release pathway
When considering models of VDAC1-mediated protein release, the molecular sizes of the released proteins (12 to 100 kDa) and the diameter of the VDAC1 pore (2.6-3.0 nm) should be considered. The VDAC1 pore can allow passage of nucleotides and small molecules but is too small for the passage of a folded protein like Cyto c. As such, we proposed the formation of a large protein-conducting channel within a VDAC1 homo-oligomer serving as the Cyto c release route. Indeed, upon apoptosis induction by various stimuli, VDAC1 undergoes conformational changes and oligomerization, followed by Cyto c release, and finally, apoptosis [29][33][36][118][129].
–
Apoptosis induction leads to VDAC1 oligomerization regardless of the cell type or apoptosis inducer used, including staurosporine (STS), curcumin, As2O3, etoposide, cisplatin, selenite, H2O2 or UV light, all affecting mitochondria yet acting via different mechanisms [28][112]. Moreover, shifting the equilibrium towards the VDAC1 oligomeric state upon over-expression of the protein in the absence of apoptosis stimuli resulted in release of pro-apoptotic proteins, leading to cell death, regardless of cell type, in a manner that could be inhibited by anti-apoptotic proteins [23][33][113][118][119][120][121][122]. The specific lipid composition of the OMM significantly enhances VDAC1 oligomerization [130], while p53 also promotes VDAC1 oligomerization [131].
–
Several VDAC1-interacting molecules inhibit both apoptosis and VDAC1 oligomerization as induced by various stimuli [28][104][112][113][119][120][122][132][133]. These include 4,4 diisothiocyanostilbene-2,2-disulfonic acid (DIDS), 4-acetamido-4-isothiocyanato-stilbene-2,2-disulfonic acid (SITS), 4,4′ diisothiocyanatodihy-drostilbene-2,2′-disulfonic acid (H2DIDS), 4,4’-dinitrostilbene-2,2’-disulfonic acid (DNDS), and diphenylamine-2-carboxylate (DPC). Similarly, the newly developed VDAC1-interacting molecules AKOS-022 and VBIT-4 prevented VDAC1 oligomerization and apoptosis as induced by various means and in several cell lines [134]. These compounds also protected against apoptosis-associated mitochondrial dysfunction, specifically restoring dissipated mitochondrial membrane potential, and thus cell energy and metabolism, decreasing ROS production, and preventing disruption of intracellular Ca2+ levels. The use of these apoptosis inhibitors thus supports the tight coupling between VDAC1 oligomerization and apoptosis induction. Inhibiting apoptosis at an early stage via prevention of VDAC1 oligomerization may be an effective approach for blocking or slowing apoptosis in neurodegenerative disorders [135][136] and various cardiovascular diseases, where enhanced apoptosis also occurs [137][138][139].
–
To conclude, it is proposed that VDAC1 exists in a dynamic equilibrium between the monomeric and oligomeric states, with apoptosis inducers or VDAC1 over-expression shifting the equilibrium towards oligomerization. Thus, the cellular VDAC1 expression level and its oligomeric state are crucial factors in the process of mitochondria-mediated apoptosis.
–
The mode of action of apoptotic stimuli and VDAC1 over-expression – a new concept
Several studies have demonstrated that the induction of apoptosis by various reagents is accompanied by an increase in the level of VDAC1 expression [54]. These include arbutin (hydroquinone-O-beta-D-glucopyranoside), a tyrosinase inhibitor that induces apoptosis in A375 human ma-lignant melanoma cell by causing VDAC1 over-expression [140]. Up-regulation of VDAC1 expression was noted in acute lymphoblastic leukemia (ALL) cell lines following prednisolone treatment [141]. Somatostatin up-regulated the expression of VDAC1 and VDAC2 in the LNCaP prostate cancer cell line [142]. Up-regulation of VDAC1 expression levels was also induced by the hepatitis E virus ORF3 protein [143]. Also, both UV irradiation and ROS were shown to up-regulate VDAC1 expression [144][145][146] that was prevented by the ROS chelator epigallocatechin [144]. Cisplatin induced VDAC1 over-expression in a cisplatin-sensitive cervix squamous cell carcinoma cell line (A431), while down-regulation of VDAC1 was noted in a cisplatin-resistant cell line (A431/Pt) [147]. Up-regulation of VDAC expression was proposed to mediate the actions of vorinostat, a histone deacetylase inhibitor that induced synergistic anti-proliferative and pro-apoptotic effects in NSCLC cells in combination with EGFR-tyrosine kinase inhibitors [148]. Finally, apoptosis induction by H2O2, etoposide, cisplatin, selenite and UV irradiation all led to enhanced VDAC1 expression levels, which was accompanied by VDAC1 oligomerization, Cyto c release and apoptosis [54][112][113].
–
As apoptosis induction by agents such as STS, As2O3, and selenite disrupt [Ca2+]i homeostasis and energy production [112][113][149], apoptosis-induced VDAC1 up-regulation is proposed to be mediated by an increase in [Ca2+]i. Indeed, we have shown that pro-apoptotic-agents induce cell death through Ca2+-dependent up-regulation of VDAC1 expression levels [54][112][113]. Direct elevation of [Ca2+]i by the Ca2+-mobilizing agents A23187, ionomycin and thapsigargin also resulted in VDAC1 over-expression, VDAC1 oligomerization and apoptosis [112][113]. In contrast, decreasing [Ca2+]i using the cell-permeable Ca2+-chelating reagent BAPTA-AM inhibited VDAC1 over-expression, VDAC1 oligomerization and apoptosis. Thus, the increase in [Ca2+]i induced by apoptosis stimuli was found to be a pre-requisite for induction of VDAC1 over-expression and apoptosis [112][113]. The over-expressed VDAC1 forms oligomers and this triggers Cyto c release and then cell death. The following new concept of apoptosis induction is thus proposed [112][113]: Apoptosis inducers à increased [Ca2+]i à enhanced VDAC1 expression levels à VDAC1 oligomerization à Cyto c release à apoptosis.
–
As such, up-regulation of the expression of VDAC1 may represent a new common mode of action of apoptosis induction.
–
VDAC1 AND CA2+ HOMEOSTASIS
Mitochondria serve as a major hub for cellular Ca2+ homeostasis, regulating oxidative phosphorylation and modulating cytosolic Ca2+ signals of cell death and secretion [150][151]. Mitochondria can rapidly sequester large amounts of Ca2+ at the expense of the membrane potential across the IMM and mediate Ca2+ efflux. Ca2+ is an essential co-factor for several rate-limiting TCA enzymes (i.e., pyruvate dehydrogenase, isocitrate dehydrogenase, and α-ketoglutarate dehydrogenase) located in the matrix, such that intra-mitochondrial Ca2+ controls energy and metabolism. To reach the matrix, Ca2+ must cross both the OMM and the IMM, in a manner mediated by several proteins. VDAC1 acts in the OMM, whereas the mitochondrial Ca2+ uniporter (MCU) [152][153] and the Na+/Ca2+ exchanger, NCLX, the major Ca2+ efflux mediator [154], are both found in the IMM.
–
The function of VDAC1 in regulation of cell Ca2+ homeostasis was recently summarized [155]. VDAC1 in the OMM is highly Ca2+-permeable and transports Ca2+ into and out of the IMS [156][157][158][159], consequently allowing Ca2+ access to IMM transporters. Ruthenium red (RuR) [157][160][161], ruthenium amine binuclear complex (Ru360) [161], the photo-reactive analogue azido ruthenium (AzRu) [162] and the lanthanides La3+and Tb3+ [160] all reduce VDAC1 conductance in the case of native but not mutated VDAC1 [160][161].
–
Competition between Ca2+ and RuR [160] suggests that VDAC1 possesses divalent cation-binding site(s). The physiological function of the VDAC1 Ca2+-binding site(s), reflected in the regulation of VDAC1 gating by physiological levels of Ca2+, prolongs a fully open state of the channel, thereby promoting metabolite exchange [156]. Thus, it has become apparent that VDAC1 both mediates Ca2+ transport and is also regulated by Ca2+ binding.
–
VDAC1 also functions in the ER/mitochondria-Ca2+ cross-talk. VDAC1 is a constituent of a supra-molecular complex composed of the IP3 receptor in the ER and VDAC1 in the OMM, linked by a chaperone called GRP75 [13][163], together with mitofusin-2 [164][165].
–
Thus, by transporting Ca2+, VDAC1 plays a fundamental role in regulating mitochondrial Ca2+ homeostasis, oxidative phosphorylation, and Ca2+ crosstalk among mitochondria, cytoplasm, and the ER.
VDAC AND OXIDATIVE STRESS
Oxidative stress results when production of ROS exceeds the capacity of mitochondrial and cellular anti-oxidant defenses to remove these toxic species. ROS act as second messengers in cell signaling and are essential for multiple biological processes in normal cells. However, ROS are also well known contributors to cell proliferation and cell death [166][167][168][169], provoking damage to multiple cellular organelles and processes [170].
–
Mitochondria are the major source of ROS formation, mostly at complex I (site IQ), complex II (site IIF) and complex III (site IIIQo) [171][172][173]. O2•- generated at complex III is released to the cytosol through VDAC1. By contrast, O2•- produced at complexes I and II is released to the matrix, where it is rapidly converted to H2O2 by superoxide dismutases located in the mitochondrial matrix (MnSOD or SOD2) and the cytosol (Cu,ZnSOD or SOD1) [174]. H2O2 acts as a cell signaling molecule that does not disrupt redox homeostasis [175] and modulates the pro-survival PI3K/Akt/mTOR HIF-1 and MAP/ERK pathways to promote tumorigenesis and metastasis [176][177][178]. H2O2 also forms the highly reactive hydroxyl radical (OH•) by the Fenton reaction. Whereas both H2O2 and O2•- react with mitochondrial and extra-mitochondrial structures, OH• is so reactive that its effects are almost completely restricted to mitochondria. O2•- and OH• can inactivate mitochondrial proteins, and damage mitochondrial DNA and lipids in the MIM [179][180]. Cytosolic ROS also activate members of the MAPK family of serine/threonine kinases, especially the c-Jun N-terminal kinase (JNK), the extracellular signal-regulated kinase (ERK 1/2), and p38, whose signaling can cause mitochondrial dysfunction. JNK translocation to mitochondria has been shown to cause mitochondrial dysfunction in several models [181][182].
–
Cells possess several anti-oxidant defense mechanisms, including the presence of various endogenous molecules, such as glutathione [183][184][185][186][187], or the expression of enzymes like superoxide dismutases (SOD1 and SOD2), catalase, and peroxidase [188]. About 1% of ROS escape elimination and can be released to the cytosol by crossing the OMM, where they can attack and modify DNA, lipids and proteins affecting cell survival [189].
–
VDAC1 has been proposed to mediate ROS release from the IMS to the cytosol. This is based on the finding that ROS release from mitochondria was decreased when HK-I and HK-II bound to VDAC1 were over-expressed in HEK cells, reducing intracellular ROS levels [190][191][192] and protecting against oxidant-induced cell death [190][193]. In tumor cells, VDAC opening or closing increases or decreases OXPHOS and subsequently increases or decreases ROS generation, respectively. Erastin, a small molecule that antagonizes the inhibitory effect of tubulin on VDAC, increases mitochondrial ΔΨ, NADH and ROS production [85]. Thus, blockage of tubulin-dependent VDAC inhibition works as a pro-oxidant anti-Warburg metabolic switch to promote cancer cell death [88][194]. By contrast, VDAC1 closure by DIDS and dextran sulfate inhibits the efflux of O2-• to the cytosol and increases the steady-state level of O2•-, sensitizing mitochondria to Ca2+-induced MPT [195].
–
Cysteine residues, often involved in redox reactions, metal coordination and thiol-disulfide interchanges, are extremely vulnerable to oxidation by ROS. VDAC has been proposed to function in physiological redox regulation via the modification of the sulfhydryl groups of VDAC [196]. In humans, VDAC1 contains two cysteines, VDAC2 contains nine cysteines and VDAC3 contains six cysteines, all of which are predicted to protrude towards the IMS and can be subjected to oxidation by ROS [4]. We have shown that for VDAC1, deletion of both cysteines does not affect channel conductance, VDAC1 oligomerization or apoptosis [53]. Cysteines contribute to the folding, function and stability of hVDAC2. VDAC3 was found to be the target of mitochondrial ROS specifically generated by complex III and was proposed to act as a sensor of the oxidative state of the IMS via cysteine residue modification [197].
–
Accumulating evidence indicates that ROS play a key role in Cyto c release from mitochondria and that this involves VDAC1. Apoptosis-inducing agents, such as inorganic arsenic compounds [198][199] and doxorubicin [200], induce apoptosis by inducing ROS generation. The inhibition of O2-•-induced apoptosis by DIDS, an inhibitor of VDAC channel activity, or by anti-VDAC1 antibodies [12][129][201], suggests that O2-• induces Cyto c release via VDAC1-dependent permeabilization of the OMM [129]. Moreover, O2-• was found to evoke Cyto c release in VDAC1-reconstituted liposomes [129]. In other studies, it was found that ROS-induced alterations of VDAC1 and/or ANT could make the PTP selective for Cyto c release, without causing further mitochondrial damage [129][202]. Moreover, it was shown that ROS induced up-regulation of VDAC1 that could be prevented by the ROS chelator, epigallocatechin [144]. It has been suggested that ROS-mediated Cyto c and SOD1 release from mitochondria involves VDAC, leading to increased susceptibility of mitochondria to oxidative stress and apoptosis [203].
–
VDAC is also affected by hypoxic conditions shown to induce cleavage at the C-terminal end of the protein (VDAC1-ΔC), with such cleavage being prevented upon silencing of HIF-1α expression [204][205]. It was proposed that hypoxia, by inducing formation of VDAC1-ΔC, confers selective protection from apoptosis that allows maintenance of ATP and cell survival in hypoxia [206].
VDAC1 AS A HUB PROTEIN – MODULATION OF VDAC1-MEDIATED APOPTOSIS AND METABOLISM VIA INTERACTING PROTEINS
As presented above, VDAC1 is crucial for many cellular processes, including metabolism, Ca2+ homeostasis, apoptosis, and other activities regulated via the interaction of VDAC1 with many proteins associated with cell survival and cellular death pathways [1][29][30][31]. Indeed, VDAC1 is considered as a hub protein, interacting with over 100 proteins that regulate the integration of mitochondrial functions with other cellular activities [207]. VDAC1 serves as an anchor protein for diverse sets of cytosolic, ER, and mitochondrial proteins [12][208] that together regulate metabolic and signaling pathways, provide energy for cellular functions, or trigger cell death. Thus, VDAC1 appears to be a convergence point for a variety of cell survival and death signals, mediated via association with ligands and proteins.
–
In support of this viewpoint, the conserved nature of VDAC1 [1] is in agreement with the finding that hub proteins are more evolutionarily conserved than are non-hub proteins [209]. VDAC1 protein-protein interaction (PPI) networks contain both hub-bottlenecks [210] (namely nodes with high degree values constituting vulnerable areas of the network) and/or bottlenecks (those with high “betweenness” centrality scores, corresponding to key intersecting nodes [211]). The VDAC1 interactome includes proteins involved in metabolism, apoptosis, signal transduction, anti-oxidation, and DNA- and RNA-associated proteins and more (Supplemental Table S1) [1][29][31][32]. Furthermore, these proteins may be located in the OMM, IMM, the IMS, the cytosol, ER, plasma membrane, and nucleus. Importantly, we have been able to develop VDAC1-based peptides which can interfere with these interactions, leading to impaired cell metabolism and apoptosis [25][26][27][212][213].
–
Interactions of VDAC1 with metabolism-related proteins
VDAC1 displays binding sites for a large number of metabolism-related proteins, such as glycerol kinase (GK), HK, c-Raf kinase, ANT, tubulin, [1][29][30][31] and the glycolytic enzyme GAPDH (glyceraldehyde 3-phosphate dehydrogenase) [214]. Mitochondrial creatine kinase (MtCK), in its octameric state, interacts with VDAC1 [35] and causes decreased affinity of VDAC1 for HK and Bax [215] (Supplemental Table S1). HK binding to VDAC1 [25][104][118][122][124][216] allows direct coupling of mitochondrially generated ATP to glucose phosphorylation. Thus, the formation of a VDAC1-HK complex coordinates glycolytic flux with the actions of the TCA cycle and ATP synthase [1][33][81].
–
The OMM protein CPT1a that catalyzes the primary step of fatty acid oxidation interacts with VDAC1 [65]. Another OMM protein interacting with VDAC1 is the TSPO, involved in the transport of cholesterol into mitochondria [217]. Aldolase, involved in gluconeogenesis and glycolysis, was also shown to interact with VDAC1 [17].
–
Interactions of VDAC1 with apoptosis-related proteins
The Bcl-2 family comprises pro-apoptotic (e.g. Bid, Bax, Bim and Bak) and anti-apoptotic (e.g. Bcl-2 and Bcl-xL) members that up- or down-regulate apoptosis, respectively [218][219]. VDAC1 function in apoptosis can be regulated by interactions with anti-apoptotic proteins, such as Bcl2 and Bcl-xL [23][25][26][117][220][221][222][223], resulting in inhibition of apoptotic pathways. Bcl-2 and Bcl-xL were shown to interact with bilayer-reconstituted VDAC1 and subsequently to reduce the channel conductance of native but not mutated VDAC1, as well as to protect against apoptosis in cells expressing native but not mutated VDAC1 [25][26]. The VDAC1 domains that interact with Bcl-2 and Bcl-xL to confer anti-apoptotic activity were identified by site-directed mutagenesis [25]. Mcl-1 has been shown to directly interact with VDAC to increase mitochondrial Ca2+ uptake and ROS generation [224]. The HK-VDAC interaction also prevents release of pro-apoptotic factors, such as Cyto c, and subsequent apoptosis. Thus, HK plays a role in tumor cell survival via inhibition of apoptosis [118]. The interaction between TSPO and VDAC is considered to play a role in the activation of the mitochondrial apoptosis pathway, given the reported grouping of TSPO molecules around VDAC, potentially reflecting TSPO polymerization [225], and the increased ROS generation by TSPO in the proximity of VDAC, leading to apoptosis induction [225][226]. Nek1 (NIMA-related protein kinase 1) phosphorylates VDAC1 on serine 193, with this leading to apoptosis inhibition [146][227]. Finally, the pro-apoptotic protein BNIP3 was shown to interact with VDAC so as to induce mitochondrial release of endonuclease G [228].
–
Interactions of VDAC1 with cytoskeletal proteins
VDAC1 interacts with several cytoskeletal proteins, such as gelsolin (Gsn), with this interaction resulting in inhibited VDAC1 channel activity and Cyto c release from liposomes through direct binding to VDAC1 in a Ca2+-dependent manner [229][230]. Tubulin was shown to associate with VDAC1 [231] and induce VDAC1 closure [86], proposed to sustain the Warburg effect [232]. It was further proposed that tubulin, VDAC1, and MtCK form a super-complex that is structurally and functionally coupled to the ATP synthasome [233]. G-actin directly and selectively binds to VDAC in yeast, [234], reducing conductance of the Neurospora crassa VDAC channel [235]. Microtubule-associated protein 2 (MAP2) was shown to bind VDAC [236]. The interaction of VDAC1 with Tctex-1/DYNLT1 (dynein light chain) was also demonstrated [237].
–
Interactions of VDAC1 with signaling proteins
Superoxide dismutase 1 (SOD1) is a predominantly cytosolic protein, with mutant SOD1 being present mostly in fractions enriched for mitochondria [238][239][240]. Mutant SOD1 associated with amyotrophic lateral sclerosis (ALS) bound to bilayer-reconstituted VDAC1 and inhibited its channel conductance [241]. Mutant SOD1 also interacted with Bcl2 protein and altered the interaction between Bcl-2 and VDAC1, thus reducing OMM permeability [242].
–
Endothelial NO synthase (eNOS) was also found to bind VDAC1. Such interactions amplified eNOS activity in an intracellular Ca2+-mediated manner [243]. These findings suggest that the interaction between VDAC and eNOS may be important for regulating eNOS activity and modulation of VDAC [243].
–
The mitochondrial anti-viral signaling protein MAVS, also known as IPS-1, VISA, or Cardif [244], and localized in the OMM, was demonstrated to mediate its pro-apoptotic activity via VDAC1 and to modulate VDAC1 protein stability via the ubiquitin-proteasome pathway [245]. VDAC was further proposed to interact with the L-type Ca2+ channel [246].
–
Several additional proteins were shown or proposed to directly interact with VDAC1 (Supplemental Table S1). These include PBP74, also known as mtHSP70/GRP75/mortalin [237], CRYAB (α-crystallin B) [247] and α-synuclein [248]. VDAC1-interacting protein complexes mediate and/or regulate metabolic, apoptotic, and other processes that may be impaired in disease.
VDAC INVOLVEMENT IN DISEASE
Mitochondria occupy a central position in cell life and death and mitochondrial dysfunction has been implicated in many diseases, including cancer, Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), diabetes, and cardiovascular diseases (CVDs). VDAC1 functions as mitochondria gatekeeper that regulates ATP production, Ca2+ homeostasis and apoptosis execution, all indispensable for proper mitochondrial function, and consequently, for cell normal physiology. Thus, the association of VDAC with various diseases is not surprising. Furthermore, VDAC over-expression is a common feature of cancer, AD, type 2 diabetes (T2D) and CVDs. The over-expression of VDAC1 in cancer [54][59], in affected regions of AD brains [97][98][99], in β-cells of T2D [101] and in CVDs [249], is a feature common to these diseases. As VDAC1 over-expression induces apoptotic cell death [58][113][119][120][122], its over-expression in CVDs, AD and T2D, may be a common mechanism in these pathologies.
–
The cancer-mitochondria-metabolism-apoptosis-VDAC1 link
Cancer is a complex disease in which cells acquire a common set of properties, including unlimited proliferation, metabolic reprograming, and resistance to anti-proliferative and apoptotic cues [78][250]. Emerging evidence indicates that metabolic reprogramming, which supports macromolecule synthesis, bioenergetics demands, and cellular survival is a characteristic of nearly all cancers [68][251]. Over the years, Otto Warburg’s view of cancer as a metabolic disease was gradually displaced with the view of cancer as a genetic disease [252]. Today, however, cancer is again being seen as a metabolic disease, primarily associated with impaired mitochondrial function and metabolism [75][79].
–
VDAC1 is highly expressed in different tumors [54][59], contributing to their metabolism via the transport of various metabolites, and the binding and channeling of mitochondrial ATP directly to HK [5]. This results in mitochondria regulating glycolytic flux with that actions of the TCA cycle and ATP synthase to fulfill the requirements of tumors for metabolites or metabolite precursors. Indeed, tumors switch their HK expression pattern predominantly to present the VDAC1-binding isoforms (HK-I, HK-II) [253]. The requirement of cancer cells for VDAC1 is demonstrated by down-regulation of VDAC1, resulting in reduced metabolite exchange between mitochondria and cytosol and inhibition of cell and tumor growth [32][58][59][90][91].
–
VDAC1 also regulates apoptosis in cancer cells by interacting with the anti-apoptotic proteins Bcl-2 and Bcl-xL [23][25][26][254] and HK [23][27], interactions that protect tumor cells from cell death [23][27]. Thus, activating mitochondria-mediated apoptosis directly or via generating stress responses [255][256][257] is a strategy to treat cancer. Indeed, a large number of anti-cancer chemotherapeutic agents exert their therapeutic action by inducing apoptosis of malignant cells [258][259][260][261][262][263], mainly by activating the Cyto c/caspase-9 pathway. These include etoposide, doxorubicin, lonidamine, betulinic acid, arsenite, CD437, and several amphiphilic cationic α-helical peptides [264]. Therefore, targeted activation of apoptosis in cancerous tissues may be exploited as a potential route to cancer therapy [265]. However, they do not act on cancer stem cells (CSCs), which are resistant to chemo- and radio-therapies [266][267][268]. The CSC hypothesis postulates that a sub-population of malignant cells constantly supply the tumor with cancerous cells. CSCs, as embryonic and somatic stem cells, have self-renewal and multi-potent differentiation abilities [269][270]. Recent studies from the Shoshan-Barmatz group [91][213] have demonstrated novel strategies for eliminating CSCs.
–
Regulation of VDAC1 expression by miRNA was demonstrated in several studies. In serum-starved cervical cancer cells, miR320a promoted mitophagy [93], while ectopic over-expression of miR320a blocked tumor cell proliferation and invasion in NSCLC, both in vitro and in vivo [271]. Another miRNA species, miR-7, was shown to inhibit VDAC1 expression, proliferation and metastasis in hepatocellular carcinoma [94], possibly by affecting the PTP [95].
–
Thus, the importance of VDAC1 for cancer cell survival is clearly reflected in the above findings, with silencing VDAC1 expression in cancer cells resulting in a multi-pronged attack on cancer hallmarks.
–
Neurodegenerative diseases, mitochondria, apoptosis, and VDAC
There is emerging evidence connecting mitochondrial dysfunction to neurodegenerative disorders [169]. In PD, Huntington’s disease (HD), ALS and AD, impaired mitochondrial function has been reported [272], with a focus on the involvement of mitochondria-mediated apoptotic death [273]. Mitochondrial dysfunction was proposed as an early event in AD pathogenesis, as reflected in reduced metabolism, increased ROS, lipid peroxidation and disruption of Ca2+-homeostasis, [274][275][276]. Moreover, mitochondria-mediated apoptosis is common to neurological disorders in which premature neuron death is implicated [273][277][278], with caspases playing dominant roles [279][280][281]. Amyloid beta (Aβ) also affects mitochondrial respiration [282] and activates Cyto c release, thereby promoting apoptosis [283].
–
Several studies suggested that VDAC malfunction is associated with AD [284][285][286][287], Down’s syndrome [287], and familial ALS [241][288]. High levels of VDAC1 were demonstrated in the dystrophic neurites of Aβ deposits in post-mortem brains of AD patients and in amyloid precursor protein (APP) transgenic mice, where Aβ-VDAC interactions are toxic to AD-affected neurons [97][98][286][287][289][290][291]. The expression of hVDAC-2 was shown to be associated with neurodegenerative diseases, including ALS [288], epilepsy [292], and AD [286].
–
As VDAC1 over-expression was shown to lead to apoptotic cell death [58][113][119][120][122] and high-levels of VDAC1 were found in the dystrophic neurites of Aβ deposits in AD post-mortem brains and APP transgenic mice [97][98][99], we propose that over-expressed VDAC is associated with neuronal cell death [291].
–
We have demonstrated that Aβ interacts directly with VDAC1, specifically with the VDAC1 N-terminal region and that VDAC1 is required for Aβ entry into the cell, as well as for Aβ-mediated apoptosis, with Aβ cell penetration and toxicity being prevented in cells depleted of VDAC1 by siRNA [291]. VDAC was also shown to interact with phosphorylated Tau, leading to mitochondrial dysfunction [290]. In addition, an increase in nitrated VDAC1 levels in AD was reported, reflecting oxidative damage to VDAC [293], and possibly affecting cell energy and metabolite homeostasis [284]. The involvement of plasmalemmal VDAC in AD was also proposed [285][289].
–
The relationship between VDAC1 expression levels and neurodegenerative disorders is also reflected in the finding that in patients and animal models of several neurodegenerative disorders, such as AD, HD, and spinocerebellar ataxias, miR-29a expression levels were reduced [294]. miR-29a was also shown to regulate cell survival of astrocytes differentially by targeting VDAC1 [295]. These findings suggest that VDAC1 down-regulation by miR-29 is an important aspect of neuronal cell survival in the brain [294]. As VDAC1 over-expression triggers apoptosis [120][121][122], and high-levels of VDAC1 were demonstrated in AD post-mortem brains and in AD-like transgenic mice [99], the reported decrease in miR-29a in AD [294] may be associated with neuronal cell death. Indeed, miR-320a-mediated down-regulation of VDAC1 expression has been proposed as a novel therapeutic target for astroglia-mediated HIV-1 neuropathogenesis [296].
–
Finally, several proteins interacting with VDAC, such as SOD1, α-synuclein and ApoE, were proposed to be involved in several neurodegenerative diseases, affecting intraneuronal Ca2+ [155]. These findings point to VDAC1 as a potential target for novel therapeutic strategies for neurodegenerative diseases.
–
T2D, metabolism, mitochondria and VDAC1
T2D is the most common metabolic disease [297]. Defective insulin secretion, insulin resistance at target tissues and a loss of functional β-cells contribute to T2D, and dysregulation of glucose homeostasis [298]. Recently, it has been shown that hyperglycemia increases VDAC1 expression in pancreatic β-cells [101] and in the kidney [102]. VDAC1 levels were increased in mouse coronary vascular endothelial cells (MCECs) isolated from diabetic mice. This was associated with increased [Ca2+]m, O2 production, and PTP opening activity [299]. Down-regulation of VDAC1 in diabetic MCECs decreased [Ca2+]m and subsequently affected PTP activity and ROS production [300]. As glucose-stimulated insulin secretion depends on the generation of ATP and other metabolites in the mitochondria [301], and VDAC1 regulates energy and metabolism, VDAC1 is thus required for insulin secretion. Recently, lncRNA-H19/miR-675 was reported to regulate high glucose-induced apoptosis by targeting VDAC1, and thus provides a novel therapeutic strategy for the treatment of diabetic cardiomyopathy [96]. These findings point to the connection between VDAC, mitochondrial function and the pathogenesis of T2D.
–
Cardiovascular diseases, mitochondria, apoptosis and VDAC
It is known that most CVDs evolve into heart failure and that the loss of cardiac myocytes plays a critical role in the pathogenesis of CVDs. Activation of mitochondria-mediated apoptosis has been implicated in ischemia/reperfusion injury [302]. VDAC levels were increased in cardiomyoblast H9c2 cells [249]. As VDAC1 over-expression is associated with apoptosis [58][113][119][120][122], it is possible that increased cardiomyocyte susceptibility to mitochondrial-mediated cell death is related to the increase in VDAC1 levels. Indeed, the effect of resveratrol against myocardial ischemia/reperfusion injury showed involvement of VDAC1 down-regulation [100].
–
The findings presented above suggest that modulating VDAC1 expression levels or its apoptotic activity are possible strategies to either activate apoptosis in cancer or inhibit apoptosis in CVDs, AD and T2D.
UNRAVELING VDAC1-BASED THERAPIES
VDAC1-based strategies are expected to be effective in various diseases characterized by altered cell metabolism and/or apoptosis and by VDAC1 over-expression. As VDAC1 over-expression induces apoptotic cell death [58][113][119][120][122], we suggest that this may be a common mechanism in the pathology of CVDs, AD and T2D. Modulating VDAC1 expression levels or its apoptotic activity are possible strategies to either activate apoptosis in cancer or inhibit apoptosis in CVDs, AD and T2D. In this review, VDAC1-based therapeutic strategies targeting tumor cells are presented. These cancer therapy strategies include siRNA altering the normal functioning of cancer cells, leading to growth arrest, and VDAC1-based peptides that impair energy homeostasis and minimize the self-defense mechanisms of these cells, and that can be used to overcome protective and pro-survival actions taken by cancer cells.
–
VDAC1-depletion using RNAi
Specifically targeting metabolism in cancer cells presents a potential therapeutic strategy. However, although glucose metabolism is increased in cancer cells, these cells mostly use the same glycolytic enzymes as do normal cells, so that the choice of glycolytic enzymes as targets for cancer treatment may increase the risk of adverse and undesirable consequences [303]. Targeting VDAC1, acting as a ‘governor’ of mitochondrial function, regulating cellular energy and metabolism, and over-expressed in cancer, offers a unique target for anti-cancer therapies. Down-regulation of VDAC1 addresses the cancer trademark of cell metabolic and energy reprogramming, leading to disrupted cancer cell energy and metabolism homeostasis.
–
VDAC1 depletion using specific siRNA (si-VDAC1) led to reduced cellular ATP levels and inhibited cell and tumor growth in cervical and lung cancers [58][59][90]. Using a sub-cutaneous and intracranial-orthotopic GBM model, we found that VDAC1 depletion resulted in inhibited tumor growth, with the residual tumor showing reversed oncogenic properties, including metabolic reprograming and inhibited proliferation, angiogenesis, EMT, invasiveness and stemness, leading to differentiation into neuron- and astrocyte-like cells [91] (Fig. 4).
–
VDAC1-based peptides as potential anti-cancer therapy
A hallmark of cancer cells is their ability to suppress pro-apoptotic pathways and/or to activate anti-apoptotic mechanisms [78][250] associated with drug resistance [304], such as the Bcl-2 family of proteins and HK, preventing the release of Cyto c from mitochondria. Since the anti-apoptotic proteins HK-I, HK-II, Bcl2 and Bcl-xL have been found to be expressed at high levels in many types of cancer [81][253][305][306][307][308][309][310] and interact with VDAC1 [1][22][23][24][25][26][27][81][82][84][118][122][124][128][220][311][312], the interaction of VDAC1with these proteins is proposed as an appropriate target to induce apoptosis.
–
We have engineered VDAC1-based peptides that interfere with the activity of the pro-survival proteins Bcl-2, Bcl-xL and HK [23][25][26][27][118][122][212]. Via point mutations, VDAC1 domains and amino acid residues important for interactions with HK, Bcl-2 and Bcl-xL were identified and cell-penetrating VDAC1-based peptides targeting these interactions were designed and tested [23][25][26][27][118][122]. These VDAC1-based peptides were found to induce cancer cell death in a panel of genetically characterized cancer cell lines, regardless of cancer type or mutation status, with perceived specificity toward cancerous cells [23][27][212]. Studies demonstrated a triple mode of action, namely energy and metabolism impairment, interference with the action of anti-apoptotic proteins, and a triggering of cell death.
–
In an ex vivo study, cell-penetrating VDAC1-based peptides were found to induce apoptotic cell death in the cancerous B-cells of peripheral blood mononuclear cells obtained from chronic lymphocytic leukemia (CLL) patients, yet spared those obtained from healthy donors, pointing to the potential of VDAC1-based peptides as an innovative and effective anti-CLL therapy.
–
In a GBM mouse model, i.v.-administered VDAC1-based peptide Tf-D-LP4 crossed the blood-brain barrier and was found to inhibit tumor growth by inducing apoptosis and over-expression of apoptotic proteins [213]. Such treatment simultaneously attacked several cancer hallmarks, causing impairment of energy and metabolic homeostasis, inhibition of tumor growth and induction of apoptosis. VDAC1-based peptides, expecting to also affect other cancers, provide the opportunity for the development of new anti-cancer therapies that will allow overcoming the chemo-resistance of cancer cells.
–
In summary, VDAC1 functions in ATP production and metabolism, Ca2+ homeostasis and apoptosis execution are indispensable for proper mitochondrial function of cancer cell, and consequently, for cell activity. These VDAC-mediated activities are regulated via interactions of VDAC1 with many proteins that are critically involved in the regulation of cell survival and cellular death pathways. VDAC1, standing at the crossroads between mitochondrial-mediated energy and metabolism and apoptosis, is a potential target for treating cancer and other diseases involving dysregulated metabolism and/or apoptosis and where VDAC1 is over-expressed. Thus, a new generation of VDAC1-based therapeutics may impact the treatment of a variety of diseases.
–
To conclude, the dysregulated cell stress response involves mitochondria dysfunction and this play a critical role in tumorigenesis, and other disease, such as Alzheimer’s disease, some cardiovascular disease and type 2 diabetes. The role of VDAC1 in Ca2+ homeostasis, energy production and oxidative stress, and with VDAC1 serving as a hub protein interacting with over 100 proteins allow it to mediate and regulate the integration of mitochondrial functions with cellular activities. Thus, VDAC1, standing at the crossroads between mitochondrial metabolite transport, apoptosis and other cell stress-associated processes, serves as the mitochondrial gatekeeper. This, together with its over-expression in cancer and other diseases, including Alzheimer’s disease, some cardiovascular diseases and type 2 diabetes, involves VDAC1 in the cell stress response and thus represents a target to modulate the biology of cancer and other diseases.
References
- V. Shoshan-Barmatz, V. De Pinto, M. Zweckstetter, Z. Raviv, N. Keinan, and N. Arbel, "VDAC, a multi-functional mitochondrial protein regulating cell life and death", Molecular Aspects of Medicine, vol. 31, pp. 227-285, 2010. http://dx.doi.org/10.1016/j.mam.2010.03.002
- A. Messina, S. Reina, F. Guarino, and V. De Pinto, "VDAC isoforms in mammals", Biochimica et Biophysica Acta (BBA) - Biomembranes, vol. 1818, pp. 1466-1476, 2012. http://dx.doi.org/10.1016/j.bbamem.2011.10.005
- V. De Pinto, F. Guarino, A. Guarnera, A. Messina, S. Reina, F.M. Tomasello, V. Palermo, and C. Mazzoni, "Characterization of human VDAC isoforms: A peculiar function for VDAC3?", Biochimica et Biophysica Acta (BBA) - Bioenergetics, vol. 1797, pp. 1268-1275, 2010. http://dx.doi.org/10.1016/j.bbabio.2010.01.031
- V. De Pinto, S. Reina, A. Gupta, A. Messina, and R. Mahalakshmi, "Role of cysteines in mammalian VDAC isoforms' function", Biochimica et Biophysica Acta (BBA) - Bioenergetics, vol. 1857, pp. 1219-1227, 2016. http://dx.doi.org/10.1016/j.bbabio.2016.02.020
- E.H. Cheng, T.V. Sheiko, J.K. Fisher, W.J. Craigen, and S.J. Korsmeyer, "VDAC2 Inhibits BAK Activation and Mitochondrial Apoptosis", Science, vol. 301, pp. 513-517, 2003. http://dx.doi.org/10.1126/science.1083995
- A. Raghavan, T. Sheiko, B.H. Graham, and W.J. Craigen, "Voltage-dependant anion channels: Novel insights into isoform function through genetic models", Biochimica et Biophysica Acta (BBA) - Biomembranes, vol. 1818, pp. 1477-1485, 2012. http://dx.doi.org/10.1016/j.bbamem.2011.10.019
- S. Wu, M.J. Sampson, W.K. Decker, and W.J. Craigen, "Each mammalian mitochondrial outer membrane porin protein is dispensable: effects on cellular respiration", Biochimica et Biophysica Acta (BBA) - Molecular Cell Research, vol. 1452, pp. 68-78, 1999. http://dx.doi.org/10.1016/S0167-4889(99)00120-2
- M.J. Sampson, W.K. Decker, A.L. Beaudet, W. Ruitenbeek, D. Armstrong, M.J. Hicks, and W.J. Craigen, "Immotile Sperm and Infertility in Mice Lacking Mitochondrial Voltage-dependent Anion Channel Type 3", Journal of Biological Chemistry, vol. 276, pp. 39206-39212, 2001. http://dx.doi.org/10.1074/jbc.M104724200
- E.J. Weeber, M. Levy, M.J. Sampson, K. Anflous, D.L. Armstrong, S.E. Brown, J.D. Sweatt, and W.J. Craigen, "The Role of Mitochondrial Porins and the Permeability Transition Pore in Learning and Synaptic Plasticity", Journal of Biological Chemistry, vol. 277, pp. 18891-18897, 2002. http://dx.doi.org/10.1074/jbc.M201649200
- G. Báthori, I. Parolini, I. Szabó, F. Tombola, A. Messina, M. Oliva, M. Sargiacomo, V. De Pinto, and M. Zoratti, "Array", Journal of Bioenergetics and Biomembranes, vol. 32, pp. 79-89, 2000. http://dx.doi.org/10.1023/A:1005516513313
- G. Bàthori, I. Parolini, F. Tombola, I. Szabò, A. Messina, M. Oliva, V. De Pinto, M. Lisanti, M. Sargiacomo, and M. Zoratti, "Porin Is Present in the Plasma Membrane Where It Is Concentrated in Caveolae and Caveolae-related Domains", Journal of Biological Chemistry, vol. 274, pp. 29607-29612, 1999. http://dx.doi.org/10.1074/jbc.274.42.29607
- V. Shoshan-Barmatz, N. Hadad, W. Feng, I. Shafir, I. Orr, M. Varsanyi, and L.M. Heilmeyer, "VDAC/porin is present in sarcoplasmic reticulum from skeletal muscle", FEBS Letters, vol. 386, pp. 205-210, 1996. http://dx.doi.org/10.1016/0014-5793(96)00442-5
- V. Shoshan-Barmatz, R. Zalk, D. Gincel, and N. Vardi, "Subcellular localization of VDAC in mitochondria and ER in the cerebellum", Biochimica et Biophysica Acta (BBA) - Bioenergetics, vol. 1657, pp. 105-114, 2004. http://dx.doi.org/10.1016/j.bbabio.2004.02.009
- R.Z. Sabirov, and P.G. Merzlyak, "Plasmalemmal VDAC controversies and maxi-anion channel puzzle", Biochimica et Biophysica Acta (BBA) - Biomembranes, vol. 1818, pp. 1570-1580, 2012. http://dx.doi.org/10.1016/j.bbamem.2011.09.024
- R. Buettner, G. Papoutsoglou, E. Scemes, D.C. Spray, and R. Dermietzel, "Evidence for secretory pathway localization of a voltage-dependent anion channel isoform", Proceedings of the National Academy of Sciences, vol. 97, pp. 3201-3206, 2000. http://dx.doi.org/10.1073/pnas.97.7.3201
- F.P. Thinnes, H. Götz, H. Kayser, R. Benz, W.E. Schmidt, H.D. Kratzin, and N. Hilschmann, "[Identification of human porins. I. Purification of a porin from human B-lymphocytes (Porin 31HL) and the topochemical proof of its expression on the plasmalemma of the progenitor cell].", Biological chemistry Hoppe-Seyler, 1989. http://www.ncbi.nlm.nih.gov/pubmed/2559744
- V. Shoshan-Barmatz, and A. Israelson, "The Voltage-dependent Anion Channel in Endoplasmic/Sarcoplasmic Reticulum: Characterization, Modulation and Possible Function", Journal of Membrane Biology, vol. 204, pp. 57-66, 2005. http://dx.doi.org/10.1007/s00232-005-0749-4
- M. Bayrhuber, T. Meins, M. Habeck, S. Becker, K. Giller, S. Villinger, C. Vonrhein, C. Griesinger, M. Zweckstetter, and K. Zeth, "Structure of the human voltage-dependent anion channel", Proceedings of the National Academy of Sciences, vol. 105, pp. 15370-15375, 2008. http://dx.doi.org/10.1073/pnas.0808115105
- S. Hiller, R.G. Garces, T.J. Malia, V.Y. Orekhov, M. Colombini, and G. Wagner, "Solution Structure of the Integral Human Membrane Protein VDAC-1 in Detergent Micelles", Science, vol. 321, pp. 1206-1210, 2008. http://dx.doi.org/10.1126/science.1161302
- R. Ujwal, D. Cascio, J. Colletier, S. Faham, J. Zhang, L. Toro, P. Ping, and J. Abramson, "The crystal structure of mouse VDAC1 at 2.3 Å resolution reveals mechanistic insights into metabolite gating", Proceedings of the National Academy of Sciences, vol. 105, pp. 17742-17747, 2008. http://dx.doi.org/10.1073/pnas.0809634105
- S. Hiller, and G. Wagner, "The role of solution NMR in the structure determinations of VDAC-1 and other membrane proteins", Current Opinion in Structural Biology, vol. 19, pp. 396-401, 2009. http://dx.doi.org/10.1016/j.sbi.2009.07.013
- S. Geula, D. Ben-Hail, and V. Shoshan-Barmatz, "Structure-based analysis of VDAC1: N-terminus location, translocation, channel gating and association with anti-apoptotic proteins", Biochemical Journal, vol. 444, pp. 475-485, 2012. http://dx.doi.org/10.1042/BJ20112079
- S. Abu-Hamad, N. Arbel, D. Calo, L. Arzoine, A. Israelson, N. Keinan, R. Ben-Romano, O. Friedman, and V. Shoshan-Barmatz, "The VDAC1 N-terminus is essential both for apoptosis and the protective effect of anti-apoptotic proteins", Journal of Cell Science, vol. 122, pp. 1906-1916, 2009. http://dx.doi.org/10.1242/jcs.040188
- Y. Shi, J. Chen, C. Weng, R. Chen, Y. Zheng, Q. Chen, and H. Tang, "Identification of the protein–protein contact site and interaction mode of human VDAC1 with Bcl-2 family proteins", Biochemical and Biophysical Research Communications, vol. 305, pp. 989-996, 2003. http://dx.doi.org/10.1016/S0006-291X(03)00871-4
- N. Arbel, D. Ben-Hail, and V. Shoshan-Barmatz, "Mediation of the Antiapoptotic Activity of Bcl-xL Protein upon Interaction with VDAC1 Protein", Journal of Biological Chemistry, vol. 287, pp. 23152-23161, 2012. http://dx.doi.org/10.1074/jbc.M112.345918
- N. Arbel, and V. Shoshan-Barmatz, "Voltage-dependent Anion Channel 1-based Peptides Interact with Bcl-2 to Prevent Antiapoptotic Activity", Journal of Biological Chemistry, vol. 285, pp. 6053-6062, 2010. http://dx.doi.org/10.1074/jbc.M109.082990
- L. Arzoine, N. Zilberberg, R. Ben-Romano, and V. Shoshan-Barmatz, "Voltage-dependent Anion Channel 1-based Peptides Interact with Hexokinase to Prevent Its Anti-apoptotic Activity", Journal of Biological Chemistry, vol. 284, pp. 3946-3955, 2009. http://dx.doi.org/10.1074/jbc.M803614200
- N. Keinan, D. Tyomkin, and V. Shoshan-Barmatz, "Oligomerization of the Mitochondrial Protein Voltage-Dependent Anion Channel Is Coupled to the Induction of Apoptosis", Molecular and Cellular Biology, vol. 30, pp. 5698-5709, 2010. http://dx.doi.org/10.1128/MCB.00165-10
- V. Shoshan-Barmatz, A. Israelson, D. Brdiczka, and S. Sheu, "The Voltage-Dependent Anion Channel (VDAC): Function in Intracellular Signalling, Cell Life and Cell Death", Current Pharmaceutical Design, vol. 12, pp. 2249-2270, 2006. http://dx.doi.org/10.2174/138161206777585111
- V. Shoshan-Barmatz, and D. Mizrachi, "VDAC1: from structure to cancer therapy", Frontiers in Oncology, vol. 2, 2012. http://dx.doi.org/10.3389/fonc.2012.00164
- V. Shoshan-Barmatz, D. Mizrachi, and N. Keinan, "Oligomerization of the Mitochondrial Protein VDAC1", Progress in Molecular Biology and Translational Science, pp. 303-334, 2013. http://dx.doi.org/10.1016/B978-0-12-386931-9.00011-8
- V. Shoshan-Barmatz, and M. Golan, "Mitochondrial VDAC1: Function in Cell Life and Death and a Target for Cancer Therapy", Current Medicinal Chemistry, vol. 19, pp. 714-735, 2012. http://dx.doi.org/10.2174/092986712798992110
- V. Shoshan-Barmatz, N. Keinan, and H. Zaid, "Uncovering the role of VDAC in the regulation of cell life and death", Journal of Bioenergetics and Biomembranes, vol. 40, pp. 183-191, 2008. http://dx.doi.org/10.1007/s10863-008-9147-9
- K. Zeth, T. Meins, and C. Vonrhein, "Approaching the structure of human VDAC1, a key molecule in mitochondrial cross-talk", Journal of Bioenergetics and Biomembranes, vol. 40, 2008. http://dx.doi.org/10.1007/s10863-008-9144-z
- U. Schlattner, M. Tokarska-Schlattner, and T. Wallimann, "Mitochondrial creatine kinase in human health and disease", Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease, vol. 1762, pp. 164-180, 2006. http://dx.doi.org/10.1016/j.bbadis.2005.09.004
- R. ZALK, A. ISRAELSON, E. GARTY, H. AZOULAY-ZOHAR, and V. SHOSHAN-BARMATZ, "Oligomeric states of the voltage-dependent anion channel and cytochrome c release from mitochondria", Biochemical Journal, vol. 386, pp. 73-83, 2005. http://dx.doi.org/10.1042/BJ20041356
- S. Geula, H. Naveed, J. Liang, and V. Shoshan-Barmatz, "Structure-based Analysis of VDAC1 Protein", Journal of Biological Chemistry, vol. 287, pp. 2179-2190, 2012. http://dx.doi.org/10.1074/jbc.M111.268920
- R. Ujwal, D. Cascio, V. Chaptal, P. Ping, and J. Abramson, "Crystal packing analysis of murine VDAC1 crystals in a lipidic environment reveals novel insights on oligomerization and orientation", Channels, vol. 3, pp. 167-170, 2009. http://dx.doi.org/10.4161/chan.3.3.9196
- U. Schlattner, M. Dolder, T. Wallimann, and M. Tokarska-Schlattner, "Mitochondrial Creatine Kinase and Mitochondrial Outer Membrane Porin Show a Direct Interaction That Is Modulated by Calcium", Journal of Biological Chemistry, vol. 276, pp. 48027-48030, 2001. http://dx.doi.org/10.1074/jbc.M106524200
- D. Ben-Hail, and V. Shoshan-Barmatz, "Purification of VDAC1 from Rat Liver Mitochondria", Cold Spring Harbor Protocols, vol. 2014, pp. pdb.prot073130, 2013. http://dx.doi.org/10.1101/pdb.prot073130
- D. Gincel, S.D. Silberberg, and V. Shoshan-Barmatz, "Array", Journal of Bioenergetics and Biomembranes, vol. 32, pp. 571-583, 2000. http://dx.doi.org/10.1023/A:1005670527340
- M. Colombini, "VDAC structure, selectivity, and dynamics", Biochimica et Biophysica Acta (BBA) - Biomembranes, vol. 1818, pp. 1457-1465, 2012. http://dx.doi.org/10.1016/j.bbamem.2011.12.026
- T. Rostovtseva, and M. Colombini, "VDAC channels mediate and gate the flow of ATP: implications for the regulation of mitochondrial function", Biophysical Journal, vol. 72, pp. 1954-1962, 1997. http://dx.doi.org/10.1016/S0006-3495(97)78841-6
- D. Gincel, and V. Shoshan-Barmatz, "Glutamate Interacts with VDAC and Modulates Opening of the Mitochondrial Permeability Transition Pore", Journal of Bioenergetics and Biomembranes, vol. 36, pp. 179-186, 2004. http://dx.doi.org/10.1023/B:JOBB.0000023621.72873.9e
- T. Rostovtseva, A. Komarov, S. Bezrukov, and M. Colombini, "VDAC Channels Differentiate between Natural Metabolites and Synthetic Molecules", Journal of Membrane Biology, vol. 187, pp. 147-156, 2002. http://dx.doi.org/10.1007/s00232-001-0159-1
- G. Yehezkel, N. Hadad, H. Zaid, S. Sivan, and V. Shoshan-Barmatz, "Nucleotide-binding Sites in the Voltage-dependent Anion Channel", Journal of Biological Chemistry, vol. 281, pp. 5938-5946, 2006. http://dx.doi.org/10.1074/jbc.M510104200
- V. Shoshan-Barmatz, and D. Gincel, "The Voltage-Dependent Anion Channel: Characterization, Modulation, and Role in Mitochondrial Function in Cell Life and Death", Cell Biochemistry and Biophysics, vol. 39, pp. 279-292, 2003. http://dx.doi.org/10.1385/CBB:39:3:279
- A. Bera, S. Ghosh, and S. Das, "Mitochondrial VDAC Can Be Phosphorylated by Cyclic AMP-Dependent Protein Kinase", Biochemical and Biophysical Research Communications, vol. 209, pp. 213-217, 1995. http://dx.doi.org/10.1006/bbrc.1995.1491
- C.P. Baines, C. Song, Y. Zheng, G. Wang, J. Zhang, O. Wang, Y. Guo, R. Bolli, E.M. Cardwell, and P. Ping, "Protein Kinase Cε Interacts With and Inhibits the Permeability Transition Pore in Cardiac Mitochondria", Circulation Research, vol. 92, pp. 873-880, 2003. http://dx.doi.org/10.1161/01.RES.0000069215.36389.8D
- S. Liberatori, B. Canas, C. Tani, L. Bini, G. Buonocore, J. Godovac‐Zimmermann, O.P. Mishra, M. Delivoria‐Papadopoulos, R. Bracci, and V. Pallini, "Proteomic approach to the identification of voltage‐dependent anion channel protein isoforms in guinea pig brain synaptosomes", PROTEOMICS, vol. 4, pp. 1335-1340, 2004. http://dx.doi.org/10.1002/pmic.200300734
- F. Palmieri, and C.L. Pierri, "Mitochondrial metabolite transport", Essays in Biochemistry, vol. 47, pp. 37-52, 2010. http://dx.doi.org/10.1042/bse0470037
- V. Shoshan-Barmatz, and D. Ben-Hail, "VDAC, a multi-functional mitochondrial protein as a pharmacological target", Mitochondrion, vol. 12, pp. 24-34, 2012. http://dx.doi.org/10.1016/j.mito.2011.04.001
- L. Aram, S. Geula, N. Arbel, and V. Shoshan-Barmatz, "VDAC1 cysteine residues: topology and function in channel activity and apoptosis", Biochemical Journal, vol. 427, pp. 445-454, 2010. http://dx.doi.org/10.1042/BJ20091690
- V. Shoshan-Barmatz, D. Ben-Hail, L. Admoni, Y. Krelin, and S.S. Tripathi, "The mitochondrial voltage-dependent anion channel 1 in tumor cells", Biochimica et Biophysica Acta (BBA) - Biomembranes, vol. 1848, pp. 2547-2575, 2015. http://dx.doi.org/10.1016/j.bbamem.2014.10.040
- M.G. Vander Heiden, N.S. Chandel, X.X. Li, P.T. Schumacker, M. Colombini, and C.B. Thompson, "Outer mitochondrial membrane permeability can regulate coupled respiration and cell survival", Proceedings of the National Academy of Sciences, vol. 97, pp. 4666-4671, 2000. http://dx.doi.org/10.1073/pnas.090082297
- M. Dolder, S. Wendt, and T. Wallimann, "Mitochondrial creatine kinase in contact sites: interaction with porin and adenine nucleotide translocase, role in permeability transition and sensitivity to oxidative damage.", Biological signals and receptors, 2001. http://www.ncbi.nlm.nih.gov/pubmed/11223643
- P.A. Gurnev, T.K. Rostovtseva, and S.M. Bezrukov, "Tubulin-blocked state of VDAC studied by polymer and ATP partitioning", FEBS Letters, vol. 585, pp. 2363-2366, 2011. http://dx.doi.org/10.1016/j.febslet.2011.06.008
- S. Abu-Hamad, S. Sivan, and V. Shoshan-Barmatz, "The expression level of the voltage-dependent anion channel controls life and death of the cell", Proceedings of the National Academy of Sciences, vol. 103, pp. 5787-5792, 2006. http://dx.doi.org/10.1073/pnas.0600103103
- T. Arif, L. Vasilkovsky, Y. Refaely, A. Konson, and V. Shoshan-Barmatz, "Silencing VDAC1 Expression by siRNA Inhibits Cancer Cell Proliferation and Tumor Growth In Vivo", Molecular Therapy - Nucleic Acids, vol. 3, pp. e159, 2014. http://dx.doi.org/10.1038/mtna.2014.9
- M.B. Rone, J. Fan, and V. Papadopoulos, "Cholesterol transport in steroid biosynthesis: Role of protein–protein interactions and implications in disease states", Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids, vol. 1791, pp. 646-658, 2009. http://dx.doi.org/10.1016/j.bbalip.2009.03.001
- Y. Aghazadeh, M.B. Rone, J. Blonder, X. Ye, T.D. Veenstra, D.B. Hales, M. Culty, and V. Papadopoulos, "Hormone-induced 14-3-3γ Adaptor Protein Regulates Steroidogenic Acute Regulatory Protein Activity and Steroid Biosynthesis in MA-10 Leydig Cells", Journal of Biological Chemistry, vol. 287, pp. 15380-15394, 2012. http://dx.doi.org/10.1074/jbc.M112.339580
- W. Yu, J. Gong, M. Ko, W.S. Garver, K. Yanagisawa, and M. Michikawa, "Altered Cholesterol Metabolism in Niemann-Pick Type C1 Mouse Brains Affects Mitochondrial Function", Journal of Biological Chemistry, vol. 280, pp. 11731-11739, 2005. http://dx.doi.org/10.1074/jbc.M412898200
- J.G. Pastorino, and J.B. Hoek, "Regulation of hexokinase binding to VDAC", Journal of Bioenergetics and Biomembranes, vol. 40, pp. 171-182, 2008. http://dx.doi.org/10.1007/s10863-008-9148-8
- A.M. Campbell, and S.H.P. Chan, "Mitochondrial membrane cholesterol, the voltage dependent anion channel (VDAC), and the Warburg effect", Journal of Bioenergetics and Biomembranes, vol. 40, pp. 193-197, 2008. http://dx.doi.org/10.1007/s10863-008-9138-x
- K. Lee, J. Kerner, and C.L. Hoppel, "Mitochondrial Carnitine Palmitoyltransferase 1a (CPT1a) Is Part of an Outer Membrane Fatty Acid Transfer Complex", Journal of Biological Chemistry, vol. 286, pp. 25655-25662, 2011. http://dx.doi.org/10.1074/jbc.M111.228692
- C. Martel, M. Allouche, D.D. Esposti, E. Fanelli, C. Boursier, C. Henry, J. Chopineau, G. Calamita, G. Kroemer, A. Lemoine, and C. Brenner, "Glycogen synthase kinase 3-mediated voltage-dependent anion channel phosphorylation controls outer mitochondrial membrane permeability during lipid accumulation", Hepatology, vol. 57, pp. 93-102, 2013. http://dx.doi.org/10.1002/hep.25967
- J. Qiu, Y. Tan, A.M. Hagenston, M. Martel, N. Kneisel, P.A. Skehel, D.J.A. Wyllie, H. Bading, and G.E. Hardingham, "Mitochondrial calcium uniporter Mcu controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals", Nature Communications, vol. 4, 2013. http://dx.doi.org/10.1038/ncomms3034
- J.B. Wenger, S.Y. Chun, D.T. Dang, H. Luesch, and L.H. Dang, "Combination therapy targeting cancer metabolism", Medical Hypotheses, vol. 76, pp. 169-172, 2011. http://dx.doi.org/10.1016/j.mehy.2010.09.008
- R.A. Cairns, I.S. Harris, and T.W. Mak, "Regulation of cancer cell metabolism", Nature Reviews Cancer, vol. 11, pp. 85-95, 2011. http://dx.doi.org/10.1038/nrc2981
- E.N. Maldonado, J. Patnaik, M.R. Mullins, and J.J. Lemasters, "Free Tubulin Modulates Mitochondrial Membrane Potential in Cancer Cells", Cancer Research, vol. 70, pp. 10192-10201, 2010. http://dx.doi.org/10.1158/0008-5472.CAN-10-2429
- S.P. Mathupala, Y.H. Ko, and P.L. Pedersen, "The pivotal roles of mitochondria in cancer: Warburg and beyond and encouraging prospects for effective therapies", Biochimica et Biophysica Acta (BBA) - Bioenergetics, vol. 1797, pp. 1225-1230, 2010. http://dx.doi.org/10.1016/j.bbabio.2010.03.025
- R.A. Nakashima, M.G. Paggi, and P.L. Pedersen, "Contributions of glycolysis and oxidative phosphorylation to adenosine 5'-triphosphate production in AS-30D hepatoma cells.", Cancer research, 1984. http://www.ncbi.nlm.nih.gov/pubmed/6498833
- J. Singleterry, A. Sreedhar, and Y. Zhao, "Components of cancer metabolism and therapeutic interventions", Mitochondrion, vol. 17, pp. 50-55, 2014. http://dx.doi.org/10.1016/j.mito.2014.05.010
- T.N. Seyfried, and L.M. Shelton, "Cancer as a metabolic disease", Nutrition & Metabolism, vol. 7, pp. 7, 2010. http://dx.doi.org/10.1186/1743-7075-7-7
- T.N. Seyfried, "Cancer as a mitochondrial metabolic disease", Frontiers in Cell and Developmental Biology, vol. 3, 2015. http://dx.doi.org/10.3389/fcell.2015.00043
- L.A. Loeb, "A mutator phenotype in cancer.", Cancer research, 2001. http://www.ncbi.nlm.nih.gov/pubmed/11309271
- R.A. Gatenby, and R.J. Gillies, "Why do cancers have high aerobic glycolysis?", Nature Reviews Cancer, vol. 4, pp. 891-899, 2004. http://dx.doi.org/10.1038/nrc1478
- D. Hanahan, and R. Weinberg, "Hallmarks of Cancer: The Next Generation", Cell, vol. 144, pp. 646-674, 2011. http://dx.doi.org/10.1016/j.cell.2011.02.013
- W.H. Koppenol, P.L. Bounds, and C.V. Dang, "Otto Warburg's contributions to current concepts of cancer metabolism", Nature Reviews Cancer, vol. 11, pp. 325-337, 2011. http://dx.doi.org/10.1038/nrc3038
- R. Majeed, and A. Hamid, "Therapeutic Targeting of Cancer Cell Metabolism: Role of Metabolic Enzymes, Oncogenes and Tumor Suppressor Genes", Journal of Cancer Science & Therapy, vol. 04, 2012. http://dx.doi.org/10.4172/1948-5956.1000156
- S.P. Mathupala, Y.H. Ko, and P.L. Pedersen, "Hexokinase II: Cancer's double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria", Oncogene, vol. 25, pp. 4777-4786, 2006. http://dx.doi.org/10.1038/sj.onc.1209603
- P.L. Pedersen, S. Mathupala, A. Rempel, J. Geschwind, and Y.H. Ko, "Mitochondrial bound type II hexokinase: a key player in the growth and survival of many cancers and an ideal prospect for therapeutic intervention", Biochimica et Biophysica Acta (BBA) - Bioenergetics, vol. 1555, pp. 14-20, 2002. http://dx.doi.org/10.1016/s0005-2728(02)00248-7
- N. Majewski, V. Nogueira, R.B. Robey, and N. Hay, "Akt Inhibits Apoptosis Downstream of BID Cleavage via a Glucose-Dependent Mechanism Involving Mitochondrial Hexokinases", Molecular and Cellular Biology, vol. 24, pp. 730-740, 2004. http://dx.doi.org/10.1128/MCB.24.2.730-740.2004
- J.G. Pastorino, J.B. Hoek, and N. Shulga, "Activation of Glycogen Synthase Kinase 3β Disrupts the Binding of Hexokinase II to Mitochondria by Phosphorylating Voltage-Dependent Anion Channel and Potentiates Chemotherapy-Induced Cytotoxicity", Cancer Research, vol. 65, pp. 10545-10554, 2005. http://dx.doi.org/10.1158/0008-5472.CAN-05-1925
- E.N. Maldonado, K.L. Sheldon, D.N. DeHart, J. Patnaik, Y. Manevich, D.M. Townsend, S.M. Bezrukov, T.K. Rostovtseva, and J.J. Lemasters, "Voltage-dependent Anion Channels Modulate Mitochondrial Metabolism in Cancer Cells", Journal of Biological Chemistry, vol. 288, pp. 11920-11929, 2013. http://dx.doi.org/10.1074/jbc.M112.433847
- T.K. Rostovtseva, K.L. Sheldon, E. Hassanzadeh, C. Monge, V. Saks, S.M. Bezrukov, and D.L. Sackett, "Tubulin binding blocks mitochondrial voltage-dependent anion channel and regulates respiration", Proceedings of the National Academy of Sciences, vol. 105, pp. 18746-18751, 2008. http://dx.doi.org/10.1073/pnas.0806303105
- N. Timohhina, R. Guzun, K. Tepp, C. Monge, M. Varikmaa, H. Vija, P. Sikk, T. Kaambre, D. Sackett, and V. Saks, "Direct measurement of energy fluxes from mitochondria into cytoplasm in permeabilized cardiac cells in situ: some evidence for mitochondrial interactosome", Journal of Bioenergetics and Biomembranes, vol. 41, pp. 259-275, 2009. http://dx.doi.org/10.1007/s10863-009-9224-8
- E.N. Maldonado, "VDAC–Tubulin, an Anti-Warburg Pro-Oxidant Switch", Frontiers in Oncology, vol. 7, 2017. http://dx.doi.org/10.3389/fonc.2017.00004
- M. Colombini, "VDAC: The channel at the interface between mitochondria and the cytosol", Molecular and Cellular Biochemistry, vol. 256, pp. 107-115, 2004. http://dx.doi.org/10.1023/B:MCBI.0000009862.17396.8d
- I. Koren, Z. Raviv, and V. Shoshan-Barmatz, "Downregulation of voltage-dependent anion channel-1 expression by RNA interference prevents cancer cell growth in vivo", Cancer Biology & Therapy, vol. 9, pp. 1046-1052, 2010. http://dx.doi.org/10.4161/cbt.9.12.11879
- T. Arif, Y. Krelin, I. Nakdimon, D. Benharroch, A. Paul, D. Dadon-Klein, and V. Shoshan-Barmatz, "VDAC1 is a molecular target in glioblastoma, with its depletion leading to reprogrammed metabolism and reversed oncogenic properties", Neuro-Oncology, vol. 19, pp. 951-964, 2017. http://dx.doi.org/10.1093/neuonc/now297
- R. Bargaje, S. Gupta, A. Sarkeshik, R. Park, T. Xu, M. Sarkar, M. Halimani, S.S. Roy, J. Yates, and B. Pillai, "Identification of Novel Targets for miR-29a Using miRNA Proteomics", PLoS ONE, vol. 7, pp. e43243, 2012. http://dx.doi.org/10.1371/journal.pone.0043243
- Q. Li, L. Zhang, H. Wan, M. Liu, X. Li, and H. Tang, "CREB1-driven expression of miR-320a promotes mitophagy by down-regulating VDAC1 expression during serum starvation in cervical cancer cells", Oncotarget, vol. 6, pp. 34924-34940, 2015. http://dx.doi.org/10.18632/oncotarget.5318
- F. Wang, Y. Qiang, L. Zhu, Y. Jiang, Y. Wang, X. Shao, L. Yin, J. Chen, and Z. Chen, "MicroRNA-7 downregulates the oncogene VDAC1 to influence hepatocellular carcinoma proliferation and metastasis", Tumor Biology, vol. 37, pp. 10235-10246, 2016. http://dx.doi.org/10.1007/s13277-016-4836-1
- A.D. Chaudhuri, D.C. Choi, S. Kabaria, A. Tran, and E. Junn, "MicroRNA-7 Regulates the Function of Mitochondrial Permeability Transition Pore by Targeting VDAC1 Expression", Journal of Biological Chemistry, vol. 291, pp. 6483-6493, 2016. http://dx.doi.org/10.1074/jbc.M115.691352
- X. Li, H. Wang, B. Yao, W. Xu, J. Chen, and X. Zhou, "lncRNA H19/miR-675 axis regulates cardiomyocyte apoptosis by targeting VDAC1 in diabetic cardiomyopathy", Scientific Reports, vol. 6, 2016. http://dx.doi.org/10.1038/srep36340
- M. Manczak, and P.H. Reddy, "Abnormal interaction of VDAC1 with amyloid beta and phosphorylated tau causes mitochondrial dysfunction in Alzheimer's disease", Human Molecular Genetics, vol. 21, pp. 5131-5146, 2012. http://dx.doi.org/10.1093/hmg/dds360
- M. Cuadrado-Tejedor, M. Vilariño, F. Cabodevilla, J. Del Río, D. Frechilla, and A. Pérez-Mediavilla, "Enhanced expression of the voltage-dependent anion channel 1 (VDAC1) in Alzheimer's disease transgenic mice: an insight into the pathogenic effects of amyloid-β.", Journal of Alzheimer's disease : JAD, 2011. http://www.ncbi.nlm.nih.gov/pubmed/20930307
- E. Pérez-Gracia, B. Torrejón-Escribano, and I. Ferrer, "Dystrophic neurites of senile plaques in Alzheimer’s disease are deficient in cytochrome c oxidase", Acta Neuropathologica, vol. 116, pp. 261-268, 2008. http://dx.doi.org/10.1007/s00401-008-0370-6
- Z. Liao, D. Liu, L. Tang, D. Yin, S. Yin, S. Lai, J. Yao, and M. He, "Long-term oral resveratrol intake provides nutritional preconditioning against myocardial ischemia/reperfusion injury: Involvement of VDAC1 downregulation", Molecular Nutrition & Food Research, vol. 59, pp. 454-464, 2015. http://dx.doi.org/10.1002/mnfr.201400730
- M. Ahmed, S.J. Muhammed, B. Kessler, and A. Salehi, "Mitochondrial proteome analysis reveals altered expression of voltage dependent anion channels in pancreatic β-cells exposed to high glucose", Islets, vol. 2, pp. 283-292, 2010. http://dx.doi.org/10.4161/isl.2.5.12639
- D. Gong, X. Chen, M. Middleditch, L. Huang, G. Vazhoor Amarsingh, S. Reddy, J. Lu, S. Zhang, K. Ruggiero, A.R.J. Phillips, and G.J.S. Cooper, "Quantitative proteomic profiling identifies new renal targets of copper(II)‐selective chelation in the reversal of diabetic nephropathy in rats", PROTEOMICS, vol. 9, pp. 4309-4320, 2009. http://dx.doi.org/10.1002/pmic.200900285
- G. Kroemer, L. Galluzzi, and C. Brenner, "Mitochondrial Membrane Permeabilization in Cell Death", Physiological Reviews, vol. 87, pp. 99-163, 2007. http://dx.doi.org/10.1152/physrev.00013.2006
- V. Shoshan-Barmatz, N. Arbel, and L. Arzoine, " VDAC, the voltage-dependent anion channel: function, regulation & mitochondrial signaling in cell life and death.", Cell Science 4:74-118., 2008.
- J.J. Lemasters, and E. Holmuhamedov, "Voltage-dependent anion channel (VDAC) as mitochondrial governator—Thinking outside the box", Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease, vol. 1762, pp. 181-190, 2006. http://dx.doi.org/10.1016/j.bbadis.2005.10.006
- M.G. Vander Heiden, X.X. Li, E. Gottleib, R.B. Hill, C.B. Thompson, and M. Colombini, "Bcl-x Promotes the Open Configuration of the Voltage-dependent Anion Channel and Metabolite Passage through the Outer Mitochondrial Membrane", Journal of Biological Chemistry, vol. 276, pp. 19414-19419, 2001. http://dx.doi.org/10.1074/jbc.M101590200
- L. Biasutto, M. Azzolini, I. Szabò, and M. Zoratti, "The mitochondrial permeability transition pore in AD 2016: An update", Biochimica et Biophysica Acta (BBA) - Molecular Cell Research, vol. 1863, pp. 2515-2530, 2016. http://dx.doi.org/10.1016/j.bbamcr.2016.02.012
- A. Antignani, and R.J. Youle, "How do Bax and Bak lead to permeabilization of the outer mitochondrial membrane?", Current Opinion in Cell Biology, vol. 18, pp. 685-689, 2006. http://dx.doi.org/10.1016/j.ceb.2006.10.004
- J.F. Lovell, L.P. Billen, S. Bindner, A. Shamas-Din, C. Fradin, B. Leber, and D.W. Andrews, "Membrane Binding by tBid Initiates an Ordered Series of Events Culminating in Membrane Permeabilization by Bax", Cell, vol. 135, pp. 1074-1084, 2008. http://dx.doi.org/10.1016/j.cell.2008.11.010
- J. Banerjee, and S. Ghosh, "Bax increases the pore size of rat brain mitochondrial voltage-dependent anion channel in the presence of tBid", Biochemical and Biophysical Research Communications, vol. 323, pp. 310-314, 2004. http://dx.doi.org/10.1016/j.bbrc.2004.08.094
- S. Shimizu, and Y. Tsujimoto, "Proapoptotic BH3-only Bcl-2 family members induce cytochromecrelease, but not mitochondrial membrane potential loss, and do not directly modulate voltage-dependent anion channel activity", Proceedings of the National Academy of Sciences, vol. 97, pp. 577-582, 2000. http://dx.doi.org/10.1073/pnas.97.2.577
- N. Keinan, H. Pahima, D. Ben-Hail, and V. Shoshan-Barmatz, "The role of calcium in VDAC1 oligomerization and mitochondria-mediated apoptosis", Biochimica et Biophysica Acta (BBA) - Molecular Cell Research, vol. 1833, pp. 1745-1754, 2013. http://dx.doi.org/10.1016/j.bbamcr.2013.03.017
- S. Weisthal, N. Keinan, D. Ben-Hail, T. Arif, and V. Shoshan-Barmatz, "Ca2+-mediated regulation of VDAC1 expression levels is associated with cell death induction", Biochimica et Biophysica Acta (BBA) - Molecular Cell Research, vol. 1843, pp. 2270-2281, 2014. http://dx.doi.org/10.1016/j.bbamcr.2014.03.021
- J. Martinou, S. Desagher, and B. Antonsson, "Cytochrome c release from mitochondria: all or nothing", Nature Cell Biology, vol. 2, pp. E41-E43, 2000. http://dx.doi.org/10.1038/35004069
- A.P. Halestrap, G.P. McStay, and S.J. Clarke, "The permeability transition pore complex: another view", Biochimie, vol. 84, pp. 153-166, 2002. http://dx.doi.org/10.1016/s0300-9084(02)01375-5
- E. DORAN, and A.P. HALESTRAP, "Cytochrome c release from isolated rat liver mitochondria can occur independently of outer-membrane rupture: possible role of contact sites", Biochemical Journal, vol. 348, pp. 343-350, 2000. http://dx.doi.org/10.1042/bj3480343
- Y. Tsujimoto, and S. Shimizu, "The voltage-dependent anion channel: an essential player in apoptosis", Biochimie, vol. 84, pp. 187-193, 2002. http://dx.doi.org/10.1016/s0300-9084(02)01370-6
- S. Abu-Hamad, H. Zaid, A. Israelson, E. Nahon, and V. Shoshan-Barmatz, "Hexokinase-I Protection against Apoptotic Cell Death Is Mediated via Interaction with the Voltage-dependent Anion Channel-1", Journal of Biological Chemistry, vol. 283, pp. 13482-13490, 2008. http://dx.doi.org/10.1074/jbc.M708216200
- T. Ghosh, N. Pandey, A. Maitra, S.K. Brahmachari, and B. Pillai, "A Role for Voltage-Dependent Anion Channel Vdac1 in Polyglutamine-Mediated Neuronal Cell Death", PLoS ONE, vol. 2, pp. e1170, 2007. http://dx.doi.org/10.1371/journal.pone.0001170
- A. Godbole, J. Varghese, A. Sarin, and M. Mathew, "VDAC is a conserved element of death pathways in plant and animal systems", Biochimica et Biophysica Acta (BBA) - Molecular Cell Research, vol. 1642, pp. 87-96, 2003. http://dx.doi.org/10.1016/s0167-4889(03)00102-2
- A. Lü, C. Dong, C. Du, and Q. Zhang, "Characterization and expression analysis of Paralichthys olivaceus voltage-dependent anion channel (VDAC) gene in response to virus infection", Fish & Shellfish Immunology, vol. 23, pp. 601-613, 2007. http://dx.doi.org/10.1016/j.fsi.2007.01.007
- H. Zaid, S. Abu-Hamad, A. Israelson, I. Nathan, and V. Shoshan-Barmatz, "The voltage-dependent anion channel-1 modulates apoptotic cell death", Cell Death & Differentiation, vol. 12, pp. 751-760, 2005. http://dx.doi.org/10.1038/sj.cdd.4401599
- A. Israelson, H. Zaid, S. Abu-Hamad, E. Nahon, and V. Shoshan-Barmatz, "Mapping the ruthenium red-binding site of the voltage-dependent anion channel-1", Cell Calcium, vol. 43, pp. 196-204, 2008. http://dx.doi.org/10.1016/j.ceca.2007.05.006
- H. AZOULAY-ZOHAR, A. ISRAELSON, S. ABU-HAMAD, and V. SHOSHAN-BARMATZ, "In self-defence: hexokinase promotes voltage-dependent anion channel closure and prevents mitochondria-mediated apoptotic cell death", Biochemical Journal, vol. 377, pp. 347-355, 2004. http://dx.doi.org/10.1042/BJ20031465
- N. Tajeddine, L. Galluzzi, O. Kepp, E. Hangen, E. Morselli, L. Senovilla, N. Araujo, G. Pinna, N. Larochette, N. Zamzami, N. Modjtahedi, A. Harel-Bellan, and G. Kroemer, "Hierarchical involvement of Bak, VDAC1 and Bax in cisplatin-induced cell death", Oncogene, vol. 27, pp. 4221-4232, 2008. http://dx.doi.org/10.1038/onc.2008.63
- F. Tomasello, A. Messina, L. Lartigue, L. Schembri, C. Medina, S. Reina, D. Thoraval, M. Crouzet, F. Ichas, V. De Pinto, and F. De Giorgi, "Outer membrane VDAC1 controls permeability transition of the inner mitochondrial membrane in cellulo during stress-induced apoptosis", Cell Research, vol. 19, pp. 1363-1376, 2009. http://dx.doi.org/10.1038/cr.2009.98
- S. Yuan, Y. Fu, X. Wang, H. Shi, Y. Huang, X. Song, L. Li, N. Song, and Y. Luo, "Voltage‐dependent anion channel 1 is involved in endostatin‐induced endothelial cell apoptosis", The FASEB Journal, vol. 22, pp. 2809-2820, 2008. http://dx.doi.org/10.1096/fj.08-107417
- S. Shimizu, M. Narita, Y. Tsujimoto, and Y. Tsujimoto, "Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC", Nature, vol. 399, pp. 483-487, 1999. http://dx.doi.org/10.1038/20959
- M. Madesh, and G. Hajnóczky, "VDAC-dependent permeabilization of the outer mitochondrial membrane by superoxide induces rapid and massive cytochrome c release", The Journal of Cell Biology, vol. 155, pp. 1003-1016, 2001. http://dx.doi.org/10.1083/jcb.200105057
- V. Betaneli, E. Petrov, and P. Schwille, "The Role of Lipids in VDAC Oligomerization", Biophysical Journal, vol. 102, pp. 523-531, 2012. http://dx.doi.org/10.1016/j.bpj.2011.12.049
- A. Vaseva, N. Marchenko, K. Ji, S. Tsirka, S. Holzmann, and U. Moll, "p53 Opens the Mitochondrial Permeability Transition Pore to Trigger Necrosis", Cell, vol. 149, pp. 1536-1548, 2012. http://dx.doi.org/10.1016/j.cell.2012.05.014
- D. Ben-Hail, and V. Shoshan-Barmatz, "VDAC1-interacting anion transport inhibitors inhibit VDAC1 oligomerization and apoptosis", Biochimica et Biophysica Acta (BBA) - Molecular Cell Research, vol. 1863, pp. 1612-1623, 2016. http://dx.doi.org/10.1016/j.bbamcr.2016.04.002
- H. Chen, W. Gao, Y. Yang, S. Guo, H. Wang, W. Wang, S. Zhang, Q. Zhou, H. Xu, J. Yao, Z. Tian, B. Li, W. Cao, Z. Zhang, and Y. Tian, "Inhibition of VDAC1 prevents Ca2+-mediated oxidative stress and apoptosis induced by 5-aminolevulinic acid mediated sonodynamic therapy in THP-1 macrophages", Apoptosis, vol. 19, pp. 1712-1726, 2014. http://dx.doi.org/10.1007/s10495-014-1045-5
- D. Ben-Hail, R. Begas-Shvartz, M. Shalev, A. Shteinfer-Kuzmine, A. Gruzman, S. Reina, V. De Pinto, and V. Shoshan-Barmatz, "Novel Compounds Targeting the Mitochondrial Protein VDAC1 Inhibit Apoptosis and Protect against Mitochondrial Dysfunction", Journal of Biological Chemistry, vol. 291, pp. 24986-25003, 2016. http://dx.doi.org/10.1074/jbc.M116.744284
- M. Obulesu, and M.J. Lakshmi, "Apoptosis in Alzheimer’s Disease: An Understanding of the Physiology, Pathology and Therapeutic Avenues", Neurochemical Research, vol. 39, pp. 2301-2312, 2014. http://dx.doi.org/10.1007/s11064-014-1454-4
- F. X. Sureda, F. Junyent, E. Verdaguer, C. Auladell, C. Pelegri, J. Vilaplana, J. Folch, A. Maria Canudas, C. Beas Zarate, M. Pallas, and A. Camins, "Antiapoptotic Drugs: A Therapautic Strategy for the Prevention of Neurodegenerative Diseases", Current Pharmaceutical Design, vol. 17, pp. 230-245, 2011. http://dx.doi.org/10.2174/138161211795049732
- S. Kostin, L. Pool, A. Elsässer, S. Hein, H.C. Drexler, E. Arnon, Y. Hayakawa, R. Zimmermann, E. Bauer, W. Klövekorn, and J. Schaper, "Myocytes Die by Multiple Mechanisms in Failing Human Hearts", Circulation Research, vol. 92, pp. 715-724, 2003. http://dx.doi.org/10.1161/01.RES.0000067471.95890.5C
- G. Kroemer, W.S. El-Deiry, P. Golstein, M.E. Peter, D. Vaux, P. Vandenabeele, B. Zhivotovsky, M.V. Blagosklonny, W. Malorni, R.A. Knight, M. Piacentini, S. Nagata, and G. Melino, "Classification of cell death: recommendations of the Nomenclature Committee on Cell Death", Cell Death & Differentiation, vol. 12, pp. 1463-1467, 2005. http://dx.doi.org/10.1038/sj.cdd.4401724
- T. Marunouchi, and K. Tanonaka, "Cell Death in the Cardiac Myocyte", Biological & Pharmaceutical Bulletin, vol. 38, pp. 1094-1097, 2015. http://dx.doi.org/10.1248/bpb.b15-00288
- J. Nawarak, R. Huang-Liu, S. Kao, H. Liao, S. Sinchaikul, S. Chen, and S. Cheng, "Proteomics analysis of A375 human malignant melanoma cells in response to arbutin treatment", Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics, vol. 1794, pp. 159-167, 2009. http://dx.doi.org/10.1016/j.bbapap.2008.09.023
- N. Jiang, S.K.Y. Kham, G.S. Koh, J.Y. Suang Lim, H. Ariffin, F.T. Chew, and A.E.J. Yeoh, "Identification of prognostic protein biomarkers in childhood acute lymphoblastic leukemia (ALL)", Journal of Proteomics, vol. 74, pp. 843-857, 2011. http://dx.doi.org/10.1016/j.jprot.2011.02.034
- Z. Liu, S. Bengtsson, M. Krogh, M. Marquez, S. Nilsson, P. James, A. Aliaya, and A. Holmberg, "Somatostatin effects on the proteome of the LNCaP cell-line", International Journal of Oncology, 2007. http://dx.doi.org/10.3892/ijo.30.5.1173
- S.M. Moin, M. Panteva, and S. Jameel, "The Hepatitis E Virus Orf3 Protein Protects Cells from Mitochondrial Depolarization and Death", Journal of Biological Chemistry, vol. 282, pp. 21124-21133, 2007. http://dx.doi.org/10.1074/jbc.M701696200
- J.Y. Jung, C.R. Han, Y.J. Jeong, H.J. Kim, H.S. Lim, K.H. Lee, H.O. Park, W.M. Oh, S.H. Kim, and W.J. Kim, "Epigallocatechin gallate inhibits nitric oxide-induced apoptosis in rat PC12 cells", Neuroscience Letters, vol. 411, pp. 222-227, 2007. http://dx.doi.org/10.1016/j.neulet.2006.09.089
- D.W. Voehringer, D.L. Hirschberg, J. Xiao, Q. Lu, M. Roederer, C.B. Lock, L.A. Herzenberg, L. Steinman, and L.A. Herzenberg, "Gene microarray identification of redox and mitochondrial elements that control resistance or sensitivity to apoptosis", Proceedings of the National Academy of Sciences, vol. 97, pp. 2680-2685, 2000. http://dx.doi.org/10.1073/pnas.97.6.2680
- Y. Chen, W.J. Craigen, and D.J. Riley, "Nek1 regulates cell death and mitochondrial membrane permeability through phosphorylation of VDAC1", Cell Cycle, vol. 8, pp. 257-267, 2009. http://dx.doi.org/10.4161/cc.8.2.7551
- A. Castagna, P. Antonioli, H. Astner, M. Hamdan, S.C. Righetti, P. Perego, F. Zunino, and P.G. Righetti, "A proteomic approach to cisplatin resistance in the cervix squamous cell carcinoma cell line A431", PROTEOMICS, vol. 4, pp. 3246-3267, 2004. http://dx.doi.org/10.1002/pmic.200400835
- A. Leone, M.S. Roca, C. Ciardiello, M. Terranova-Barberio, C. Vitagliano, G. Ciliberto, R. Mancini, E. Di Gennaro, F. Bruzzese, and A. Budillon, "Vorinostat synergizes with EGFR inhibitors in NSCLC cells by increasing ROS via up-regulation of the major mitochondrial porin VDAC1 and modulation of the c-Myc-NRF2-KEAP1 pathway", Free Radical Biology and Medicine, vol. 89, pp. 287-299, 2015. http://dx.doi.org/10.1016/j.freeradbiomed.2015.07.155
- Y. Anis, " Involvement of Ca2+ in the apoptotic process – friends or foes.", Pathways 2:2-7., 2006.
- M.J. Berridge, M.D. Bootman, and H.L. Roderick, "Calcium signalling: dynamics, homeostasis and remodelling", Nature Reviews Molecular Cell Biology, vol. 4, pp. 517-529, 2003. http://dx.doi.org/10.1038/nrm1155
- R. Rizzuto, D. De Stefani, A. Raffaello, and C. Mammucari, "Mitochondria as sensors and regulators of calcium signalling", Nature Reviews Molecular Cell Biology, vol. 13, pp. 566-578, 2012. http://dx.doi.org/10.1038/nrm3412
- J.M. Baughman, F. Perocchi, H.S. Girgis, M. Plovanich, C.A. Belcher-Timme, Y. Sancak, X.R. Bao, L. Strittmatter, O. Goldberger, R.L. Bogorad, V. Koteliansky, and V.K. Mootha, "Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter", Nature, vol. 476, pp. 341-345, 2011. http://dx.doi.org/10.1038/nature10234
- D. De Stefani, A. Raffaello, E. Teardo, I. Szabò, and R. Rizzuto, "A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter", Nature, vol. 476, pp. 336-340, 2011. http://dx.doi.org/10.1038/nature10230
- R. Palty, W.F. Silverman, M. Hershfinkel, T. Caporale, S.L. Sensi, J. Parnis, C. Nolte, D. Fishman, V. Shoshan-Barmatz, S. Herrmann, D. Khananshvili, and I. Sekler, "NCLX is an essential component of mitochondrial Na + /Ca 2+ exchange", Proceedings of the National Academy of Sciences, vol. 107, pp. 436-441, 2009. http://dx.doi.org/10.1073/pnas.0908099107
- V. Shoshan-Barmatz, S. De, and A. Meir, "The Mitochondrial Voltage-Dependent Anion Channel 1, Ca2+ Transport, Apoptosis, and Their Regulation", Frontiers in Oncology, vol. 7, 2017. http://dx.doi.org/10.3389/fonc.2017.00060
- G. Báthori, G. Csordás, C. Garcia-Perez, E. Davies, and G. Hajnóczky, "Ca2+-dependent Control of the Permeability Properties of the Mitochondrial Outer Membrane and Voltage-dependent Anion-selective Channel (VDAC)", Journal of Biological Chemistry, vol. 281, pp. 17347-17358, 2006. http://dx.doi.org/10.1074/jbc.M600906200
- D. GINCEL, H. ZAID, and V. SHOSHAN-BARMATZ, "Calcium binding and translocation by the voltage-dependent anion channel: a possible regulatory mechanism in mitochondrial function", Biochemical Journal, vol. 358, pp. 147-155, 2001. http://dx.doi.org/10.1042/bj3580147
- E. Rapizzi, P. Pinton, G. Szabadkai, M.R. Wieckowski, G. Vandecasteele, G. Baird, R.A. Tuft, K.E. Fogarty, and R. Rizzuto, "Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria", The Journal of Cell Biology, vol. 159, pp. 613-624, 2002. http://dx.doi.org/10.1083/jcb.200205091
- W. Tan, and M. Colombini, "VDAC closure increases calcium ion flux", Biochimica et Biophysica Acta (BBA) - Biomembranes, vol. 1768, pp. 2510-2515, 2007. http://dx.doi.org/10.1016/j.bbamem.2007.06.002
- A. Israelson, S. Abu-Hamad, H. Zaid, E. Nahon, and V. Shoshan-Barmatz, "Localization of the voltage-dependent anion channel-1 Ca2+-binding sites", Cell Calcium, vol. 41, pp. 235-244, 2007. http://dx.doi.org/10.1016/j.ceca.2006.06.005
- D. Gincel, N. Vardi, and V. Shoshan-Barmatz, "Retinal voltage-dependent anion channel: characterization and cellular localization.", Investigative ophthalmology & visual science, 2002. http://www.ncbi.nlm.nih.gov/pubmed/12091402
- A. Israelson, L. Arzoine, S. Abu-hamad, V. Khodorkovsky, and V. Shoshan-Barmatz, "A Photoactivable Probe for Calcium Binding Proteins", Chemistry & Biology, vol. 12, pp. 1169-1178, 2005. http://dx.doi.org/10.1016/j.chembiol.2005.08.006
- G. Csordás, C. Renken, P. Várnai, L. Walter, D. Weaver, K.F. Buttle, T. Balla, C.A. Mannella, and G. Hajnóczky, "Structural and functional features and significance of the physical linkage between ER and mitochondria", The Journal of Cell Biology, vol. 174, pp. 915-921, 2006. http://dx.doi.org/10.1083/jcb.200604016
- O.M. de Brito, and L. Scorrano, "Mitofusin 2 tethers endoplasmic reticulum to mitochondria", Nature, vol. 456, pp. 605-610, 2008. http://dx.doi.org/10.1038/nature07534
- O.M. de Brito, and L. Scorrano, "Mitofusin-2 regulates mitochondrial and endoplasmic reticulum morphology and tethering: The role of Ras", Mitochondrion, vol. 9, pp. 222-226, 2009. http://dx.doi.org/10.1016/j.mito.2009.02.005
- T.M. Buttke, and P.A. Sandstrom, "Oxidative stress as a mediator of apoptosis", Immunology Today, vol. 15, pp. 7-10, 1994. http://dx.doi.org/10.1016/0167-5699(94)90018-3
- G. FISKUM, "Mitochondrial Participation in Ischemic and Traumatic Neural Cell Death", Journal of Neurotrauma, vol. 17, pp. 843-855, 2000. http://dx.doi.org/10.1089/neu.2000.17.843
- M. Patel, B.J. Day, J.D. Crapo, I. Fridovich, and J.O. McNamara, "Requirement for Superoxide in Excitotoxic Cell Death", Neuron, vol. 16, pp. 345-355, 1996. http://dx.doi.org/10.1016/S0896-6273(00)80052-5
- J. Yuan, and B.A. Yankner, "Apoptosis in the nervous system", Nature, vol. 407, pp. 802-809, 2000. http://dx.doi.org/10.1038/35037739
- R.L. Auten, and J.M. Davis, "Oxygen Toxicity and Reactive Oxygen Species: The Devil Is in the Details", Pediatric Research, vol. 66, pp. 121-127, 2009. http://dx.doi.org/10.1203/PDR.0b013e3181a9eafb
- Q. Chen, E.J. Vazquez, S. Moghaddas, C.L. Hoppel, and E.J. Lesnefsky, "Production of Reactive Oxygen Species by Mitochondria", Journal of Biological Chemistry, vol. 278, pp. 36027-36031, 2003. http://dx.doi.org/10.1074/jbc.M304854200
- C.L. Quinlan, A.L. Orr, I.V. Perevoshchikova, J.R. Treberg, B.A. Ackrell, and M.D. Brand, "Mitochondrial Complex II Can Generate Reactive Oxygen Species at High Rates in Both the Forward and Reverse Reactions", Journal of Biological Chemistry, vol. 287, pp. 27255-27264, 2012. http://dx.doi.org/10.1074/jbc.M112.374629
- D.L. Tribble, D.P. Jones, and D.E. Edmondson, "Effect of hypoxia on tert-butylhydroperoxide-induced oxidative injury in hepatocytes.", Molecular pharmacology, 1988. http://www.ncbi.nlm.nih.gov/pubmed/3419429
- I. Fridovich, "Superoxide Anion Radical (O·̄2), Superoxide Dismutases, and Related Matters", Journal of Biological Chemistry, vol. 272, pp. 18515-18517, 1997. http://dx.doi.org/10.1074/jbc.272.30.18515
- E.A. Veal, A.M. Day, and B.A. Morgan, "Hydrogen Peroxide Sensing and Signaling", Molecular Cell, vol. 26, pp. 1-14, 2007. http://dx.doi.org/10.1016/j.molcel.2007.03.016
- J. Clerkin, R. Naughton, C. Quiney, and T. Cotter, "Mechanisms of ROS modulated cell survival during carcinogenesis", Cancer Letters, vol. 266, pp. 30-36, 2008. http://dx.doi.org/10.1016/j.canlet.2008.02.029
- G. Giles, "The Redox Regulation of Thiol Dependent Signaling Pathways in Cancer", Current Pharmaceutical Design, vol. 12, pp. 4427-4443, 2006. http://dx.doi.org/10.2174/138161206779010549
- M. Ushio-Fukai, and Y. Nakamura, "Reactive oxygen species and angiogenesis: NADPH oxidase as target for cancer therapy", Cancer Letters, vol. 266, pp. 37-52, 2008. http://dx.doi.org/10.1016/j.canlet.2008.02.044
- L.C. Schenkel, and M. Bakovic, "Formation and Regulation of Mitochondrial Membranes", International Journal of Cell Biology, vol. 2014, pp. 1-13, 2014. http://dx.doi.org/10.1155/2014/709828
- Y. Zhang, O. Marcillat, C. Giulivi, L. Ernster, and K.J. Davies, "The oxidative inactivation of mitochondrial electron transport chain components and ATPase.", The Journal of biological chemistry, 1990. http://www.ncbi.nlm.nih.gov/pubmed/2168888
- H. Kamata, S. Honda, S. Maeda, L. Chang, H. Hirata, and M. Karin, "Reactive Oxygen Species Promote TNFα-Induced Death and Sustained JNK Activation by Inhibiting MAP Kinase Phosphatases", Cell, vol. 120, pp. 649-661, 2005. http://dx.doi.org/10.1016/j.cell.2004.12.041
- Y. Son, Y. Cheong, N. Kim, H. Chung, D.G. Kang, and H. Pae, "Mitogen-Activated Protein Kinases and Reactive Oxygen Species: How Can ROS Activate MAPK Pathways?", Journal of Signal Transduction, vol. 2011, pp. 1-6, 2011. http://dx.doi.org/10.1155/2011/792639
- K.B. Beckman, and B.N. Ames, "Oxidative decay of DNA.", The Journal of biological chemistry, 1997. http://www.ncbi.nlm.nih.gov/pubmed/9289489
- D.E. Handy, and J. Loscalzo, "Redox Regulation of Mitochondrial Function", Antioxidants & Redox Signaling, vol. 16, pp. 1323-1367, 2012. http://dx.doi.org/10.1089/ars.2011.4123
- R.J. Mailloux, S.L. McBride, and M. Harper, "Unearthing the secrets of mitochondrial ROS and glutathione in bioenergetics", Trends in Biochemical Sciences, vol. 38, pp. 592-602, 2013. http://dx.doi.org/10.1016/j.tibs.2013.09.001
- L. Sena, and N. Chandel, "Physiological Roles of Mitochondrial Reactive Oxygen Species", Molecular Cell, vol. 48, pp. 158-167, 2012. http://dx.doi.org/10.1016/j.molcel.2012.09.025
- D. Trachootham, W. Lu, M.A. Ogasawara, N.R. Valle, and P. Huang, "Redox Regulation of Cell Survival", Antioxidants & Redox Signaling, vol. 10, pp. 1343-1374, 2008. http://dx.doi.org/10.1089/ars.2007.1957
- S.C. Barber, R.J. Mead, and P.J. Shaw, "Oxidative stress in ALS: A mechanism of neurodegeneration and a therapeutic target", Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease, vol. 1762, pp. 1051-1067, 2006. http://dx.doi.org/10.1016/j.bbadis.2006.03.008
- M. Ott, V. Gogvadze, S. Orrenius, and B. Zhivotovsky, "Mitochondria, oxidative stress and cell death", Apoptosis, vol. 12, pp. 913-922, 2007. http://dx.doi.org/10.1007/s10495-007-0756-2
- A. Ahmad, S. Ahmad, B.K. Schneider, C.B. Allen, L. Chang, and C.W. White, "Elevated expression of hexokinase II protects human lung epithelial-like A549 cells against oxidative injury", American Journal of Physiology-Lung Cellular and Molecular Physiology, vol. 283, pp. L573-L584, 2002. http://dx.doi.org/10.1152/ajplung.00410.2001
- W.S. da-Silva, A. Gómez-Puyou, M.T. de Gómez-Puyou, R. Moreno-Sanchez, F.G. De Felice, L. de Meis, M.F. Oliveira, and A. Galina, "Mitochondrial Bound Hexokinase Activity as a Preventive Antioxidant Defense", Journal of Biological Chemistry, vol. 279, pp. 39846-39855, 2004. http://dx.doi.org/10.1074/jbc.M403835200
- L. Sun, S. Shukair, T.J. Naik, F. Moazed, and H. Ardehali, "Glucose Phosphorylation and Mitochondrial Binding Are Required for the Protective Effects of Hexokinases I and II", Molecular and Cellular Biology, vol. 28, pp. 1007-1017, 2008. http://dx.doi.org/10.1128/MCB.00224-07
- J.M. Bryson, P.E. Coy, K. Gottlob, N. Hay, and R.B. Robey, "Increased Hexokinase Activity, of Either Ectopic or Endogenous Origin, Protects Renal Epithelial Cells against Acute Oxidant-induced Cell Death", Journal of Biological Chemistry, vol. 277, pp. 11392-11400, 2002. http://dx.doi.org/10.1074/jbc.M110927200
- A. Tikunov, C.B. Johnson, P. Pediaditakis, N. Markevich, J.M. Macdonald, J.J. Lemasters, and E. Holmuhamedov, "Closure of VDAC causes oxidative stress and accelerates the Ca2+-induced mitochondrial permeability transition in rat liver mitochondria", Archives of Biochemistry and Biophysics, vol. 495, pp. 174-181, 2010. http://dx.doi.org/10.1016/j.abb.2010.01.008
- D. Han, F. Antunes, R. Canali, D. Rettori, and E. Cadenas, "Voltage-dependent Anion Channels Control the Release of the Superoxide Anion from Mitochondria to Cytosol", Journal of Biological Chemistry, vol. 278, pp. 5557-5563, 2003. http://dx.doi.org/10.1074/jbc.M210269200
- U. Saeed, L. Durgadoss, R.K. Valli, D.C. Joshi, P.G. Joshi, and V. Ravindranath, "Knockdown of Cytosolic Glutaredoxin 1 Leads to Loss of Mitochondrial Membrane Potential: Implication in Neurodegenerative Diseases", PLoS ONE, vol. 3, pp. e2459, 2008. http://dx.doi.org/10.1371/journal.pone.0002459
- S. Reina, V. Checchetto, R. Saletti, A. Gupta, D. Chaturvedi, C. Guardiani, F. Guarino, M.A. Scorciapino, A. Magrì, S. Foti, M. Ceccarelli, A.A. Messina, R. Mahalakshmi, I. Szabo, and V. De Pinto, "VDAC3 as a sensor of oxidative state of the intermembrane space of mitochondria: the putative role of cysteine residue modifications", Oncotarget, vol. 7, pp. 2249-2268, 2016. http://dx.doi.org/10.18632/oncotarget.6850
- W. Ding, L.G. Hudson, and K.J. Liu, "Inorganic arsenic compounds cause oxidative damage to DNA and protein by inducing ROS and RNS generation in human keratinocytes", Molecular and Cellular Biochemistry, vol. 279, pp. 105-112, 2005. http://dx.doi.org/10.1007/s11010-005-8227-y
- H. Shi, L.G. Hudson, W. Ding, S. Wang, K.L. Cooper, S. Liu, Y. Chen, X. Shi, and K.J. Liu, "Arsenite Causes DNA Damage in Keratinocytes Via Generation of Hydroxyl Radicals", Chemical Research in Toxicology, vol. 17, pp. 871-878, 2004. http://dx.doi.org/10.1021/tx049939e
- R.D. Olson, R.C. Boerth, J.G. Gerber, and A.S. Nies, "Mechanism of adriamycin cardiotoxicity: Evidence for oxidative stress", Life Sciences, vol. 29, pp. 1393-1401, 1981. http://dx.doi.org/10.1016/0024-3205(81)90001-1
- E. Simamura, K. Hirai, H. Shimada, J. Koyama, Y. Niwa, and S. Shimizu, "Furanonaphthoquinones cause apoptosis of cancer cells by inducing the production of reactive oxygen species by the mitochondrial voltage-dependent anion channel", Cancer Biology & Therapy, vol. 5, pp. 1523-1529, 2006. http://dx.doi.org/10.4161/cbt.5.11.3302
- M. Le Bras, M. Clément, S. Pervaiz, and C. Brenner, "Reactive oxygen species and the mitochondrial signaling pathway of cell death.", Histology and histopathology, 2005. http://www.ncbi.nlm.nih.gov/pubmed/15578439
- Q. Li, E.F. Sato, X. Zhu, and M. Inoue, "A simultaneous release of SOD1 with cytochrome c regulates mitochondria-dependent apoptosis", Molecular and Cellular Biochemistry, vol. 322, pp. 151-159, 2008. http://dx.doi.org/10.1007/s11010-008-9952-9
- M.C. Brahimi-Horn, S. Lacas-Gervais, R. Adaixo, K. Ilc, M. Rouleau, A. Notte, M. Dieu, C. Michiels, T. Voeltzel, V. Maguer-Satta, J. Pelletier, M. Ilie, P. Hofman, B. Manoury, A. Schmidt, S. Hiller, J. Pouysségur, and N.M. Mazure, "Local Mitochondrial-Endolysosomal Microfusion Cleaves Voltage-Dependent Anion Channel 1 To Promote Survival in Hypoxia", Molecular and Cellular Biology, vol. 35, pp. 1491-1505, 2015. http://dx.doi.org/10.1128/MCB.01402-14
- N.M. Mazure, "News about VDAC1 in Hypoxia", Frontiers in Oncology, vol. 6, 2016. http://dx.doi.org/10.3389/fonc.2016.00193
- M.C. Brahimi-Horn, D. Ben-Hail, M. Ilie, P. Gounon, M. Rouleau, V. Hofman, J. Doyen, B. Mari, V. Shoshan-Barmatz, P. Hofman, J. Pouysségur, and N.M. Mazure, "Expression of a Truncated Active Form of VDAC1 in Lung Cancer Associates with Hypoxic Cell Survival and Correlates with Progression to Chemotherapy Resistance", Cancer Research, vol. 72, pp. 2140-2150, 2012. http://dx.doi.org/10.1158/0008-5472.CAN-11-3940
- J. Ko, W. Gu, I. Lim, T. Zhou, and H. Bang, "Expression Profiling of Mitochondrial Voltage-Dependent Anion Channel-1 Associated Genes Predicts Recurrence-Free Survival in Human Carcinomas", PLoS ONE, vol. 9, pp. e110094, 2014. http://dx.doi.org/10.1371/journal.pone.0110094
- V. De Pinto, A. Messina, D.J. Lane, and A. Lawen, "Voltage‐dependent anion‐selective channel (VDAC) in the plasma membrane", FEBS Letters, vol. 584, pp. 1793-1799, 2010. http://dx.doi.org/10.1016/j.febslet.2010.02.049
- B. Manna, T. Bhattacharya, B. Kahali, and T.C. Ghosh, "Evolutionary constraints on hub and non-hub proteins in human protein interaction network: Insight from protein connectivity and intrinsic disorder", Gene, vol. 434, pp. 50-55, 2009. http://dx.doi.org/10.1016/j.gene.2008.12.013
- C. Grills, P.V. Jithesh, J. Blayney, S. Zhang, and D.A. Fennell, "Gene Expression Meta-Analysis Identifies VDAC1 as a Predictor of Poor Outcome in Early Stage Non-Small Cell Lung Cancer", PLoS ONE, vol. 6, pp. e14635, 2011. http://dx.doi.org/10.1371/journal.pone.0014635
- H. Yu, P.M. Kim, E. Sprecher, V. Trifonov, and M. Gerstein, "The Importance of Bottlenecks in Protein Networks: Correlation with Gene Essentiality and Expression Dynamics", PLoS Computational Biology, vol. 3, pp. e59, 2007. http://dx.doi.org/10.1371/journal.pcbi.0030059
- T. Prezma, A. Shteinfer, L. Admoni, Z. Raviv, I. Sela, I. Levi, and V. Shoshan-Barmatz, "VDAC1-based peptides: novel pro-apoptotic agents and potential therapeutics for B-cell chronic lymphocytic leukemia", Cell Death & Disease, vol. 4, pp. e809-e809, 2013. http://dx.doi.org/10.1038/cddis.2013.316
- A. Shteinfer-Kuzmine, T. Arif, Y. Krelin, S.S. Tripathi, A. Paul, and V. Shoshan-Barmatz, "Mitochondrial VDAC1-based peptides: Attacking oncogenic properties in glioblastoma", Oncotarget, vol. 8, pp. 31329-31346, 2017. http://dx.doi.org/10.18632/oncotarget.15455
- R.L. Patterson, D.B. van Rossum, A.I. Kaplin, R.K. Barrow, and S.H. Snyder, "Inositol 1,4,5-trisphosphate receptor/GAPDH complex augments Ca 2+ release via locally derived NADH", Proceedings of the National Academy of Sciences, vol. 102, pp. 1357-1359, 2005. http://dx.doi.org/10.1073/pnas.0409657102
- M. Vyssokikh, L. Zorova, D. Zorov, G. Heimlich, J. Jürgensmeier, D. Schreiner, and D. Brdiczka, "The intra-mitochondrial cytochrome c distribution varies correlated to the formation of a complex between VDAC and the adenine nucleotide translocase: this affects Bax-dependent cytochrome c release", Biochimica et Biophysica Acta (BBA) - Molecular Cell Research, vol. 1644, pp. 27-36, 2004. http://dx.doi.org/10.1016/j.bbamcr.2003.10.007
- D. Neumann, J. Bückers, L. Kastrup, S.W. Hell, and S. Jakobs, "Two-color STED microscopy reveals different degrees of colocalization between hexokinase-I and the three human VDAC isoforms", PMC Biophysics, vol. 3, 2010. http://dx.doi.org/10.1186/1757-5036-3-4
- T. Azarashvili, O. Krestinina, Y. Baburina, I. Odinokova, D. Grachev, V. Papadopoulos, V. Akatov, J. Lemasters, and G. Reiser, "Combined effect of G3139 and TSPO ligands on Ca2+-induced permeability transition in rat brain mitochondria", Archives of Biochemistry and Biophysics, vol. 587, pp. 70-77, 2015. http://dx.doi.org/10.1016/j.abb.2015.10.012
- R.J. Youle, and A. Strasser, "The BCL-2 protein family: opposing activities that mediate cell death", Nature Reviews Molecular Cell Biology, vol. 9, pp. 47-59, 2008. http://dx.doi.org/10.1038/nrm2308
- N.N. Danial, "BCL-2 Family Proteins: Critical Checkpoints of Apoptotic Cell Death", Clinical Cancer Research, vol. 13, pp. 7254-7263, 2007. http://dx.doi.org/10.1158/1078-0432.ccr-07-1598
- T.J. Malia, and G. Wagner, "NMR Structural Investigation of the Mitochondrial Outer Membrane Protein VDAC and Its Interaction with Antiapoptotic Bcl-xL", Biochemistry, vol. 46, pp. 514-525, 2006. http://dx.doi.org/10.1021/bi061577h
- S. Shimizu, A. Konishi, T. Kodama, and Y. Tsujimoto, "BH4 domain of antiapoptotic Bcl-2 family members closes voltage-dependent anion channel and inhibits apoptotic mitochondrial changes and cell death", Proceedings of the National Academy of Sciences, vol. 97, pp. 3100-3105, 2000. http://dx.doi.org/10.1073/pnas.97.7.3100
- T. Sugiyama, S. Shimizu, Y. Matsuoka, Y. Yoneda, and Y. Tsujimoto, "Activation of mitochondrial voltage-dependent anion channel by apro-apoptotic BH3-only protein Bim", Oncogene, vol. 21, pp. 4944-4956, 2002. http://dx.doi.org/10.1038/sj.onc.1205621
- H. Yamagata, S. Shimizu, Y. Nishida, Y. Watanabe, W.J. Craigen, and Y. Tsujimoto, "Requirement of voltage-dependent anion channel 2 for pro-apoptotic activity of Bax", Oncogene, vol. 28, pp. 3563-3572, 2009. http://dx.doi.org/10.1038/onc.2009.213
- H. Huang, K. Shah, N.A. Bradbury, C. Li, and C. White, "Mcl-1 promotes lung cancer cell migration by directly interacting with VDAC to increase mitochondrial Ca2+ uptake and reactive oxygen species generation", Cell Death & Disease, vol. 5, pp. e1482-e1482, 2014. http://dx.doi.org/10.1038/cddis.2014.419
- . Leo Veenman, . Vassilios Papadopoulos, and . Moshe Gavish, "Channel-Like Functions of the 18-kDa Translocator Protein (TSPO): Regulation of Apoptosis and Steroidogenesis as Part of the Host-Defense Response", Current Pharmaceutical Design, vol. 13, pp. 2385-2405, 2007. http://dx.doi.org/10.2174/138161207781368710
- L. Veenman, Y. Shandalov, and M. Gavish, "VDAC activation by the 18 kDa translocator protein (TSPO), implications for apoptosis", Journal of Bioenergetics and Biomembranes, vol. 40, pp. 199-205, 2008. http://dx.doi.org/10.1007/s10863-008-9142-1
- Y. Chen, M. Gaczynska, P. Osmulski, R. Polci, and D.J. Riley, "Phosphorylation by Nek1 regulates opening and closing of voltage dependent anion channel 1", Biochemical and Biophysical Research Communications, vol. 394, pp. 798-803, 2010. http://dx.doi.org/10.1016/j.bbrc.2010.03.077
- X. Zhang, X. Bian, and J. Kong, "The Proapoptotic Protein BNIP3 Interacts with VDAC to Induce Mitochondrial Release of Endonuclease G", PLoS ONE, vol. 9, pp. e113642, 2014. http://dx.doi.org/10.1371/journal.pone.0113642
- H. Kusano, S. Shimizu, R.C. Koya, H. Fujita, S. Kamada, N. Kuzumaki, and Y. Tsujimoto, "Human gelsolin prevents apoptosis by inhibiting apoptotic mitochondrial changes via closing VDAC", Oncogene, vol. 19, pp. 4807-4814, 2000. http://dx.doi.org/10.1038/sj.onc.1203868
- H. Qiao, and J.R. McMillan, "Gelsolin segment 5 inhibits HIV‐induced T‐cell apoptosis via Vpr‐binding to VDAC", FEBS Letters, vol. 581, pp. 535-540, 2007. http://dx.doi.org/10.1016/j.febslet.2006.12.057
- M. Carreá, N. Andreá, G. Carles, H. Borghi, L. Brichese, C. Briand, and D. Braguer, "Tubulin Is an Inherent Component of Mitochondrial Membranes That Interacts with the Voltage-dependent Anion Channel", Journal of Biological Chemistry, vol. 277, pp. 33664-33669, 2002. http://dx.doi.org/10.1074/jbc.M203834200
- T.K. Rostovtseva, and S.M. Bezrukov, "VDAC inhibition by tubulin and its physiological implications", Biochimica et Biophysica Acta (BBA) - Biomembranes, vol. 1818, pp. 1526-1535, 2012. http://dx.doi.org/10.1016/j.bbamem.2011.11.004
- V. Saks, R. Guzun, N. Timohhina, K. Tepp, M. Varikmaa, C. Monge, N. Beraud, T. Kaambre, A. Kuznetsov, L. Kadaja, M. Eimre, and E. Seppet, "Structure–function relationships in feedback regulation of energy fluxes in vivo in health and disease: Mitochondrial Interactosome", Biochimica et Biophysica Acta (BBA) - Bioenergetics, vol. 1797, pp. 678-697, 2010. http://dx.doi.org/10.1016/j.bbabio.2010.01.011
- I. Roman, J. Figys, G. Steurs, and M. Zizi, "Direct measurement of VDAC–actin interaction by surface plasmon resonance", Biochimica et Biophysica Acta (BBA) - Biomembranes, vol. 1758, pp. 479-486, 2006. http://dx.doi.org/10.1016/j.bbamem.2006.03.019
- X. Xu, J. Forbes, and M. Colombini, "Actin Modulates the Gating of Neurospora crassa VDAC", Journal of Membrane Biology, vol. 180, pp. 73-81, 2001. http://dx.doi.org/10.1007/s002320010060
- M. Lindén, and G. Karlsson, "Identification of Porin as a Binding Site for MAP2", Biochemical and Biophysical Research Communications, vol. 218, pp. 833-836, 1996. http://dx.doi.org/10.1006/bbrc.1996.0148
- C. Schwarzer, S. Barnikol-Watanabe, F.P. Thinnes, and N. Hilschmann, "Voltage-dependent anion-selective channel (VDAC) interacts with the dynein light chain Tctex1 and the heat-shock protein PBP74", The International Journal of Biochemistry & Cell Biology, vol. 34, pp. 1059-1070, 2002. http://dx.doi.org/10.1016/s1357-2725(02)00026-2
- D. Bergemalm, P.A. Jonsson, K.S. Graffmo, P.M. Andersen, T. Brännström, A. Rehnmark, and S.L. Marklund, "Overloading of Stable and Exclusion of Unstable Human Superoxide Dismutase-1 Variants in Mitochondria of Murine Amyotrophic Lateral Sclerosis Models", The Journal of Neuroscience, vol. 26, pp. 4147-4154, 2006. http://dx.doi.org/10.1523/JNEUROSCI.5461-05.2006
- H. Deng, Y. Shi, Y. Furukawa, H. Zhai, R. Fu, E. Liu, G.H. Gorrie, M.S. Khan, W. Hung, E.H. Bigio, T. Lukas, M.C. Dal Canto, T.V. O'Halloran, and T. Siddique, "Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria", Proceedings of the National Academy of Sciences, vol. 103, pp. 7142-7147, 2006. http://dx.doi.org/10.1073/pnas.0602046103
- J. Liu, C. Lillo, P. Jonsson, C.V. Velde, C.M. Ward, T.M. Miller, J.R. Subramaniam, J.D. Rothstein, S. Marklund, P.M. Andersen, T. Brännström, O. Gredal, P.C. Wong, D.S. Williams, and D.W. Cleveland, "Toxicity of Familial ALS-Linked SOD1 Mutants from Selective Recruitment to Spinal Mitochondria", Neuron, vol. 43, pp. 5-17, 2004. http://dx.doi.org/10.1016/j.neuron.2004.06.016
- A. Israelson, N. Arbel, S. Da Cruz, H. Ilieva, K. Yamanaka, V. Shoshan-Barmatz, and D.W. Cleveland, "Misfolded Mutant SOD1 Directly Inhibits VDAC1 Conductance in a Mouse Model of Inherited ALS", Neuron, vol. 67, pp. 575-587, 2010. http://dx.doi.org/10.1016/j.neuron.2010.07.019
- W. Tan, N. Naniche, A. Bogush, S. Pedrini, D. Trotti, and P. Pasinelli, "Small peptides against the mutant SOD1/Bcl-2 toxic mitochondrial complex restore mitochondrial function and cell viability in mutant SOD1-mediated ALS.", The Journal of neuroscience : the official journal of the Society for Neuroscience, 2013. http://www.ncbi.nlm.nih.gov/pubmed/23843527
- J. Sun, and J.K. Liao, "Functional interaction of endothelial nitric oxide synthase with a voltage-dependent anion channel", Proceedings of the National Academy of Sciences, vol. 99, pp. 13108-13113, 2002. http://dx.doi.org/10.1073/pnas.202260999
- L. Xu, Y. Wang, K. Han, L. Li, Z. Zhai, and H. Shu, "VISA Is an Adapter Protein Required for Virus-Triggered IFN-β Signaling", Molecular Cell, vol. 19, pp. 727-740, 2005. http://dx.doi.org/10.1016/j.molcel.2005.08.014
- K. Guan, Z. Zheng, T. Song, X. He, C. Xu, Y. Zhang, S. Ma, Y. Wang, Q. Xu, Y. Cao, J. Li, X. Yang, X. Ge, C. Wei, and H. Zhong, "MAVS Regulates Apoptotic Cell Death by Decreasing K48-Linked Ubiquitination of Voltage-Dependent Anion Channel 1", Molecular and Cellular Biology, vol. 33, pp. 3137-3149, 2013. http://dx.doi.org/10.1128/MCB.00030-13
- H.M. Viola, A.M. Adams, S.M.K. Davies, S. Fletcher, A. Filipovska, and L.C. Hool, "Impaired functional communication between the L-type calcium channel and mitochondria contributes to metabolic inhibition in the mdx heart", Proceedings of the National Academy of Sciences, vol. 111, 2014. http://dx.doi.org/10.1073/pnas.1402544111
- A. Mitra, T. Basak, K. Datta, S. Naskar, S. Sengupta, and S. Sarkar, "Role of α-crystallin B as a regulatory switch in modulating cardiomyocyte apoptosis by mitochondria or endoplasmic reticulum during cardiac hypertrophy and myocardial infarction", Cell Death & Disease, vol. 4, pp. e582-e582, 2013. http://dx.doi.org/10.1038/cddis.2013.114
- L. Lu, C. Zhang, Q. Cai, Q. Lu, C. Duan, Y. Zhu, and H. Yang, "Voltage-dependent anion channel involved in the α-synuclein-induced dopaminergic neuron toxicity in rats", Acta Biochimica et Biophysica Sinica, vol. 45, pp. 170-178, 2013. http://dx.doi.org/10.1093/abbs/gms114
- A.F. Branco, S.L. Pereira, A.C. Moreira, J. Holy, V.A. Sardão, and P.J. Oliveira, "Isoproterenol Cytotoxicity is Dependent on the Differentiation State of the Cardiomyoblast H9c2 Cell Line", Cardiovascular Toxicology, vol. 11, pp. 191-203, 2011. http://dx.doi.org/10.1007/s12012-011-9111-5
- S. Fulda, "Tumor resistance to apoptosis", International Journal of Cancer, vol. 124, pp. 511-515, 2008. http://dx.doi.org/10.1002/ijc.24064
- N. Pavlova, and C. Thompson, "The Emerging Hallmarks of Cancer Metabolism", Cell Metabolism, vol. 23, pp. 27-47, 2016. http://dx.doi.org/10.1016/j.cmet.2015.12.006
- P.P. Hsu, and D.M. Sabatini, "Cancer Cell Metabolism: Warburg and Beyond", Cell, vol. 134, pp. 703-707, 2008. http://dx.doi.org/10.1016/j.cell.2008.08.021
- P.L. Pedersen, "Voltage dependent anion channels (VDACs): a brief introduction with a focus on the outer mitochondrial compartment’s roles together with hexokinase-2 in the “Warburg effect” in cancer", Journal of Bioenergetics and Biomembranes, vol. 40, pp. 123-126, 2008. http://dx.doi.org/10.1007/s10863-008-9165-7
- Y. Shi, C. Jiang, Q. Chen, and H. Tang, "One-step on-column affinity refolding purification and functional analysis of recombinant human VDAC1", Biochemical and Biophysical Research Communications, vol. 303, pp. 475-482, 2003. http://dx.doi.org/10.1016/s0006-291x(03)00359-0
- K. Debatin, D. Poncet, and G. Kroemer, "Chemotherapy: targeting the mitochondrial cell death pathway", Oncogene, vol. 21, pp. 8786-8803, 2002. http://dx.doi.org/10.1038/sj.onc.1206039
- I.M. Ghobrial, T.E. Witzig, and A.A. Adjei, "Targeting Apoptosis Pathways in Cancer Therapy", CA: A Cancer Journal for Clinicians, vol. 55, pp. 178-194, 2005. http://dx.doi.org/10.3322/canjclin.55.3.178
- W. Hu, and J.J. Kavanagh, "Anticancer therapy targeting the apoptotic pathway", The Lancet Oncology, vol. 4, pp. 721-729, 2003. http://dx.doi.org/10.1016/S1470-2045(03)01277-4
- S.H. Kaufmann, and D.L. Vaux, "Alterations in the apoptotic machinery and their potential role in anticancer drug resistance", Oncogene, vol. 22, pp. 7414-7430, 2003. http://dx.doi.org/10.1038/sj.onc.1206945
- R. Kim, M. Emi, K. Tanabe, Y. Uchida, and K. Arihiro, "The role of apoptotic or nonapoptotic cell death in determining cellular response to anticancer treatment", European Journal of Surgical Oncology (EJSO), vol. 32, pp. 269-277, 2006. http://dx.doi.org/10.1016/j.ejso.2005.12.006
- N. Hail, "Mitochondria: A novel target for the chemoprevention of cancer", Apoptosis, vol. 10, pp. 687-705, 2005. http://dx.doi.org/10.1007/s10495-005-0792-8
- M. Gallego, B. Joseph, T.H. Hemström, S. Tamiji, L. Mortier, G. Kroemer, P. Formstecher, B. Zhivotovsky, and P. Marchetti, "Apoptosis-inducing factor determines the chemoresistance of non-small-cell lung carcinomas", Oncogene, vol. 23, pp. 6282-6291, 2004. http://dx.doi.org/10.1038/sj.onc.1207835
- K. Debatin, "Activation of Apoptosis Pathways by Anticancer Drugs", Advances in Experimental Medicine and Biology, pp. 237-244, 1999. http://dx.doi.org/10.1007/978-1-4615-4811-9_25
- T.J. Preston, A. Abadi, L. Wilson, and G. Singh, "Mitochondrial contributions to cancer cell physiology: potential for drug development", Advanced Drug Delivery Reviews, vol. 49, pp. 45-61, 2001. http://dx.doi.org/10.1016/S0169-409X(01)00127-2
- P. Costantini, "Mitochondrion as a Novel Target of Anticancer Chemotherapy", Journal of the National Cancer Institute, vol. 92, pp. 1042-1053, 2000. http://dx.doi.org/10.1093/jnci/92.13.1042
- V. Gogvadze, S. Orrenius, and B. Zhivotovsky, "Mitochondria as targets for cancer chemotherapy", Seminars in Cancer Biology, vol. 19, pp. 57-66, 2009. http://dx.doi.org/10.1016/j.semcancer.2008.11.007
- J.E. Visvader, and G.J. Lindeman, "Cancer Stem Cells: Current Status and Evolving Complexities", Cell Stem Cell, vol. 10, pp. 717-728, 2012. http://dx.doi.org/10.1016/j.stem.2012.05.007
- J. Chen, Y. Li, T. Yu, R.M. McKay, D.K. Burns, S.G. Kernie, and L.F. Parada, "A restricted cell population propagates glioblastoma growth after chemotherapy", Nature, vol. 488, pp. 522-526, 2012. http://dx.doi.org/10.1038/nature11287
- P. Valent, D. Bonnet, R. De Maria, T. Lapidot, M. Copland, J.V. Melo, C. Chomienne, F. Ishikawa, J.J. Schuringa, G. Stassi, B. Huntly, H. Herrmann, J. Soulier, A. Roesch, G.J. Schuurhuis, S. Wöhrer, M. Arock, J. Zuber, S. Cerny-Reiterer, H.E. Johnsen, M. Andreeff, and C. Eaves, "Cancer stem cell definitions and terminology: the devil is in the details", Nature Reviews Cancer, vol. 12, pp. 767-775, 2012. http://dx.doi.org/10.1038/nrc3368
- J.T. Lee, and M. Herlyn, "Old disease, new culprit: Tumor stem cells in cancer", Journal of Cellular Physiology, vol. 213, pp. 603-609, 2007. http://dx.doi.org/10.1002/jcp.21252
- N.Y. Frank, T. Schatton, and M.H. Frank, "The therapeutic promise of the cancer stem cell concept", Journal of Clinical Investigation, vol. 120, pp. 41-50, 2010. http://dx.doi.org/10.1172/JCI41004
- G. Zhang, G. Jiang, C. Wang, K. Zhong, J. Zhang, Q. Xue, X. Li, H. Jin, and B. Li, "Decreased expression of microRNA-320a promotes proliferation and invasion of non-small cell lung cancer cells by increasing VDAC1 expression", Oncotarget, vol. 7, pp. 49470-49480, 2016. http://dx.doi.org/10.18632/oncotarget.9943
- E. Lezi, and R.H. Swerdlow, "Mitochondria in Neurodegeneration", Advances in Experimental Medicine and Biology, pp. 269-286, 2011. http://dx.doi.org/10.1007/978-94-007-2869-1_12
- M.P. Mattson, M. Gleichmann, and A. Cheng, "Mitochondria in Neuroplasticity and Neurological Disorders", Neuron, vol. 60, pp. 748-766, 2008. http://dx.doi.org/10.1016/j.neuron.2008.10.010
- M. Manczak, B.S. Park, Y. Jung, and P.H. Reddy, "Differential Expression of Oxidative Phosphorylation Genes in Patients With Alzheimer's Disease: Implications for Early Mitochondrial Dysfunction and Oxidative Damage", NeuroMolecular Medicine, vol. 5, pp. 147-162, 2004. http://dx.doi.org/10.1385/NMM:5:2:147
- O. Benek, L. Aitken, L. Hroch, K. Kuca, F. Gunn-Moore, and K. Musilek, "A Direct Interaction Between Mitochondrial Proteins and Amyloid-β Peptide and its Significance for the Progression and Treatment of Alzheimer’s Disease", Current Medicinal Chemistry, vol. 22, pp. 1056-1085, 2015. http://dx.doi.org/10.2174/0929867322666150114163051
- L. Devi, and M. Ohno, "Mitochondrial dysfunction and accumulation of the β-secretase-cleaved C-terminal fragment of APP in Alzheimer's disease transgenic mice", Neurobiology of Disease, vol. 45, pp. 417-424, 2012. http://dx.doi.org/10.1016/j.nbd.2011.09.001
- L. Petrozzi, G. Ricci, N.J. Giglioli, G. Siciliano, and M. Mancuso, "Mitochondria and Neurodegeneration", Bioscience Reports, vol. 27, pp. 87-104, 2007. http://dx.doi.org/10.1007/s10540-007-9038-z
- E. Radi, P. Formichi, C. Battisti, and A. Federico, "Apoptosis and oxidative stress in neurodegenerative diseases.", Journal of Alzheimer's disease : JAD, 2014. http://www.ncbi.nlm.nih.gov/pubmed/25056458
- F.G. Gervais, D. Xu, G.S. Robertson, J.P. Vaillancourt, Y. Zhu, J. Huang, A. LeBlanc, D. Smith, M. Rigby, M.S. Shearman, E.E. Clarke, H. Zheng, L.H. Van Der Ploeg, S.C. Ruffolo, N.A. Thornberry, S. Xanthoudakis, R.J. Zamboni, S. Roy, and D.W. Nicholson, "Involvement of Caspases in Proteolytic Cleavage of Alzheimer’s Amyloid-β Precursor Protein and Amyloidogenic Aβ Peptide Formation", Cell, vol. 97, pp. 395-406, 1999. http://dx.doi.org/10.1016/s0092-8674(00)80748-5
- M. Li, V.O. Ona, C. Guégan, M. Chen, V. Jackson-Lewis, L.J. Andrews, A.J. Olszewski, P.E. Stieg, J. Lee, S. Przedborski, and R.M. Friedlander, "Functional Role of Caspase-1 and Caspase-3 in an ALS Transgenic Mouse Model", Science, vol. 288, pp. 335-339, 2000. http://dx.doi.org/10.1126/science.288.5464.335
- R.M. Friedlander, "Apoptosis and Caspases in Neurodegenerative Diseases", New England Journal of Medicine, vol. 348, pp. 1365-1375, 2003. http://dx.doi.org/10.1056/NEJMra022366
- K. Hirai, G. Aliev, A. Nunomura, H. Fujioka, R.L. Russell, C.S. Atwood, A.B. Johnson, Y. Kress, H.V. Vinters, M. Tabaton, S. Shimohama, A.D. Cash, S.L. Siedlak, P.L. Harris, P.K. Jones, R.B. Petersen, G. Perry, and M.A. Smith, "Mitochondrial abnormalities in Alzheimer's disease.", The Journal of neuroscience : the official journal of the Society for Neuroscience, 2001. http://www.ncbi.nlm.nih.gov/pubmed/11312286
- A.M. Aleardi, G. Benard, O. Augereau, M. Malgat, J.C. Talbot, J.P. Mazat, T. Letellier, J. Dachary-Prigent, G.C. Solaini, and R. Rossignol, "Gradual Alteration of Mitochondrial Structure and Function by β-Amyloids: Importance of Membrane Viscosity Changes, Energy Deprivation, Reactive Oxygen Species Production, and Cytochrome c Release", Journal of Bioenergetics and Biomembranes, vol. 37, pp. 207-225, 2005. http://dx.doi.org/10.1007/s10863-005-6631-3
- I. Ferrer, "Altered mitochondria, energy metabolism, voltage-dependent anion channel, and lipid rafts converge to exhaust neurons in Alzheimer’s disease", Journal of Bioenergetics and Biomembranes, vol. 41, pp. 425-431, 2009. http://dx.doi.org/10.1007/s10863-009-9243-5
- C.M. Ramírez, M. González, M. Díaz, R. Alonso, I. Ferrer, G. Santpere, B. Puig, G. Meyer, and R. Marin, "VDAC and ERα interaction in caveolae from human cortex is altered in Alzheimer's disease", Molecular and Cellular Neuroscience, vol. 42, pp. 172-183, 2009. http://dx.doi.org/10.1016/j.mcn.2009.07.001
- P.H. Reddy, "Is the mitochondrial outermembrane protein VDAC1 therapeutic target for Alzheimer's disease?", Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease, vol. 1832, pp. 67-75, 2013. http://dx.doi.org/10.1016/j.bbadis.2012.09.003
- B.C. Yoo, M. Fountoulakis, N. Cairns, and G. Lubec, "Changes of voltage-dependent anion-selective channel proteins VDAC1 and VDAC2 brain levels in patients with Alzheimer's disease and Down Syndrome", ELECTROPHORESIS, vol. 22, pp. 172-179, 2001. http://dx.doi.org/10.1002/1522-2683(200101)22:1<172::AID-ELPS172>3.0.CO;2-P
- K. Fukada, F. Zhang, A. Vien, N.R. Cashman, and H. Zhu, "Mitochondrial Proteomic Analysis of a Cell Line Model of Familial Amyotrophic Lateral Sclerosis", Molecular & Cellular Proteomics, vol. 3, pp. 1211-1223, 2004. http://dx.doi.org/10.1074/mcp.M400094-MCP200
- R. Marin, C.M. Ramírez, R. Marin, C.M. Ramírez, M. González, E. González-Muñoz, A. Zorzano, M. Camps, R. Alonso, and M. Díaz, "Voltage-dependent anion channel (VDAC) participates in amyloid beta-induced toxicity and interacts with plasma membrane estrogen receptor α in septal and hippocampal neurons", Molecular Membrane Biology, vol. 24, pp. 148-160, 2007. http://dx.doi.org/10.1080/09687860601055559
- P.H. Reddy, "Amyloid beta-induced glycogen synthase kinase 3β phosphorylated VDAC1 in Alzheimer's disease: Implications for synaptic dysfunction and neuronal damage", Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease, vol. 1832, pp. 1913-1921, 2013. http://dx.doi.org/10.1016/j.bbadis.2013.06.012
- A. Smilansky, L. Dangoor, I. Nakdimon, D. Ben-Hail, D. Mizrachi, and V. Shoshan-Barmatz, "The Voltage-dependent Anion Channel 1 Mediates Amyloid β Toxicity and Represents a Potential Target for Alzheimer Disease Therapy", Journal of Biological Chemistry, vol. 290, pp. 30670-30683, 2015. http://dx.doi.org/10.1074/jbc.M115.691493
- W. Jiang, B. Du, Z. Chi, L. Ma, S. Wang, X. Zhang, W. Wu, X. Wang, G. Xu, and C. Guo, "Preliminary explorations of the role of mitochondrial proteins in refractory epilepsy: Some Findings From Comparative Proteomics", Journal of Neuroscience Research, vol. 85, pp. 3160-3170, 2007. http://dx.doi.org/10.1002/jnr.21384
- R. Sultana, H.F. Poon, J. Cai, W.M. Pierce, M. Merchant, J.B. Klein, W.R. Markesbery, and D.A. Butterfield, "Identification of nitrated proteins in Alzheimer's disease brain using a redox proteomics approach", Neurobiology of Disease, vol. 22, pp. 76-87, 2006. http://dx.doi.org/10.1016/j.nbd.2005.10.004
- R. Roshan, S. Shridhar, M.A. Sarangdhar, A. Banik, M. Chawla, M. Garg, V.P. Singh, and B. Pillai, "Brain-specific knockdown of miR-29 results in neuronal cell death and ataxia in mice", RNA, vol. 20, pp. 1287-1297, 2014. http://dx.doi.org/10.1261/rna.044008.113
- C.M. Stary, X. Sun, Y. Ouyang, L. Li, and R.G. Giffard, "miR-29a differentially regulates cell survival in astrocytes from cornu ammonis 1 and dentate gyrus by targeting VDAC1", Mitochondrion, vol. 30, pp. 248-254, 2016. http://dx.doi.org/10.1016/j.mito.2016.08.013
- M. Fatima, B. Prajapati, K. Saleem, R. Kumari, C. Mohindar Singh Singal, and P. Seth, "Novel insights into role of miR-320a-VDAC1 axis in astrocyte-mediated neuronal damage in neuroAIDS", Glia, vol. 65, pp. 250-263, 2016. http://dx.doi.org/10.1002/glia.23089
- P. Zimmet, K.G.M.M. Alberti, and J. Shaw, "Global and societal implications of the diabetes epidemic", Nature, vol. 414, pp. 782-787, 2001. http://dx.doi.org/10.1038/414782a
- S. Bolen, L. Feldman, J. Vassy, L. Wilson, H. Yeh, S. Marinopoulos, C. Wiley, E. Selvin, R. Wilson, E.B. Bass, and F.L. Brancati, "Systematic Review: Comparative Effectiveness and Safety of Oral Medications for Type 2 Diabetes Mellitus", Annals of Internal Medicine, vol. 147, pp. 386, 2007. http://dx.doi.org/10.7326/0003-4819-147-6-200709180-00178
- K. Sasaki, R. Donthamsetty, M. Heldak, Y. Cho, B.T. Scott, and A. Makino, "VDAC: old protein with new roles in diabetes", American Journal of Physiology-Cell Physiology, vol. 303, pp. C1055-C1060, 2012. http://dx.doi.org/10.1152/ajpcell.00087.2012
- A.H. Truong, S. Murugesan, K.D. Youssef, and A. Makino, "Mitochondrial Ion Channels in Metabolic Disease", Vascular Ion Channels in Physiology and Disease, pp. 397-419, 2016. http://dx.doi.org/10.1007/978-3-319-29635-7_18
- B.B. Lowell, and G.I. Shulman, "Mitochondrial Dysfunction and Type 2 Diabetes", Science, vol. 307, pp. 384-387, 2005. http://dx.doi.org/10.1126/science.1104343
- X. Jiang, and X. Wang, "Cytochrome c Promotes Caspase-9 Activation by Inducing Nucleotide Binding to Apaf-1", Journal of Biological Chemistry, vol. 275, pp. 31199-31203, 2000. http://dx.doi.org/10.1074/jbc.C000405200
- N. Hay, "Reprogramming glucose metabolism in cancer: can it be exploited for cancer therapy?", Nature Reviews Cancer, vol. 16, pp. 635-649, 2016. http://dx.doi.org/10.1038/nrc.2016.77
- R.W. Johnstone, A.A. Ruefli, and S.W. Lowe, "Apoptosis: a link between cancer genetics and chemotherapy.", Cell, 2002. http://www.ncbi.nlm.nih.gov/pubmed/11832206
- T. Takehara, "Expression and role of Bcl-xL in human hepatocellular carcinomas", Hepatology, vol. 34, pp. 55-61, 2001. http://dx.doi.org/10.1053/jhep.2001.25387
- Z. Ding, X. Yang, A. Pater, and S. Tang, "Resistance to Apoptosis Is Correlated with the Reduced Caspase-3 Activation and Enhanced Expression of Antiapoptotic Proteins in Human Cervical Multidrug-Resistant Cells", Biochemical and Biophysical Research Communications, vol. 270, pp. 415-420, 2000. http://dx.doi.org/10.1006/bbrc.2000.2432
- R. GROBHOLZ, H. ZENTGRAF, K. KÖHRMANN, and U. BLEYL, "Bax, Bcl‐2, Fas and Fas‐L antigen expression in human seminoma: Correlation with the apoptotic index", APMIS, vol. 110, pp. 724-732, 2002. http://dx.doi.org/10.1034/j.1600-0463.2002.1101006.x
- M. Krajewska, S.F. Moss, S. Krajewski, K. Song, P.R. Holt, and J.C. Reed, "Elevated expression of Bcl-X and reduced Bak in primary colorectal adenocarcinomas.", Cancer research, 1996. http://www.ncbi.nlm.nih.gov/pubmed/8625322
- P.L. Pedersen, "Warburg, me and Hexokinase 2: Multiple discoveries of key molecular events underlying one of cancers’ most common phenotypes, the “Warburg Effect”, i.e., elevated glycolysis in the presence of oxygen", Journal of Bioenergetics and Biomembranes, vol. 39, pp. 211-222, 2007. http://dx.doi.org/10.1007/s10863-007-9094-x
- S.P. Mathupala, Y.H. Ko, and P.L. Pedersen, "Hexokinase-2 bound to mitochondria: Cancer's stygian link to the “Warburg effect” and a pivotal target for effective therapy", Seminars in Cancer Biology, vol. 19, pp. 17-24, 2009. http://dx.doi.org/10.1016/j.semcancer.2008.11.006
- S. Shimizu, T. Ide, T. Yanagida, and Y. Tsujimoto, "Electrophysiological Study of a Novel Large Pore Formed by Bax and the Voltage-dependent Anion Channel That Is Permeable to Cytochrome c", Journal of Biological Chemistry, vol. 275, pp. 12321-12325, 2000. http://dx.doi.org/10.1074/jbc.275.16.12321
- J.G. Pastorino, N. Shulga, and J.B. Hoek, "Mitochondrial Binding of Hexokinase II Inhibits Bax-induced Cytochrome c Release and Apoptosis", Journal of Biological Chemistry, vol. 277, pp. 7610-7618, 2002. http://dx.doi.org/10.1074/jbc.M109950200
SUPPLEMENTAL INFORMATION
 Download Supplemental Information
Download Supplemental Information
ACKNOWLEDGMENTS
This research was supported by grants from the Israel Science Foundation (307/13), by Ezra and Yafa Yeruham and Sima and Philip Needleman research funds to VSB, and R01CA184456 (NCI), COBRE Project GM103542 and ACS 13-041-01-IRG funding to ENM.
COPYRIGHT
© 2017

VDAC1 at the crossroads of cell metabolism, apoptosis and cell stress by Shoshan-Barmatz et al. is licensed under a Creative Commons Attribution 4.0 International License.


