Reviews:
Cell Stress, Vol. 3, No. 7, pp. 208 - 220; doi: 10.15698/cst2019.07.191
The role of epigenetics in hypothalamic energy balance control: implications for obesity

1 Neuronal Control of Metabolism Laboratory, Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), 08036 Barcelona, Spain.
2 Centro de Investigación Biomédica en Red (CIBER) de Diabetes y Enfermedades Metabólicas Asociadas (CIBERDEM), 08036 Barcelona, Spain.
Keywords: obesity, hypothalamus, epigenetic, energy balance, AgRP neurons, POMC neurons.
Abbreviatons:
5-mC – 5-methylcytosine,
α-MSH – α-melanocyte-stimulating hormone,
AgRP – agouti-related protein,
AMPK – AMP-activated protein kinase,
ARC – arcuate nucleus,
DNMT – DNA methyltransferases,
GABA – γ-aminobutyric acid,
GWAS – genome-wide association studies,
HAT – histone acetyltransferase,
HDAC – histone deacetylase,
LH – lateral hypothalamus,
lncRNA – long ncRNA,
MCR – melanocortin receptor,
miRNA – micro RNA,
mRNA – messenger RNA,
ncRNA – non-coding RNA,
NPY – neuropeptide Y,
POMC – pro-opimelanocortin,
PTM – post-translational modification,
PVN – paraventricular nucleus,
snoRNA – small nucleolar RNA,
snRNA – small nuclear RNA,
T2D – type 2 diabetes,
VMH – ventromedial hypothalamus.
Received originally: 24/02/2019 Received in revised form: 15/04/2019
Accepted: 08/05/2019
Published: 05/06/2019
Correspondence:
Arnaud Obri, Neuronal Control of Metabolism Laboratory, Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), Centre Esther Koplowitz (CEK), C/ Rosselló 149-153, 5th floor, 08036 Barcelona, Spain, Tel: +34932275707 ext 2910; obri@clinic.cat
Conflict of interest statement: The authors declare no competing interests.
Please cite this article as: Arnaud Obri and Marc Claret (2019). The role of epige-netics in hypothalamic energy balance control: implications for obesity. Cell Stress 3(7): 208-220. doi: 10.15698/cst2019.07.191
Abstract
Despite enormous social and scientific efforts, obesity rates continue to increase worldwide. While genetic factors contribute to obesity development, genetics alone cannot explain the current epidemic. Obesity is essentially the consequence of complex genetic-environmental interactions. Evidence suggests that contemporary lifestyles trigger epigenetic changes, which can dysregulate energy balance and thus contribute to obesity. The hypothalamus plays a pivotal role in the regulation of body weight, through a sophisticated network of neuronal systems. Alterations in the activity of these neuronal pathways have been implicated in the pathophysiology of obesity. Here, we review the current knowledge on the central control of energy balance with a focus on recent studies linking epigenetic mechanisms in the hypothalamus to the development of obesity and metabolic disorders.
INTRODUCTION
The prevalence of obesity continues to increase worldwide. This trend is of concern because of its dramatic economic impact, concomitant decreased lifespan and increased comorbidities, such as hypertension, cardiovascular diseases, type 2 diabetes (T2D) and cancer [1][2][3]. Only in Europe alone, the (direct and indirect) cost of obesity is estimated to be around 81 billion of euros per year.
–
The most basic definition of obesity refers to an excessive and/or abnormal accumulation of fat. Obesity is considered to be the consequence of an imbalance between energy intake and expenditure [6]. Thus, altered feeding behavior (chronic overeating) and a sedentary lifestyle (chronic low energy expenditure) are important contributors to the development of overweight and obesity.
–
The brain, and in particular the hypothalamus, plays an essential role in maintaining energy homeostasis. Specific neuronal circuits in the hypothalamus sense and decode multiple nutritional, hormonal and metabolic cues to fine-tune food intake and energy expenditure. However, environmental factors, such as the diet, physical activity or exposure to certain chemicals, can impair the hypothalamic mechanisms controlling appetite and energy balance [7][8][9][10][11]. At the molecular level, epigenetic processes might play a fundamental role in the complex interactions between environment and energy imbalance seen in obesity [12].
–
In this review, we discuss epigenetic determinants in hypothalamic pathways controlling energy homeostasis and its association with the development of obesity and metabolic syndrome.
OBESITY IS AT THE INTERPLAY BETWEEN GENETIC AND ENVIRONMENTAL FACTORS
Energy imbalance is an important contributor of body weight gain; however, the pathophysiology of obesity has proven to be much more complex. Research into monogenic obesity has resulted in the identification of single genes that dramatically affect body weight through hypothalamic pathways [14][15][16][17]. Yet, mutations in those genes are relatively rare and account for only ≈5% of obese patients [18]. Genome-wide association studies (GWAS) have emerged as a valuable tool to identify novel genetic factors contributing to obesity [19][20]. GWAS have identified numerous single nucleotide polymorphisms (SNPs) associated with body mass index (BMI), and again have highlighted the importance of neuronal pathways to obesity [19][21]. However, genetics alone cannot explain the rather recent and steadfast increase in worldwide obesity rates. At this point, it is clear that obesity stems from the interaction of susceptibility genes with multiple environmental factors (Figure 1).
–
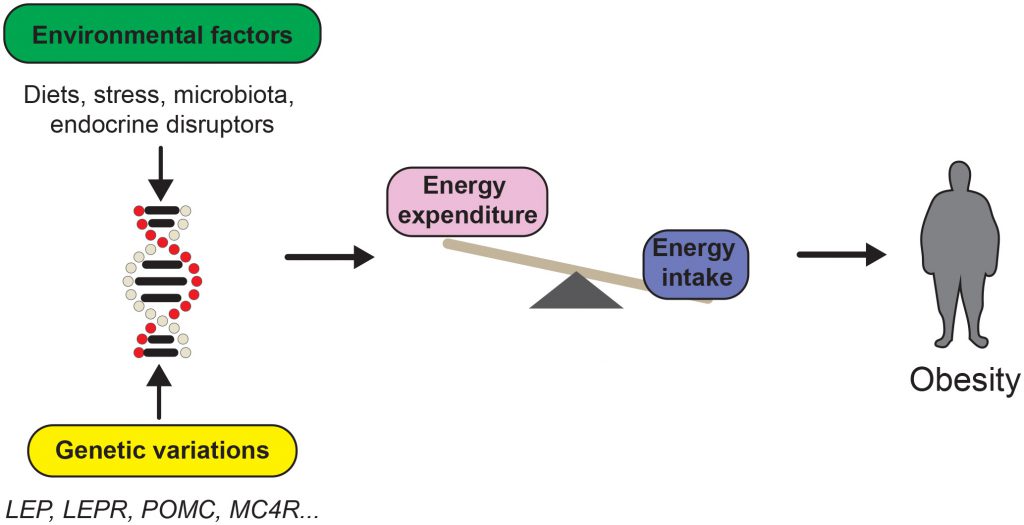 | FIGURE 1: Obesity is at the interplay between genetic and environmental factors. The development of obesity is influenced by genetic and environmental factors. The study of monogenic obesity has led the discovery of several obesity susceptibility genes such as Proopimelanocortin (POMC), Melanocortin receptor 4 (MC4R) or Leptin (LEP) among others. However, diverse environmental factors such as the diet, combined with genetic variations, also influence energy balance control. |
–
There are critical periods in the prenatal and perinatal phases that are particularly susceptible to the impact of environmental factors. The metabolic imprinting during those periods, might influence the future development of obesity from infancy to the onset of adulthood. Epidemiological studies in humans have shown that maternal obesity and diabetes during fetal life and lactation are risk factors for the future development of obesity [22][23][24]. Likewise, maternal undernutrition was also shown to influence offspring predisposition to metabolic disorders [25][26].
–
In the hypothalamus, maternal nutritional insults during development have been shown to affect the function of hypothalamic circuits that regulate energy balance. For instance, maternal undernutrition in rodents alters the activity of appetite and satiety centers in the offspring [27][28]. This is accompanied by an impairment in neuronal proliferation, axonal elongation and neuropeptide expression in the hypothalamus [29][30][31]. Similarly, maternal overnutrition has been shown to damage axonal projections in the hypothalamus [32][33][34]. Hormonal imbalance during pregnancy can also lead to defects in hypothalamic circuits [34][35]. It is unclear in what way these environmental factors influence the function of brain pathways controlling energy balance. However, over the past two decades, several studies have underscored the importance of epigenetic gene regulation.
REGULATION OF ENERGY HOMEOSTASIS BY THE BRAIN
The brain acts like the central processing unit of a computer to control energy homeostasis. It integrates internal metabolic signals (i.e. nutrients and hormones) and external sensory cues regarding food availability and palatability [36][37]. These signals provide information about the type of circulating fuels available in the organism, as well as the amount of energy stored and needed. The areas governing energy balance include important parts of the limbic system, midbrain, brainstem and cortex. However, ≈80 years of research have unquestionably shown that the hypothalamus is the quintessential brain region in the control of homeostatic food intake and energy homeostasis [38].
–
Arcuate nucleus of the hypothalamus
The arcuate nucleus (ARC) resides in the medial-basal part of the tuberal hypothalamus, on both sides of the third ventricle (3V) so it receives input from other regions of the brain through the cerebrospinal fluid [39]. The ARC is also in direct contact with the median eminence, an area of the brain where the blood-brain barrier is semi-permeable [40]. This strategic position within the brain, allows the ARC to sense the circulating levels of nutrients and hormones. In the ARC, there are two subsets of neurons playing critical functions in the regulation of appetite and energy expenditure: i) orexigeneic neurons that co-express agouti-related protein (AgRP) and neuropeptide Y (NPY) (hereafter AgRP neurons) and ii) anorexigeneic neurons expressing pro-opimelanocortin (POMC) (hereafter POMC neurons).
–
AgRP neurons and the orexigenic pathway
AgRP neurons are stimulated by fasting and send intra-ARC projections to POMC neurons and other hypothalamic structures such as paraventricular nucleus (PVN), lateral hypothalamus (LH) and parabrachial hypothalamus (PBN) [41][42]. AgRP is an orexigenic (i.e. appetite-stimulating) neuropeptide that is exclusively expressed in the ARC and acts as an inverse agonist to melanocortin receptors MC3R and MC4R [43][44]. AgRP inhibits α-melanocyte-stimulating hormone (α-MSH) signaling exclusively in the PVN to regulate feeding [45]. Conversely, NPY is widely expressed outside the ARC and exerts its orexigenic effect through NPY receptors (NPY1R to NPY5R) [46]. AgRP and NPY are both primary drivers to initiate food intake, as central injection of either neuropeptide causes hyperphagia [43][47][48]. However, deletion of Agrp or Npy genes in mice have shown no effect on food intake and body weight [49]. Several studies have consolidated our understanding of the function of AgRP neurons. For instance, ablation of AgRP neurons in adult mice results in severe anorexia [42][50], while acute activation of those neurons by optogenetic or chemogenetic means leads to a robust increase in food intake [51][52]. In addition, AgRP neurons can inhibit other neurons via γ-aminobutyric acid (GABA) action in the ARC (i.e. POMC neurons) and in other areas of the brain [53][54][55][56]. Remarkably, GABAergic inputs from AgRP neurons can modulate food intake by acting in the PVN [53]. These discoveries highlight the importance of GABA signals from AgRP neurons in the regulation of energy balance, through the inhibition of anorexigenic neuronal populations all over the brain [57].
–
POMC neurons and the central melanocortin system
POMC neurons project mainly to the PVN but also to the LH, the ventromedial hypothalamus (VMH) and dorsomedial nucleus (DMN) [58]. POMC neurons produce the prohormone POMC, which expression is restricted to the ARC and the nucleus of the solitary tract (NTS) of the brainstem [59][60]. POMC precursor is cleaved into diverse neuropeptides including α-MSH, which binds MC4Rs resulting in a reduction in appetite and enhanced energy expenditure [44][61][62][63]. Consistently, Pomc or Mc4r deficiency causes hyperphagia and obesity in both mice and humans [16][17][64][65][66]. In addition, β-endorphin (a POMC-derived neuropeptide) is released from ARC neurons and regulates feeding after binding to the opioid receptor [67]. Recent investigations using optogenetic and chemogenetic approaches have confirmed the role of POMC neurons in feeding control and energy homeostasis [52][68]. Acute chemogenetic stimulation of POMC neurons in the dark phase (a natural feeding period) suppresses food intake whereas consumption of a meal increases their activity, which supports the role for α-MSH in short-term feeding control [69]. However, prolonged activation of POMC neurons is necessary to suppress food intake during the light phase, suggesting that α-MSH might be as well involved in long-term regulation of energy homeostasis [52][68]. Indeed, deep brain imaging studies have shown that POMC neurons are gradually and persistently depolarized by leptin [37]. Altogether, these data point towards a role of α-MSH in long- and short-term energy balance. At this time, the mechanisms underlying these two distinct effects remain unknown.
–
POMC, AgRP and MC4R-expressing neurons constitute the central melanocortin system. This is arguably the best-characterized neuronal network involved in energy balance control. The melanocortin system is characteristically composed of fibers that express both agonists (α-MSH) and antagonists (AgRP) of the melanocortin receptors and receives inputs from hormones, nutrients and afferent neural circuits [15][70][71][72][73][74].
–
In addition to neuropeptides, hypothalamic neurons can respond to nutrients by modifying the synthesis and/or activity of cellular energy sensors. In the last decades, many evidences have shown that hypothalamic AMP-activated protein kinase (AMPK) is a nutrient and energy sensor that controls whole-body energy homeostasis [75][76][77]. Nonetheless, in the hypothalamus AMPK integrates the orexigenic and anorexigenic pathways [78]. Genetic evidences have shown that mice lacking Ampk in POMC or AgRP neurons display an impaired energy balance alongside alteration on body weight and glucose homestasis [75].
–
The term epigenetics was defined by the developmental biologist Conrad Waddington in 1942 [79]. Currently, the accepted definition of epigenetics is “stably heritable phenotype resulting from changes in a chromosome without alterations in the DNA sequence” [80]. In general, it is acknowledged that epigenetic is an additional regulatory layer for gene expression control. There are several epigenetic modifications that can change chromatin structure, including DNA methylation, post translational modification of histone tails and regulatory RNAs.
–
DNA methylation
DNA methylation was the first epigenetic mark discovered [81]. It is a stable covalent modification that mostly occurs on DNA regions where a cytosine is followed by a guanine (CpG) and is catalyzed by a family of enzymes called DNA methyltransferases (DNMT). These enzymes add a methyl group on the 5th carbon of a cytosine to generate a 5-methylcytosine (5-mC). In mammals, there are three DNMTs: DNMT1, DNMT3a and DNMT3b. DNMT1 maintains DNA methylation during replication [82]. DNMT3a and DNMT3b are responsible for de novo methylation [83]. DNA methylation has many biological functions including X chromosome inactivation, the monoallelic expression of imprinted genes and transcriptional repression of transposon-derived sequences [84][85].
–
For many years, DNA methylation was considered a permanent epigenetic modification that could not be removed. However, this concept has been challenged in recent years with the discovery of ten eleven translocation (TET) enzymes, which catalyze crucial steps for an oxidative demethylation reaction thus providing a mechanistic basis for an active DNA demethylation pathway [86][87]. In the brain, DNA methylation appears to be particularly important. On one hand, DNMT1 and DNMT3a are highly expressed in post-mitotic neurons when compared to other cell types and double knock-out mice for these proteins show alterations in neuronal plasticity [88]. On the other hand, 5-mC is very abundant in the brain, particularly in the hypothalamus, cortex and hippocampus [89].
–
DNA methylation is not only restricted to CpG dinucleotides. Several relatively recent studies have revealed high levels of methylation in non-CpG cytosines (mCH, where methylated cytosine is followed by an adenine, thymidine or cytosine) [90][91][92]. In the brain, methylation of mCH sites is very dynamic when compared to CpG islands and it occurs during central nervous system maturation in the early years of life [90]. These novel insights have raised new and exciting questions regarding the functional role of mCH in the brain.
–
Histone post-translational modifications
Chromatin is a complex of DNA wrapped around a nucleosome, which is composed of canonical histones H2A, H2B, H3 and H4 [93]. Nonetheless, DNA has to be accessible to allow molecular processes like transcription, DNA repair and replication. Compelling evidence have shown that post-translational modifications (PTMs) of histone tails can modulate chromatin structure and hence transcriptional activity. Histone tails can undergo a large variety of PTMs including acetylation and methylation among others [94].
–
Histone acetylation is defined by the addition of an acetyl group on a lysine or an arginine residue of histone tails [95]. It is catalyzed by specific histone acetyltransferases (HAT) [96]. Histone acetylation is reversible, as histone deacetylases (HDACs) can remove the acetyl groups from histone tails. HDACs are classified in four classes (I, II, III and IV) according to their functions and DNA sequence [97]. Histone acetylation plays a role in chromatin assembly and participates in the regulation of gene expression [98]. At the molecular level, it is believed that histone acetylation increases the accessibility of transcription factors to DNA by lowering the affinity between histones and DNA [98].
–
Histone methylation occurs mainly on arginine, lysine and histidine [99][100]. Specific histone methyltransferases (HMT) catalyze the addition of one or more methyl groups to histone tails [101]. SET-domain containing, and DOT1-like methyltransferases are specific for lysine, while N-methyltransferases (PRMT) are specific for arginine [102]. Histone methylation was believed to be irreversible until the discovery of the H3K4 lysine-specific demethylase 1A (KDM1A or LSD1) [99][103]. Since then, many other demethylases have been identified [103][104]. Overall, histone acetylation and methylation are the most studied histone PTMs, due to their effects in gene expression, and are systematically used to map chromatin structure across the genome.
–
Non-coding RNA
Non-coding RNAs (ncRNAs) are relatively new concept in epigenetics. These molecules make up the majority of the transcriptome but, unlike messenger RNA (mRNA), ncRNAs are transcribed from DNA but not translated into protein. There are three different types of ncRNAs [105][106]: (i) small nuclear RNAs (snRNAs) and small nucleolar RNAs (snoRNAs); (ii) interference RNA, including micro RNAs (miRNAs); and (iii) long ncRNAs (lncRNAs).
–
snRNAs and snoRNAs are involved in the processing and regulation of other RNAs such as mRNA and ribosomal RNA (rRNA). miRNAs are short ncRNAs (≈22 nucleotides in length) that regulate gene expression via mRNA silencing [105]. Normally, miRNAs bind to complementary mRNA target sequences and either inhibit their translation or cause the degradation of the mRNA [105]. lncRNAs are large RNA molecules localized in the cytoplasm or the nucleus with a length of more than 200 nucleotides. Despite lncRNAs being thought to account for the majority of the ncRNA transcriptome, their discovery is still at a preliminary stage and few lncRNAs have been characterized in detail so far. However, it is clear that lncRNAs are important regulators of gene expression through a wide variety of mechanisms [106][107].
EPIGENETICS OF ENERGY BALANCE CONTROL IN THE HYPOTHALAMUS
Numerous studies have revealed that epigenetic mechanisms are involved in many aspects of metabolic dysfunction. Genomic data indicate that obesity and T2D are associated with altered DNA methylation patterns at specific loci [108][109]. Similarly, growing evidence links PTMs of histone tails with metabolic disease, especially T2D [110]. Moreover, epigenetic modifications can explain the molecular mechanisms underlying fetal programming and its association with metabolic disorders [12][111].
–
The epigenetic-dependent regulation of metabolism is reciprocal, as many metabolites and nutrients can serve as substrates/co-factors for epigenetic-modifying enzymes. Therefore, changes in the concentration of particular metabolites should be considered as a novel signaling cue implicated in the control of gene expression (Figure 2). For instance, acetyl-CoA derived from glucose and fatty acid metabolism directly impacts the chromatin architecture by modulating the activity of chromatin-modifying enzymes [112][113]. Moreover, nutrient sensors such as AMPK directly regulate epigenetic processes. For example, activated AMPK can modify the activity of several HATs and therefore impact histone acetylation [114][115]. Yet, AMPK impact on epigenetic is not limited to histone acetylation as it has been shown to also influence DNA methylation and other histone PTMs [116]. In this context, it seems reasonable that diet insults may cause epigenetic perturbations in the neurons of the brain governing energy balance.
–
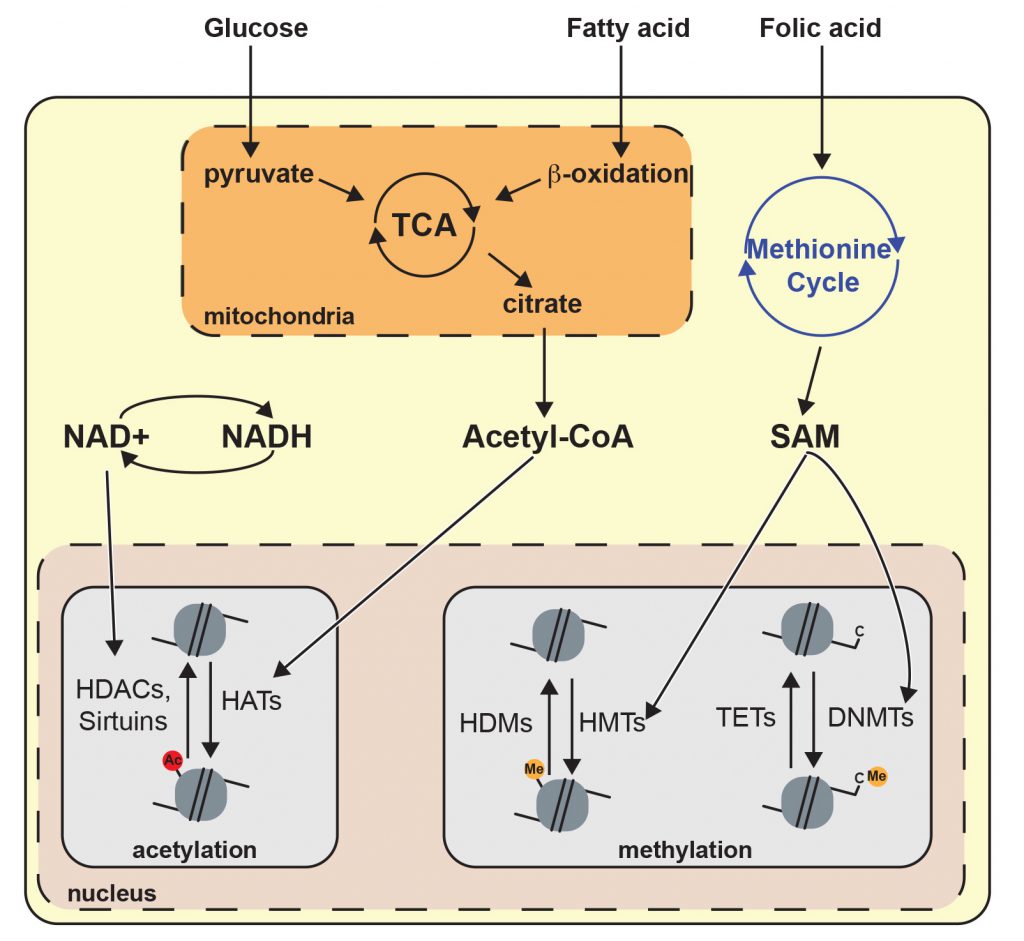 | FIGURE 2: Metabolites influence chromatin architecture. Glucose and fatty acid catabolism produce acetyl-CoA through metabolic pathways including tricarboxylic acid cycle (TCA) and β-oxidation. Acetyl-CoA regulates histone acetylation because it is important for the enzymatic activity of histone acetyltransferases. NAD+ is produced by oxidative pathways and is a relevant cofactor for histone deacetylation mediated by sirtuins. The methionine cycle is the principal producer of S-adenosyl methionine (SAM), which is a cofactor for histone/DNA methyltransferase (HMT or DNMT). Histone deacetylase (HDAC), ten-eleven translocation (TET), histone demethylase (HDM). |
–
Hypothalamic DNA methylation and energy balance control
An excellent example of the influence of epigenetic gene regulation is shown by the Agouti (Avy) mouse model, in which genetically identical mice can have a completely different phenotype in terms of both color and size. The Agouti gene promotes a yellow mouse coat color and affects energy metabolism through inhibition of the melanocortin signaling [117]. This gene is regulated in part by the methylation status of a transposable element (IAP) located in its promoter. Accordingly, mice showing a methylated IAP have a normal body weight and are brown while an unmethylated promoter generates yellow mice that are prone to obesity [118]. In Avy mice, maternal supplementation with folic acid, a form of vitamin B9 critical for DNA and protein methylation, results in a shift towards the lean phenotype in the offspring [118]. Conversely, fetal or neonatal exposure to the endocrine disruptor bisphenol A is associated with higher body weight and unmethylation of the Agouti gene [119].
–
Maternal undernutrition has been shown to decrease the activity of hypothalamic DNMTs [120]. Maternal stress, which predisposed the female offspring to binge eating (BE)-like behavior, also altered the expression of hypothalamic DNMTs, causing hypomethylation of hypothalamic miR-1a and downstream dysregulation of the melanocortin system [121]. Moreover, these alterations could be reverted by a methyl-balanced diet during puberty [121].
–
Numerous reports have evaluated the impact of DNA methylation on the expression of key metabolic genes in the hypothalamus. For instance, overfeeding altered the methylation status of Pomc promoter in rat [122]. Similarly, maternal undernutrition changed the methylation of Pomc promoter in sheep [111][123]. In addition, Pomc promoter methylation was decreased in a model of rats resistant to diet-induced obesity [124]. More importantly, methylation of CpGs at the intron2–exon3 junction of POMC gene is higher in obese children as compared to normal-weight individuals. Insulin signaling in the hypothalamus might also be affected by DNA methylation [125]. Plagemann and colleagues reported that methylation in the promoter region of Insulin receptor (InsR) is higher in the hypothalamus of rats coming from small litters, suggesting that increased glucose levels due to overfeeding in neonates might be the cause [126]. Alterations in the methylation of Npy promoter were also observed in the PVN of mice fed on a cafeteria diet [127]. Moreover, genetic studies have shown that deletion of the DNA methyltransferase Dnmt3a in PVN Sim1-neurons leads to obesity [128].
–
Methylated DNA recruits various proteins with a methyl-CpG-binding domain (MBD), such as methyl-CpG-binding protein 2 (MeCP2). A genetic study revealed an important role for MeCP2 in the regulation of energy metabolism, as mice lacking Mecp2 in Sim1 neurons developed an obese phenotype [129]. Similarly, Mecp2 deletion in POMC neurons results in increased body weight, fat mass, leptin resistance and food intake [130]. Altogether, these studies have highlighted the crucial role of DNA methylation in the hypothalamic regulation of energy metabolism.
–
Hypothalamic histone PTMs and energy balance control
The activity of most chromatin modifiers is influenced by metabolites. Glucose and fatty acid catabolism produce acetyl-CoA, which is an essential acetyl group donor in histone acetylation reactions. Thus, acetyl-CoA links energy metabolism with epigenetic gene regulation [131]. In addition, NAD+ is a common molecule in various oxidative pathways and it is also an obligate cofactor for sirtuin-dependent histone deacetylation (Figure 2) [132]. Hence, fluctuating NAD+ levels could contribute to histone deacetylation by sirtuin. Nonetheless, the connection between histone PTMs in the hypothalamus and obesity predisposition has not been sufficiently explored and remains largely unknown.
–
Some of the few available studies have focus on the function of histone acetylation in hypothalamic neurons. The first evidence emerged from studies on the (NAD+)-dependent class III deacetylase sirtuin 1 (SIRT1). This particular enzyme regulates gene expression by deacetylation of proteins including transcription factors and histones. Importantly, SIRT1 levels are high in the hypothalamus including the ARC and VMH [132]. SIRT1 in the hypothalamus is believed to act as a nutrient sensor, as lack of Sirt1 in SF1 or POMC neurons causes hypersensitivity to high-fat diet and decreased energy expenditure [133][134]. On the contrary, in orexigenic AgRP neurons SIRT1 deficiency suppresses food intake on a standard diet [135][136]. Interestingly, Sirt1 expression was also shown to be affected by aging in the ARC [137], and it has been suggested that conditional knock-in of Sirt1 in AgRP and POMC neurons could protect against aging-associated obesity by inhibiting feeding and stimulating energy expenditure [138]. Nevertheless, the data accumulated so far on the role of SIRT1 in the hypothalamus do not distinguish if its action is mediated through chromatin remodeling or other processes.
–
Diet insults seem to modulate the hypothalamic expression of several HDACs, such as Hdac3, Hdac4 and Hdac5 [139]. Indeed, HDAC5 is necessary for correct leptin signaling in hypothalamic neurons. Specifically, HDAC5 regulates the localization of STAT3, a crucial transcription factor that mediates leptin signaling in neurons [140].
–
Hypothalamic ncRNAs and energy balance control
Many recent studies have suggested that miRNAs might be important regulators of energy balance by modulating the melanocortin system. The first evidence that miRNAs are involved in the hypothalamic control of energy balance came from the observation that expression of Dicer, a key gene for miRNAs maturation, is modulated by nutritional status in the hypothalamus [141]. Interestingly, most POMC and AgRP neurons express Dicer. Deletion of Dicer in POMC neurons causes post-natal neurodegeneration resulting in increased appetite, obesity and T2D (Figure 3) [141][142][143]. In agreement with these observations, brain- and ARC-specific deletion of Dicer causes similar metabolic alterations [144][145]. A recent study has shown that miR-103/107 is potentially involved in the maturation of hypothalamic Pomc progenitors [143]. These observations were in accordance with previous studies reporting the importance of miRNAs in neuronal development [146]. In fact, many hypothalamic miRNAs are expressed during development in mice and pig [147][148]. In the last years, significant effort has been made to identify hypothalamus-specific miRNAs. So far, it has been shown that expression of let-7c, miR-7a, miR-7b, miR-124a, miR-125a, miR-136, miR-138, miR-212, miR-338, miR-451, mir-200a/b and mir-429 is enriched in the hypothalamus [149][150][151][152]. Among them, miR-7a displays an interesting pattern of expression that seems to be specific for AgRP and POMC neurons, but its exact function remains elusive [149].
–
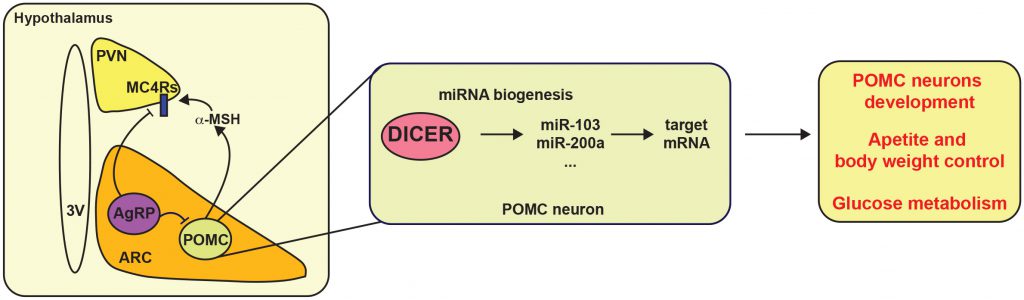 | FIGURE 3: Hypothalamic miRNAs control energy balance. POMC and AgRP neurons from the arcuate nucleus (ARC) send projections to the paraventricular nucleus (PVN) to control energy balance. Dicer and several miRNAs, such as miR-103 and miR-200a, have been proposed to control appetite and body weight likely via POMC neurons. Additionally, miRNA biogenesis is also important for the development of POMC neurons. Melanocortin receptor 4 (MC4R). |
–
In the hypothalamus, the physiological role of lncRNAs remain elusive and only few studies have focused on them. Brain cytoplasmic RNA 1 (BC1) has been well characterized in neurosecretory axon terminals from the hypothalamus and it regulates protein translation by its binding to the ribosome [153]. Many other lncRNAs have been found to be expressed exclusively in the hypothalamus, yet their function remains completely unknown [154][155]. One recent study has investigated the relationship between energy availability and lncRNAs in the hypothalamus, revealing that the pattern of expression of many lncRNA is regulated by fasting [156]. Recently, a study in rodents has highlighted the importance of the Snord116 genomic cluster, a locus encoding multiple ncRNAs, in the hyperphagia observed in Prader-Willi syndrome [157] These data have provided notable information, yet one of the biggest challenges in the field will be to elucidate their precise functions in energy balance control.
CONCLUSIONS AND FUTURE DIRECTIONS
Obesity is the result of disrupted energy balance, which is partially the consequence of alterations in the hypothalamic melanocortin circuitry. Indeed, various obesity susceptibility genes have been identified and some of them belong to the central melanocortin system. Therefore, a better understanding of the precise mechanisms implicated in the melanocortin control of energy balance is a fundamental requisite for the development of more effective anti-obesity therapeutic strategies. Importantly, genetics alone cannot explain the current obesity epidemics. Overfeeding and the prevailing obesogenic environment can impair the sophisticated hypothalamic circuits that regulate energy homeostasis, and current evidence underscores the importance of epigenetic gene regulation in this process. Indeed, metabolites derived from the diet are necessary for the function of many chromatin modifying enzymes. The reversible nature of most epigenetic modifications makes them very attractive targets for possible anti-obesity intervention and prevention strategies.
–
The hypothalamus is a complex neuronal network with a remarkable variety of cell populations. It is therefore very likely that AgRP and POMC neurons might have exclusive epigenomes. To date, analysis of epigenetic signatures has been performed in specific gene promoters and whole hypothalamic samples. This represents a major limitation, as it might mask the diversity of cell-specific epigenetic marks. New methodologies such as single-cell technology or laser dissection have been proposed to tackle this limitation. However, the high cost or low yield of these approaches do not overcome this challenge. In moving forward, more studies are necessary to elucidate the role of epigenetics upon appetite and energy balance control in a neuron-specific manner. These answers will be crucial, not only to improve our understanding of the gene-environment interactions, but also for the development of potential epigenetic-based future therapies aimed at controlling food intake and body weight.
REFERENCES
- (NCD-RisC) N (2016). Trends in adult body-mass index in 200 countries from 1975 to 2014: a pooled analysis of 1698 population-based measurement studies with 19·2 million participants. Lancet Lond Engl 387(10026): 1377–96. doi: 10.1016/s0140-6736(16)30054-x
- Calle EE, Rodriguez C, Walker-Thurmond K, and Thun MJ (2003). Overweight, Obesity, and Mortality from Cancer in a Prospectively Studied Cohort of U.S. Adults. New Engl J Medicine 348(17): 1625–1638. doi: 10.1056/nejmoa021423
- Isomaa B, Almgren P, Tuomi T, Forsén B, Lahti K, Nissén M, Taskinen M-R, and Groop L (2001). Cardiovascular Morbidity and Mortality Associated With the Metabolic Syndrome. Diabetes Care 24(4): 683–689. doi: 10.2337/diacare.24.4.683
- Cuschieri S, and Mamo J (2016). Getting to grips with the obesity epidemic in Europe. Sage Open Med 4: 2050312116670406. doi: 10.1177/2050312116670406
- Lette M, Bemelmans W, Breda J, Slobbe L, Dias J, and Boshuizen H (2016). Health care costs attributable to overweight calculated in a standardized way for three European countries. European J Heal Econ 17(1): 61–69. doi: 10.1007/s10198-014-0655-8
- Hall KD, Heymsfield SB, Kemnitz JW, Klein S, Schoeller DA, and Speakman JR (2012). Energy balance and its components: implications for body weight regulation. Am J Clin Nutrition 95(4): 989–994. doi: 10.3945/ajcn.112.036350
- Nadal A, Quesada I, Tudurí E, Nogueiras R, and Alonso-Magdalena P (2017). Endocrine-disrupting chemicals and the regulation of energy balance. Nat Rev Endocrinol 13(9): 536–546. doi: 10.1038/nrendo.2017.51
- Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, Griffin NW, Lombard V, Henrissat B, Bain JR, Muehlbauer MJ, Ilkayeva O, Semenkovich CF, Funai K, Hayashi DK, Lyle BJ, Martini MC, Ursell LK, Clemente JC, Treuren W, Walters WA, Knight R, Newgard CB, Heath AC, and Gordon JI (2013). Gut Microbiota from Twins Discordant for Obesity Modulate Metabolism in Mice. Science 341(6150): 1241214. doi: 10.1126/science.1241214
- Santacruz A, Marcos A, Wärnberg J, Martí A, Martin-Matillas M, Campoy C, Moreno LA, Veiga O, Redondo-Figuero C, Garagorri JM, Azcona C, Delgado M, García-Fuentes M, Collado MC, Sanz Y, and Group E (2009). Interplay Between Weight Loss and Gut Microbiota Composition in Overweight Adolescents. Obesity 17(10): 1906–1915. doi: 10.1038/oby.2009.112
- Sinha R, and Jastreboff AM (2013). Stress as a Common Risk Factor for Obesity and Addiction. Biol Psychiat 73(9): 827–835. doi: 10.1016/j.biopsych.2013.01.032
- Adam TC, and Epel ES (2007). Stress, eating and the reward system. Physiol Behav 91(4): 449–458. doi: 10.1016/j.physbeh.2007.04.011
- Waterland RA (2014). Epigenetic Mechanisms Affecting Regulation of Energy Balance: Many Questions, Few Answers. Nutrition 34(1): 337–355. doi: 10.1146/annurev-nutr-071813-105315
- Hall KD, Sacks G, Chandramohan D, Chow CC, Wang CY, Gortmaker SL, and Swinburn BA (2011). Quantification of the effect of energy imbalance on bodyweight. Lancet 378(9793): 826–837. doi: 10.1016/s0140-6736(11)60812-x
- Montague CT, Farooqi SI, Whitehead JP, Soos MA, Rau H, Wareham NJ, Sewter CP, Digby JE, Mohammed SN, Hurst JA, Cheetham CH, Earley AR, Barnett AH, Prins JB, and O'Rahilly S (1997). Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature 387(6636): 43185. doi: 10.1038/43185
- Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, and Friedman JM (1994). Positional cloning of the mouse obese gene and its human homologue. Nature 372(6505): 425–432. doi: 10.1038/372425a0
- Yeo G, Farooqi SI, Aminian S, Halsall DJ, Stanhope RG, and O'Rahilly S (1998). A frameshift mutation in MC4R associated with dominantly inherited human obesity. Nat Genet 20(2): 111–112. doi: 10.1038/2404
- Krude H, Biebermann H, Luck W, Horn R, Brabant G, and Grüters A (1998). Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat Genet 19(2): 155–157. doi: 10.1038/509
- Bell CG, Walley AJ, and Froguel P (2005). The genetics of human obesity. Nat Rev Genet 6(3): nrg1556. doi: 10.1038/nrg1556
- Locke AE et al. (2015). Genetic studies of body mass index yield new insights for obesity biology. Nature 518(7538): 197. doi: 10.1038/nature14177
- Shungin D et al. (2015). New genetic loci link adipose and insulin biology to body fat distribution. Nature 518(7538): 187. doi: 10.1038/nature14132
- Akiyama M, Okada Y, Kanai M, Takahashi A, Momozawa Y, Ikeda M, Iwata N, Ikegawa S, Hirata M, Matsuda K, Iwasaki M, Yamaji T, Sawada N, Hachiya T, Tanno K, Shimizu A, Hozawa A, Minegishi N, Tsugane S, Yamamoto M, Kubo M, and Kamatani Y (2017). Genome-wide association study identifies 112 new loci for body mass index in the Japanese population. Nat Genet 49(10): 1458–1467. doi: 10.1038/ng.3951
- Butruille L, Marousez L, Pourpe C, Oger F, Lecoutre S, Catheline D, Görs S, Metges CC, Guinez C, Laborie C, Deruelle P, Eeckhoute J, Breton C, Legrand P, Lesage J, and Eberlé D (2019). Maternal high-fat diet during suckling programs visceral adiposity and epigenetic regulation of adipose tissue stearoyl-CoA desaturase-1 in offspring. Int J Obesity. doi: 10.1038/s41366-018-0310-z
- Godfrey KM, Reynolds RM, Prescott SL, Nyirenda M, Jaddoe VW, Eriksson JG, and Broekman BF (2017). Influence of maternal obesity on the long-term health of offspring. Lancet Diabetes Endocrinol 5(1): 53–64. doi: 10.1016/s2213-8587(16)30107-3
- Reynolds R, Osmond C, Phillips D, and Godfrey K (2010). Maternal BMI, parity, and pregnancy weight gain: influences on offspring adiposity in young adulthood. J Clin Endocrinol Metabolism 95(12): 5365–9. doi: 10.1210/jc.2010-0697
- Ravelli A, van der Meulen J, Michels R, Osmond C, Barker D, Hales C, and Bleker O (1998). Glucose tolerance in adults after prenatal exposure to famine. Lancet 351(9097): 173–177. doi: 10.1016/s0140-6736(97)07244-9
- Barker D (2006). The Developmental Origins of Insulin Resistance. Horm Res Paediat 64(Suppl 3): 2–7. doi: 10.1159/000089311
- Vickers MH, Breier BH, Cutfield WS, Hofman PL, and Gluckman PD (2000). Fetal origins of hyperphagia, obesity, and hypertension and postnatal amplification by hypercaloric nutrition. Am J Physiol-endoc M 279(1): E83–E87. doi: 10.1152/ajpendo.2000.279.1.e83
- Yura S, Itoh H, Sagawa N, Yamamoto H, Masuzaki H, Nakao K, Kawamura M, Takemura M, Kakui K, Ogawa Y, and Fujii S (2005). Role of premature leptin surge in obesity resulting from intrauterine undernutrition. Cell Metab 1(6): 371–378. doi: 10.1016/j.cmet.2005.05.005
- Breton C, Lukaszewski M-A, Risold P-Y, Enache M, Guillemot J, Rivière G, Delahaye F, Lesage J, Dutriez-Casteloot I, Laborie C, and Vieau D (2009). Maternal prenatal undernutrition alters the response of POMC neurons to energy status variation in adult male rat offspring. Am J Physiol-endoc M 296(3): E462–E472. doi: 10.1152/ajpendo.90740.2008
- López M, Seoane L, Tovar S, García M, Nogueiras R, Diéguez C, and Señarís R (2005). A possible role of neuropeptide Y, agouti-related protein and leptin receptor isoforms in hypothalamic programming by perinatal feeding in the rat. Diabetologia 48(1): 140–148. doi: 10.1007/s00125-004-1596-z
- Shin B, Dai Y, Thamotharan M, Gibson CL, and Devaskar SU (2012). Pre- and postnatal calorie restriction perturbs early hypothalamic neuropeptide and energy balance. J Neurosci Res 90(6): 1169–1182. doi: 10.1002/jnr.23013
- Bouret SG, Gorski JN, Patterson CM, Chen S, Levin BE, and Simerly RB (2008). Hypothalamic Neural Projections Are Permanently Disrupted in Diet-Induced Obese Rats. Cell Metab 7(2): 179–185. doi: 10.1016/j.cmet.2007.12.001
- Chen H, Simar D, Lambert K, Mercier J, and Morris MJ (2008). Maternal and Postnatal Overnutrition Differentially Impact Appetite Regulators and Fuel Metabolism. Endocrinology 149(11): 5348–5356. doi: 10.1210/en.2008-0582
- Vogt MC, Paeger L, Hess S, Steculorum SM, Awazawa M, Hampel B, Neupert S, Nicholls HT, Mauer J, Hausen CA, Predel R, Kloppenburg P, Horvath TL, and Brüning JC (2014). Neonatal Insulin Action Impairs Hypothalamic Neurocircuit Formation in Response to Maternal High-Fat Feeding. Cell 156(3): 495–509. doi: 10.1016/j.cell.2014.01.008
- Steculorum SM, Collden G, Coupe B, Croizier S, Lockie S, Andrews ZB, Jarosch F, Klussmann S, and Bouret SG (2015). Neonatal ghrelin programs development of hypothalamic feeding circuits. J Clin Invest 125(2): 846–858. doi: 10.1172/jci73688
- Chen Y, Lin Y-C, Kuo T-W, and Knight ZA (2015). Sensory Detection of Food Rapidly Modulates Arcuate Feeding Circuits. Cell 160(5): 829–41. doi: 10.1016/j.cell.2015.01.033
- Beutler LR, Chen Y, Ahn JS, Lin Y-C, Essner RA, and Knight ZA (2017). Dynamics of Gut-Brain Communication Underlying Hunger. Neuron 96(2): 461-475.e5. doi: 10.1016/j.neuron.2017.09.043
- Timper K, and Brüning JC (2017). Hypothalamic circuits regulating appetite and energy homeostasis: pathways to obesity. Dis Model Mech 10(6): 679–689. doi: 10.1242/dmm.026609
- Hsu TM, Hahn JD, Konanur VR, Lam A, and Kanoski SE (2015). Hippocampal GLP-1 Receptors Influence Food Intake, Meal Size, and Effort-Based Responding for Food through Volume Transmission. Neuropsychopharmacol 40(2): 327. doi: 10.1038/npp.2014.175
- Cone R, Cowley M, Butler A, Fan W, Marks D, and Low M (2001). The arcuate nucleus as a conduit for diverse signals relevant to energy homeostasis. Int J Obesity 25(S5): 0801913. doi: 10.1038/sj.ijo.0801913
- Betley NJ, Cao Z, Ritola KD, and Sternson SM (2013). Parallel, Redundant Circuit Organization for Homeostatic Control of Feeding Behavior. Cell 155(6): 1337–50. doi: 10.1016/j.cell.2013.11.002
- Gropp E, Shanabrough M, Borok E, Xu AW, Janoschek R, Buch T, Plum L, Balthasar N, Hampel B, Waisman A, Barsh GS, Horvath TL, and Brüning JC (2005). Agouti-related peptide–expressing neurons are mandatory for feeding. Nat Neurosci 8(10): 1289–1291. doi: 10.1038/nn1548
- Ollmann MM, Wilson BD, Yang Y-K, Kerns JA, Chen Y, Gantz I, and Barsh GS (1997). Antagonism of Central Melanocortin Receptors in Vitro and in Vivo by Agouti-Related Protein. Science 278(5335): 135–138. doi: 10.1126/science.278.5335.135
- Fan W, Boston BA, Kesterson RA, Hruby VJ, and Cone RD (1997). Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature 385(6612): 385165a0. doi: 10.1038/385165a0
- Garfield AS, Li C, Madara JC, Shah BP, Webber E, Steger JS, Campbell JN, Gavrilova O, Lee CE, Olson DP, Elmquist JK, Tannous BA, Krashes MJ, and Lowell BB (2015). A neural basis for melanocortin-4 receptor-regulated appetite. Nat Neurosci 18(6): 863–871. doi: 10.1038/nn.4011
- Adrian T, Allen J, Bloom S, Ghatei M, Rossor M, Roberts G, Crow T, Tatemoto K, and Polak J (1983). Neuropeptide Y distribution in human brain. Nature 306(5943): 584–586. doi: 10.1038/306584a0
- Stanley B, and Leibowitz S (1985). Neuropeptide Y injected in the paraventricular hypothalamus: a powerful stimulant of feeding behavior. Proc National Acad Sci 82(11): 3940–3943. doi: 10.1073/pnas.82.11.3940
- Small C, Liu Y, Stanley S, Connoley I, Kennedy A, Stock M, and Bloom S (2003). Chronic CNS administration of Agouti-related protein (Agrp) reduces energy expenditure. Int J Obesity 27(4): 0802253. doi: 10.1038/sj.ijo.0802253
- Qian S, Chen H, Weingarth D, Trumbauer ME, Novi DE, Guan X, Yu H, Shen Z, Feng Y, Frazier E, Chen A, Camacho RE, Shearman LP, Gopal-Truter S, MacNeil DJ, der Ploeg LH, and Marsh DJ (2002). Neither Agouti-Related Protein nor Neuropeptide Y Is Critically Required for the Regulation of Energy Homeostasis in Mice. Mol Cell Biol 22(14): 5027–5035. doi: 10.1128/mcb.22.14.5027-5035.2002
- Luquet S, Perez FA, Hnasko TS, and Palmiter RD (2005). NPY/AgRP Neurons Are Essential for Feeding in Adult Mice but Can Be Ablated in Neonates. Science 310(5748): 683–685. doi: 10.1126/science.1115524
- Krashes MJ, Koda S, Ye C, Rogan SC, Adams AC, Cusher DS, Maratos-Flier E, Roth BL, and Lowell BB (2011). Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J Clin Invest. 121(4): 1424–1428. doi: 10.1172/jci46229
- Aponte Y, Atasoy D, and ernson S (2011). AGRP neurons are sufficient to orchestrate feeding behavior rapidly and without training. Nat Neurosci 14(3): 351. doi: 10.1038/nn.2739
- Atasoy D, Betley NJ, Su HH, and Sternson SM (2012). Deconstruction of a neural circuit for hunger. Nature 488(7410): 172. doi: 10.1038/nature11270
- Tong Q, Ye C-P, Jones JE, Elmquist JK, and Lowell BB (2008). Synaptic release of GABA by AgRP neurons is required for normal regulation of energy balance. Nat Neurosci 11(9): nn.2167. doi: 10.1038/nn.2167
- Krashes MJ, Shah BP, Koda S, and Lowell BB (2013). Rapid versus Delayed Stimulation of Feeding by the Endogenously Released AgRP Neuron Mediators GABA, NPY, and AgRP. Cell Metab 18(4): 588–95. doi: 10.1016/j.cmet.2013.09.009
- Wu Q, Boyle MP, and Palmiter RD (2009). Loss of GABAergic Signaling by AgRP Neurons to the Parabrachial Nucleus Leads to Starvation. Cell 137(7): 1225–1234. doi: 10.1016/j.cell.2009.04.022
- Wu Q, Clark MS, and Palmiter RD (2012). Deciphering a neuronal circuit that mediates appetite. Nature 483(7391): 594. doi: 10.1038/nature10899
- Waterson MJ, and Horvath TL (2015). Neuronal Regulation of Energy Homeostasis: Beyond the Hypothalamus and Feeding. Cell Metab 22(6): 962–970. doi: 10.1016/j.cmet.2015.09.026
- Balthasar N, Dalgaard LT, Lee CE, Yu J, Funahashi H, Williams T, Ferreira M, Tang V, McGovern RA, Kenny CD, Christiansen LM, Edelstein E, Choi B, Boss O, Aschkenasi C, Zhang C, Mountjoy K, Kishi T, Elmquist JK, and Lowell BB (2005). Divergence of Melanocortin Pathways in the Control of Food Intake and Energy Expenditure. Cell 123(3): 493–505. doi: 10.1016/j.cell.2005.08.035
- Padilla SL, Reef D, and Zeltser LM (2012). Defining POMC Neurons Using Transgenic Reagents: Impact of Transient Pomc Expression in Diverse Immature Neuronal Populations. Endocrinology 153(3): 1219–1231. doi: 10.1210/en.2011-1665
- Mountjoy K, Robbins L, Mortrud M, and Cone R (1992). The cloning of a family of genes that encode the melanocortin receptors. Science 257(5074): 1248–1251. doi: 10.1126/science.1325670
- Mountjoy K, Mortrud M, Low M, Simerly R, and Cone R (1994). Localization of the melanocortin-4 receptor (MC4-R) in neuroendocrine and autonomic control circuits in the brain. Mol Endocrinol 8(10): 1298–1308. doi: 10.1210/me.8.10.1298
- Shimizu H, Shargill NS, Bray GA, Yen TT, and Gesellchen PD (1989). Effects of MSH on food intake, body weight and coat color of the yellow obese mouse. Life Sci 45(6): 543–552. doi: 10.1016/0024-3205(89)90105-7
- Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD, Smith FJ, Campfield LA, Burn P, and Lee F (1997). Targeted Disruption of the Melanocortin-4 Receptor Results in Obesity in Mice. Cell 88(1): 131–141. doi: 10.1016/s0092-8674(00)81865-6
- Yaswen L, Diehl N, Brennan MB, and Hochgeschwender U (1999). Obesity in the mouse model of pro-opiomelanocortin deficiency responds to peripheral melanocortin. Nat Med 5(9): nm0999_1066. doi: 10.1038/12506
- Kim G, Shi G, Somlo D, Haataja L, Song S, Long Q, Nillni EA, Low MJ, Arvan P, Myers MG, and Qi L (2018). Hypothalamic ER–associated degradation regulates POMC maturation, feeding, and age-associated obesity. J Clin Invest 128(3): 1125–1140. doi: 10.1172/jci96420
- Koch M, Varela L, Kim J, Kim J, Hernández-Nuño F, Simonds SE, Castorena CM, Vianna CR, Elmquist JK, Morozov YM, Rakic P, Bechmann I, Cowley MA, Szigeti-Buck K, Dietrich MO, Gao X-B, Diano S, and Horvath TL (2015). Hypothalamic POMC neurons promote cannabinoid-induced feeding. Nature 519(7541): 45. doi: 10.1038/nature14260
- Zhan C, Zhou J, Feng Q, Zhang J, Lin S, Bao J, Wu P, and Luo M (2013). Acute and Long-Term Suppression of Feeding Behavior by POMC Neurons in the Brainstem and Hypothalamus, Respectively. J Neurosci 33(8): 3624–3632. doi: 10.1523/jneurosci.2742-12.2013
- Steculorum SM, Ruud J, Karakasilioti I, Backes H, Ruud L, Timper K, Hess ME, Tsaousidou E, Mauer J, Vogt MC, Paeger L, Bremser S, Klein AC, Morgan DA, Frommolt P, Brinkkötter PT, Hammerschmidt P, Benzing T, Rahmouni K, Wunderlich TF, Kloppenburg P, and Brüning JC (2016). AgRP Neurons Control Systemic Insulin Sensitivity via Myostatin Expression in Brown Adipose Tissue. Cell. 165(1): 125–38. doi: 10.1016/j.cell.2016.02.044
- Turton M, O'Shea D, Gunn I, Beak S, Edwards C, Meeran K, Choi S, Taylor G, Heath M, Lambert P, Wilding J, Smith D, Ghatei M, Herbert J, and Bloom S (1996). A role for glucagon-like peptide-1 in the central regulation of feeding. Nature 379(6560): 69–72. doi: 10.1038/379069a0
- Tschöp M, Smiley DL, and Heiman ML (2000). Ghrelin induces adiposity in rodents. Nature 407(6806): 908. doi: 10.1038/35038090
- Brüning JC, Gautam D, Burks DJ, Gillette J, Schubert M, Orban PC, Klein R, Krone W, Müller-Wieland D, and Kahn RC (2000). Role of Brain Insulin Receptor in Control of Body Weight and Reproduction. Science 289(5487): 2122–2125. doi: 10.1126/science.289.5487.2122
- Batterham RL, Cowley MA, Small CJ, Herzog H, Cohen MA, Dakin CL, Wren AM, Brynes AE, Low MJ, Ghatei MA, Cone RD, and Bloom SR (2002). Gut hormone PYY3-36 physiologically inhibits food intake. Nature 418(6898): 650. doi: 10.1038/nature00887
- Mosialou I, Shikhel S, Liu J-M, Maurizi A, Luo N, He Z, Huang Y, Zong H, Friedman RA, Barasch J, Lanzano P, Deng L, Leibel RL, Rubin M, Nickolas T, Chung W, Zeltser LM, Williams KW, Pessin JE, and Kousteni S (2017). MC4R-dependent suppression of appetite by bone-derived lipocalin 2. Nature 543(7645): 385. doi: 10.1038/nature21697
- Claret M, Smith MA, Batterham RL, Selman C, Choudhury AI, Fryer L, Clements M, Al-Qassab H, Heffron H, Xu AW, Speakman JR, Barsh GS, Viollet B, Vaulont S, Ashford M, Carling D, and Withers DJ (2007). AMPK is essential for energy homeostasis regulation and glucose sensing by POMC and AgRP neurons. J Clin Invest 117(8): 2325–2336. doi: 10.1172/jci31516
- Minokoshi Y, Alquier T, Furukawa N, Kim Y-B, Lee A, Xue B, Mu J, Foufelle F, Ferré P, Birnbaum MJ, Stuck BJ, and Kahn BB (2004). AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature 428(6982): 569. doi: 10.1038/nature02440
- López M, Lage R, Saha AK, Pérez-Tilve D, Vázquez MJ, Varela L, Sangiao-Alvarellos S, Tovar S, Raghay K, Rodríguez-Cuenca S, Deoliveira RM, Castañeda T, Datta R, Dong JZ, Culler M, Sleeman MW, Álvarez CV, Gallego R, Lelliott CJ, Carling D, Tschöp MH, Diéguez C, and Vidal-Puig A (2008). Hypothalamic Fatty Acid Metabolism Mediates the Orexigenic Action of Ghrelin. Cell Metab 7(5): 389–399. doi: 10.1016/j.cmet.2008.03.006
- López M, Nogueiras R, Tena-Sempere M, and Diéguez C (2016). Hypothalamic AMPK: a canonical regulator of whole-body energy balance. Nat Rev Endocrinol 12(7): 421–432. doi: 10.1038/nrendo.2016.67
- Jr LE, and Waddington C (1943). Organisers and Genes. Am Midl Nat 30(3): 811. doi: 10.2307/2421224
- Berger SL, Kouzarides T, Shiekhattar R, and Shilatifard A (2009). An operational definition of epigenetics. Gene Dev 23(7): 781–783. doi: 10.1101/gad.1787609
- HOTCHKISS R (1948). The quantitative separation of purines, pyrimidines, and nucleosides by paper chromatography. J Biological Chem 175(1): 315–32. 18873306
- Hermann A, Goyal R, and Jeltsch A (2004). The Dnmt1 DNA-(cytosine-C5)-methyltransferase Methylates DNA Processively with High Preference for Hemimethylated Target Sites. J Biol Chem 279(46): 48350–48359. doi: 10.1074/jbc.m403427200
- Okano M, Bell DW, Haber DA, and Li E (1999). DNA Methyltransferases Dnmt3a and Dnmt3b Are Essential for De Novo Methylation and Mammalian Development. Cell 99(3): 247–257. doi: 10.1016/s0092-8674(00)81656-6
- Ng H-H, and Adrian B (1999). DNA methylation and chromatin modification. Curr Opin Genet Dev 9(2): 158–163. doi: 10.1016/s0959-437x(99)80024-0
- Schübeler D (2015). Function and information content of DNA methylation. Nature 517(7534): 321–326. doi: 10.1038/nature14192
- Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, and Zhang Y (2011). Tet Proteins Can Convert 5-Methylcytosine to 5-Formylcytosine and 5-Carboxylcytosine. Science 333(6047): 1300–1303. doi: 10.1126/science.1210597
- Tahiliani M, Koh K, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, and Rao A (2009). Conversion of 5-Methylcytosine to 5-Hydroxymethylcytosine in Mammalian DNA by MLL Partner TET1. Science 324(5929): 930–935. doi: 10.1126/science.1170116
- Feng J, Zhou Y, Campbell SL, Le T, Li E, Sweatt DJ, Silva AJ, and Fan G (2010). Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat Neurosci 13(4): 423. doi: 10.1038/nn.2514
- Münzel M, Globisch D, Brückl T, Wagner M, Welzmiller V, Michalakis S, Müller M, Biel M, and Carell T (2010). Quantification of the Sixth DNA Base Hydroxymethylcytosine in the Brain. Angewandte Chemie Int Ed 49(31): 5375–5377. doi: 10.1002/anie.201002033
- Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND, Lucero J, Huang Y, Dwork AJ, Schultz MD, Yu M, Tonti-Filippini J, Heyn H, Hu S, Wu JC, Rao A, Esteller M, He C, Haghighi FG, Sejnowski TJ, Behrens MM, and Ecker JR (2013). Global Epigenomic Reconfiguration During Mammalian Brain Development. Science 341(6146): 1237905. doi: 10.1126/science.1237905
- Lister R, Pelizzola M, Dowen RH, Hawkins DR, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo Q-M, Edsall L, Antosiewicz-Bourget J, Stewart R, Ruotti V, Millar HA, Thomson JA, Ren B, and Ecker JR (2009). Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462(7271): 315. doi: 10.1038/nature08514
- Ziller MJ, Müller F, Liao J, Zhang Y, Gu H, Bock C, Boyle P, Epstein CB, Bernstein BE, Lengauer T, Gnirke A, and Meissner A (2011). Genomic Distribution and Inter-Sample Variation of Non-CpG Methylation across Human Cell Types. Plos Genet 7(12): e1002389. doi: 10.1371/journal.pgen.1002389
- Luger K, Mäder AW, Richmond RK, Sargent DF, and Richmond TJ (1997). Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389(6648): 251. doi: 10.1038/38444
- Zhao Y, and Garcia BA (2015). Comprehensive Catalog of Currently Documented Histone Modifications. Csh Perspect Biol 7(9): a025064. doi: 10.1101/cshperspect.a025064
- Allfrey V, Faulkner R, and Mirsky A (1964). Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc National Acad Sci 51(5): 786–794. doi: 10.1073/pnas.51.5.786
- Roth SY, Denu JM, and Allis DC (2001). HISTONE ACETYLTRANSFERASES. Annu Rev Biochem 70(1): 81–120. doi: 10.1146/annurev.biochem.70.1.81
- Yang X-J, and Seto E (2007). HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene 26(37): 1210599. doi: 10.1038/sj.onc.1210599
- Bannister AJ, and Kouzarides T (2011). Regulation of chromatin by histone modifications. Cell Res 21(3): 381. doi: 10.1038/cr.2011.22
- Byvoet P, Shepherd GR, Hardin JM, and Noland BJ (1972). The distribution and turnover of labeled methyl groups in histone fractions of cultured mammalian cells. Arch Biochem Biophys 148(2): 558–567. doi: 10.1016/0003-9861(72)90174-9
- Patel DJ (2016). A Structural Perspective on Readout of Epigenetic Histone and DNA Methylation Marks. Csh Perspect Biol 8(3): a018754. doi: 10.1101/cshperspect.a018754
- Hyun K, Jeon J, Park K, and Kim J (2017). Writing, erasing and reading histone lysine methylations. Exp Mol Medicine 49(4): e324. doi: 10.1038/emm.2017.11
- Rea S, Eisenhaber F, O'Carroll D, Strahl BD, Sun Z-W, Schmid M, Opravil S, Mechtler K, Ponting CP, Allis DC, and Jenuwein T (2000). Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406(6796): 593. doi: 10.1038/35020506
- Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, and Shi Y (2004). Histone Demethylation Mediated by the Nuclear Amine Oxidase Homolog LSD1. Cell 119(7): 941–53. doi: 10.1016/j.cell.2004.12.012
- Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, and Zhang Y (2005). Histone demethylation by a family of JmjC domain-containing proteins. Nature 439(7078): nature04433. doi: 10.1038/nature04433
- Gebert LF, and MacRae IJ (2018). Regulation of microRNA function in animals. Nat Rev Mol Cell Bio 20(1): 1–17. doi: 10.1038/s41580-018-0045-7
- Mercer TR, Dinger ME, and Mattick JS (2009). Long non-coding RNAs: insights into functions. Nat Rev Genet 10(3): 155–159. doi: 10.1038/nrg2521
- Ulitsky I, Shkumatava A, Jan CH, Sive H, and Bartel DP (2011). Conserved Function of lincRNAs in Vertebrate Embryonic Development despite Rapid Sequence Evolution. Cell 147(7): 1537–50. doi: 10.1016/j.cell.2011.11.055
- Kuroda A, Rauch TA, Todorov I, Ku H, Al-Abdullah IH, Kandeel F, Mullen Y, Pfeifer GP, and Ferreri K (2009). Insulin Gene Expression Is Regulated by DNA Methylation. Plos One 4(9): e6953. doi: 10.1371/journal.pone.0006953
- Barres R, and Zierath JR (2011). DNA methylation in metabolic disorders. Am J Clin Nutrition 93(4): 897S-900S. doi: 10.3945/ajcn.110.001933
- Portela A, and Esteller M (2010). Epigenetic modifications and human disease. Nat Biotechnol 28(10): 1057. doi: 10.1038/nbt.1685
- Stevens A, Begum G, and White A (2011). Epigenetic changes in the hypothalamic pro-opiomelanocortin gene: A mechanism linking maternal undernutrition to obesity in the offspring? Eur J Pharmacol 660(1): 194–201. doi: 10.1016/j.ejphar.2010.10.111
- Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, and Thompson CB (2009). ATP-Citrate Lyase Links Cellular Metabolism to Histone Acetylation. Science 324(5930): 1076–1080. doi: 10.1126/science.1164097
- McDonnell E, Crown SB, Fox DB, Kitir B, Ilkayeva OR, Olsen CA, Grimsrud PA, and Hirschey MD (2016). Lipids Reprogram Metabolism to Become a Major Carbon Source for Histone Acetylation. Cell Reports 17(6): 1463–1472. doi: 10.1016/j.celrep.2016.10.012
- Bungard D, Fuerth BJ, Zeng P-Y, Faubert B, Maas NL, Viollet B, Carling D, Thompson CB, Jones RG, and Berger SL (2010). Signaling Kinase AMPK Activates Stress-Promoted Transcription via Histone H2B Phosphorylation. Science 329(5996): 1201–1205. doi: 10.1126/science.1191241
- Fulco M, Cen Y, Zhao P, Hoffman EP, McBurney MW, Sauve AA, and Sartorelli V (2008). Glucose Restriction Inhibits Skeletal Myoblast Differentiation by Activating SIRT1 through AMPK-Mediated Regulation of Nampt. Dev Cell 14(5): 661–673. doi: 10.1016/j.devcel.2008.02.004
- Gongol B, Sari I, Bryant T, Rosete G, and Marin T (2018). AMPK: An Epigenetic Landscape Modulator. Int J Mol Sci 19(10): 3238. doi: 10.3390/ijms19103238
- Lu D, Willard D, Patel IR, Kadwell S, Overton L, Kost T, Luther M, Chen W, Woychik RP, Wilkison WO, and Cone RD (1994). Agouti protein is an antagonist of the melanocyte-stimulating-hormone receptor. Nature 371(6500): 371799a0. doi: 10.1038/371799a0
- Wolff GL, Kodell RL, Moore SR, and Cooney CA (1998). Maternal epigenetics and methyl supplements affect agouti gene expression in Avy/a mice. Faseb J 12(11): 949–957. doi: 10.1096/fasebj.12.11.949
- Dolinoy DC, Huang D, and Jirtle RL (2007). Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc National Acad Sci 104(32): 13056–13061. doi: 10.1073/pnas.0703739104
- Begum G, Stevens A, Smith E, Connor K, Challis JR, Bloomfield F, and White A (2012). Epigenetic changes in fetal hypothalamic energy regulating pathways are associated with maternal undernutrition and twinning. Faseb J 26(4): 1694–1703. doi: 10.1096/fj.11-198762
- Schroeder M, Jakovcevski M, Polacheck T, Lebow M, Drori Y, Engel M, Ben-Dor S, and Chen A (2017). A Methyl-Balanced Diet Prevents CRF-Induced Prenatal Stress-Triggered Predisposition to Binge Eating-like Phenotype. Cell Metab 25(6): 1269-1281.e6. doi: 10.1016/j.cmet.2017.05.001
- Plagemann A, Harder T, Brunn M, Harder A, Roepke K, Wittrock-Staar M, Ziska T, Schellong K, Rodekamp E, Melchior K, and Dudenhausen JW (2009). Hypothalamic proopiomelanocortin promoter methylation becomes altered by early overfeeding: an epigenetic model of obesity and the metabolic syndrome. J Physiology 587(20): 4963–4976. doi: 10.1113/jphysiol.2009.176156
- Stevens A, Begum G, Cook A, Connor K, Rumball C, Oliver M, Challis J, Bloomfield F, and White A (2010). Epigenetic Changes in the Hypothalamic Proopiomelanocortin and Glucocorticoid Receptor Genes in the Ovine Fetus after Periconceptional Undernutrition. Endocrinology 151(8): 3652–3664. doi: 10.1210/en.2010-0094
- Cifani C, Bonaventura MV, Pucci M, Giusepponi ME, Romano A, Francesco A, Maccarrone M, and D'Addario C (2015). Regulation of hypothalamic neuropeptides gene expression in diet induced obesity resistant rats: possible targets for obesity prediction? Front Neurosci-switz 9: 187. doi: 10.3389/fnins.2015.00187
- Kuehnen P, Mischke M, Wiegand S, Sers C, Horsthemke B, Lau S, Keil T, Lee Y-A, Grueters A, and Krude H (2012). An Alu Element–Associated Hypermethylation Variant of the POMC Gene Is Associated with Childhood Obesity. Plos Genet 8(3): e1002543. doi: 10.1371/journal.pgen.1002543
- Plagemann A, Roepke K, Harder T, Brunn M, Harder A, Wittrock-Staar M, Ziska T, Schellong K, Rodekamp E, Melchior K, and Dudenhausen JW (2010). Epigenetic malprogramming of the insulin receptor promoter due to developmental overfeeding. J Perinat Med 38(4): 393–400. doi: 10.1515/jpm.2010.051
- Lazzarino G, Andreoli M, Rossetti M, Stoker C, Tschopp M, Luque E, and Ramos J (2017). Cafeteria diet differentially alters the expression of feeding-related genes through DNA methylation mechanisms in individual hypothalamic nuclei. Mol Cell Endocrinol 450: 113–125. doi: 10.1016/j.mce.2017.05.005
- Kohno D, Lee S, Harper MJ, Kim K, Sone H, Sasaki T, Kitamura T, Fan G, and Elmquist JK (2014). Dnmt3a in Sim1 Neurons Is Necessary for Normal Energy Homeostasis. J Neurosci.34(46): 15288–15296. doi: 10.1523/jneurosci.1316-14.2014
- Fyffe SL, Neul JL, Samaco RC, Chao H-T, Ben-Shachar S, Moretti P, McGill BE, Goulding EH, Sullivan E, Tecott LH, and Zoghbi HY (2008). Deletion of Mecp2 in Sim1-Expressing Neurons Reveals a Critical Role for MeCP2 in Feeding Behavior, Aggression, and the Response to Stress. Neuron 59(6): 947–58. doi: 10.1016/j.neuron.2008.07.030
- Wang X, Lacza Z, Sun YE, and Han W (2014). Leptin resistance and obesity in mice with deletion of methyl-CpG-binding protein 2 (MeCP2) in hypothalamic pro-opiomelanocortin (POMC) neurons. Diabetologia 57(1): 236–245. doi: 10.1007/s00125-013-3072-0
- Donohoe DR, and Bultman SJ (2012). Metaboloepigenetics: Interrelationships between energy metabolism and epigenetic control of gene expression. J Cell Physiol 227(9): 3169–3177. doi: 10.1002/jcp.24054
- Ramadori G, Lee CE, Bookout AL, Lee S, Williams KW, Anderson J, Elmquist JK, and Coppari R (2008). Brain SIRT1: Anatomical Distribution and Regulation by Energy Availability. J Neurosci 28(40): 9989–9996. doi: 10.1523/jneurosci.3257-08.2008
- Ramadori G, Fujikawa T, Anderson J, Berglund ED, Frazao R, Michán S, Vianna CR, Sinclair DA, Elias CF, and Coppari R (2011). SIRT1 Deacetylase in SF1 Neurons Protects against Metabolic Imbalance. Cell Metab 14(3): 301–12. doi: 10.1016/j.cmet.2011.06.014
- Ramadori G, Fujikawa T, Fukuda M, Anderson J, Morgan DA, Mostoslavsky R, Stuart RC, Perello M, Vianna CR, Nillni EA, Rahmouni K, and Coppari R (2010). SIRT1 Deacetylase in POMC Neurons Is Required for Homeostatic Defenses against Diet-Induced Obesity. Cell Metab 12(1): 78–87. doi: 10.1016/j.cmet.2010.05.010
- Dietrich MO, Antunes C, Geliang G, Liu Z-W, Borok E, Nie Y, Xu AW, Souza DO, Gao Q, Diano S, Gao X-B, and Horvath TL (2010). Agrp Neurons Mediate Sirt1's Action on the Melanocortin System and Energy Balance: Roles for Sirt1 in Neuronal Firing and Synaptic Plasticity. J Neurosci 30(35): 11815–11825. doi: 10.1523/jneurosci.2234-10.2010
- Çakir I, Perello M, Lansari O, Messier NJ, Vaslet CA, and Nillni EA (2009). Hypothalamic Sirt1 Regulates Food Intake in a Rodent Model System. Plos One 4(12): e8322. doi: 10.1371/journal.pone.0008322
- Lafontaine-Lacasse M, Richard D, and Picard F (2010). Effects of age and gender on Sirt 1 mRNA expressions in the hypothalamus of the mouse. Neurosci Lett 480(1): 1–3. doi: 10.1016/j.neulet.2010.01.008
- Sasaki T, Kikuchi O, Shimpuku M, Susanti V, Yokota-Hashimoto H, Taguchi R, Shibusawa N, Sato T, Tang L, Amano K, Kitazumi T, Kuroko M, Fujita Y, Maruyama J, Lee Y, Kobayashi M, Nakagawa T, Minokoshi Y, Harada A, Yamada M, and Kitamura T (2014). Hypothalamic SIRT1 prevents age-associated weight gain by improving leptin sensitivity in mice. Diabetologia 57(4): 819–831. doi: 10.1007/s00125-013-3140-5
- Funato H, Oda S, Yokofujita J, Igarashi H, and Kuroda M (2011). Fasting and High-Fat Diet Alter Histone Deacetylase Expression in the Medial Hypothalamus. Plos One 6(4): e18950. doi: 10.1371/journal.pone.0018950
- Kabra DG, Pfuhlmann K, García-Cáceres C, Schriever SC, García V, Kebede A, Fuente-Martin E, Trivedi C, Heppner K, Uhlenhaut HN, Legutko B, Kabra UD, Gao Y, Yi C-X, Quarta C, Clemmensen C, Finan B, Müller TD, Meyer CW, Paez-Pereda M, Stemmer K, Woods SC, Perez-Tilve D, Schneider R, Olson EN, Tschöp MH, and Pfluger PT (2016). Hypothalamic leptin action is mediated by histone deacetylase 5. Nat Commun 7(1): 10782. doi: 10.1038/ncomms10782
- Schneeberger M, Altirriba J, García A, Esteban Y, Castaño C, García-Lavandeira M, Alvarez CV, Gomis R, and Claret M (2013). Deletion of miRNA processing enzyme Dicer in POMC-expressing cells leads to pituitary dysfunction, neurodegeneration and development of obesity. Mol Metab 2(2): 74–85. doi: 10.1016/j.molmet.2012.10.001
- Greenman Y, Kuperman Y, Drori Y, Asa SL, Navon I, Forkosh O, Gil S, Stern N, and Chen A (2013). Postnatal ablation of POMC neurons induces an obese phenotype characterized by decreased food intake and enhanced anxiety-like behavior. Mol Endocrinol Baltim Md 27(7): 1091–102. doi: 10.1210/me.2012-1344
- Croizier S, Park S, Maillard J, and Bouret SG (2018). Central Dicer-miR-103/107 controls developmental switch of POMC progenitors into NPY neurons and impacts glucose homeostasis. Elife 7: e40429. doi: 10.7554/elife.40429
- Mang GM, Pradervand S, Du N-H, Arpat A, Preitner F, Wigger L, Gatfield D, and Franken P (2015). A Neuron-Specific Deletion of the MicroRNA-Processing Enzyme DICER Induces Severe but Transient Obesity in Mice. Plos One 10(1): e0116760. doi: 10.1371/journal.pone.0116760
- Vinnikov IA, Hajdukiewicz K, Reymann J, Beneke J, Czajkowski R, Roth LC, Novak M, Roller A, Dörner N, Starkuviene V, Theis FJ, Erfle H, Schütz G, Grinevich V, and Konopka W (2014). Hypothalamic miR-103 Protects from Hyperphagic Obesity in Mice. J Neurosci 34(32): 10659–10674. doi: 10.1523/jneurosci.4251-13.2014
- Díaz NF, Cruz-Reséndiz MS, Flores-Herrera H, García-López G, and Molina-Hernández A (2014). MicroRNAs in central nervous system development. Rev Neuroscience 25(5): 675–686. doi: 10.1515/revneuro-2014-0014
- Zhang L, Cai Z, Wei S, Zhou H, Zhou H, Jiang X, and Xu N (2013). MicroRNA Expression Profiling of the Porcine Developing Hypothalamus and Pituitary Tissue. Int J Mol Sci 14(10): 20326–20339. doi: 10.3390/ijms141020326
- Doubi-Kadmiri S, Benoit C, Benigni X, Beaumont G, Vacher C-M, Taouis M, Baroin-Tourancheau A, and Amar L (2016). Substantial and robust changes in microRNA transcriptome support postnatal development of the hypothalamus in rat. Sci Rep 6(1): 24896. doi: 10.1038/srep24896
- Bak M, Silahtaroglu A, Møller M, Christensen M, Rath MF, Skryabin B, Tommerup N, and Kauppinen S (2008). MicroRNA expression in the adult mouse central nervous system. Rna 14(3): 432–444. doi: 10.1261/rna.783108
- Farh K, Grimson A, Jan C, Lewis BP, Johnston WK, Lim LP, Burge CB, and Bartel DP (2005). The Widespread Impact of Mammalian MicroRNAs on mRNA Repression and Evolution. Science 310(5755): 1817–1821. doi: 10.1126/science.1121158
- Amar L, Benoit C, Beaumont G, Vacher CM, Crepin D, Taouis M, and Baroin-Tourancheau A (2012). MicroRNA expression profiling of hypothalamic arcuate and paraventricular nuclei from single rats using Illumina sequencing technology. J Neurosci Meth 209(1): 134–143. doi: 10.1016/j.jneumeth.2012.05.033
- Crépin D, Benomar Y, Riffault L, Amine H, Gertler A, and Taouis M (2014). The over-expression of miR-200a in the hypothalamus of ob/ob mice is linked to leptin and insulin signaling impairment. Mol Cell Endocrinol 384(1–2): 1–11. doi: 10.1016/j.mce.2013.12.016
- Wang H, Iacoangeli A, Popp S, Muslimov IA, Imataka H, Sonenberg N, Lomakin IB, and Tiedge H (2002). Dendritic BC1 RNA: Functional Role in Regulation of Translation Initiation. J Neurosci 22(23): 10232–10241. doi: 10.1523/jneurosci.22-23-10232.2002
- Ellis BC, Molloy PL, and Graham LD (2012). CRNDE: A Long Non-Coding RNA Involved in CanceR, Neurobiology, and DEvelopment. Frontiers Genetics 3: 270. doi: 10.3389/fgene.2012.00270
- Kawashima M, Tamiya G, Oka A, Hohjoh H, Juji T, Ebisawa T, Honda Y, Inoko H, and Tokunaga K (2006). Genomewide Association Analysis of Human Narcolepsy and a New Resistance Gene. Am J Hum Genetics 79(2): 252–263. doi: 10.1086/505539
- Jiang H, Modise T, Helm R, Jensen RV, and Good DJ (2015). Characterization of the hypothalamic transcriptome in response to food deprivation reveals global changes in long noncoding RNA, and cell cycle response genes. Genes Nutrition 10(6): 48. doi: 10.1007/s12263-015-0496-9
- Polex-Wolf J, Lam B, Larder R, Tadross J, Rimmington D, Bosch F, Cenzano V, Ayuso E, Ma M, Rainbow K, Coll AP, O'Rahilly S, and Yeo G (2018). Hypothalamic loss of Snord116 recapitulates the hyperphagia of Prader-Willi syndrome. J Clin Invest 128(3): 960–969. doi: 10.1172/jci97007
ACKNOWLEDGMENTS
We thank Dr. P. Mera and Dr. R. Had-dad-Tóvolli for valuable suggestions and critical reading of the manuscript. This work has been supported by CERCA Programme/Generalitat de Catalunya. A.O. is supported by the Beatriu de Pinós programme (2016BP00136) from the Generalitat de Catalunya. M.C. is a recipient of a Miguel Servet 2 contract (MSII15/00025) from Instituto de Salud Carlos III and co-financed by Fondo Europeo de Desarrollo Regional (FEDER), Unión Europea, “Una manera de hacer Europa”.
COPYRIGHT
© 2019

The role of epigenetics in hypothalamic energy balance control: implications for obesity by Obri and Claret is licensed under a Creative Commons Attribution 4.0 International License.


