Research Articles:
Cell Stress, Vol. 5, No. 12, pp. 176 - 182; doi: 10.15698/cst2021.12.260
p38 regulates the tumor suppressor PDCD4 via the TSC-mTORC1 pathway
1 Center of Pathobiochemistry and Genetics, Institute of Medical Genetics, Medical University of Vienna, Vienna, Austria.
2 Clinical Division of Endocrinology and Metabolism, Department of Internal Medicine III, Medical University of Vienna, Vienna, Austria.
3 Department of Internal Medicine III, Division of Nephrology and Dialysis, Medical University of Vienna, Vienna, Austria.
Keywords: macrophages, cancer, rapamycin, MK2, PDCD4, p38, mTORC1, TSC1, TSC2.
Abbreviations:
BMDM – bone marrow-derived macrophage;
CK2 – casein kinase 2;
LPS – lipopolysaccharide;
MAPK – mitogen-activated kinase;
MK2 – MAP kinase-activated protein kinase 2;
mTORC1 – mammalian target of rapamycin complex 1;
PDCD4 – programmed cell death protein 4;
PI3K – phosphoinositide 3-kinase;
RSK – ribosomal S6 kinase;
TSC – tuberous sclerosis complex.
Received originally: 21/05/2021 Received in revised form: 08/11/2021
Accepted: 12/11/2021
Published: 23/11/2021
Correspondence:
T. Weichhart, Center of Pathobiochemistry and Genetics, Institute of Medical Genetics, Medical University of Vienna, Vienna, Austria; thomas.weichhart@meduniwien.ac.at
Conflict of interest statement: The authors declare no conflicts of interest.
Please cite this article as: Clarissa Braun, Karl Katholnig, Christopher Kaltenecker, Monika Linke, Nyamdelger Sukhbaatar, Markus Hengstschläger and Thomas Weichhart (2021). p38 regulates the tumor suppressor PDCD4 via the TSC-mTORC1 pathway. Cell Stress 5(12): 176-182. doi: 10.15698/cst2021.12.260
Abstract
Programmed cell death protein 4 (PDCD4) exerts critical functions as tumor suppressor and in immune cells to regulate inflammatory processes. The phosphoinositide 3-kinase (PI3K) promotes degradation of PDCD4 via mammalian target of rapamycin complex 1 (mTORC1). However, additional pathways that may regulate PDCD4 expression are largely ill-defined. In this study, we have found that activation of the mitogen-activated protein kinase p38 promoted degradation of PDCD4 in macrophages and fibroblasts. Mechanistically, we identified a pathway from p38 and its substrate MAP kinase-activated protein kinase 2 (MK2) to the tuberous sclerosis complex (TSC) to regulate mTORC1-dependent degradation of PDCD4. Moreover, we provide evidence that TSC1 and TSC2 regulate PDCD4 expression via an additional mechanism independent of mTORC1. These novel data extend our knowledge of how PDCD4 expression is regulated by stress- and nutrient-sensing pathways.
INTRODUCTION
Programmed cell death protein 4 (PDCD4) is an RNA-binding tumor suppressor protein that is vital for inhibiting carcinogenesis, tumor progression and invasion [1]. Low PDCD4 expression promotes neoplastic transformation [2]. The activity of the PDCD4 protein seems to be mainly determined by its stabilization [3].
–
Recent data showed that PDCD4 is also a modifier of inflammatory processes in macrophages [4][5][6][7]. Cellular PDCD4 levels remain stable throughout the process of monocyte/macrophage differentiation [8], but are upregulated upon starvation or induction of apoptosis [9]. Interestingly, macrophages reduce PDCD4 expression in cancer cells by mTOR-mediated proteasomal degradation [10]. In contrast to starvation, mitogenic signals such as growth factors or pathogen-associated molecules such as lipopolysaccharide (LPS) lead to the ubiquitination of PDCD4 by F-box/WD repeat-containing protein 1A (βTRCP) ubiquitin ligases and its subsequent degradation by the proteasome [11]. Mechanistically, mitogenic signals activate the phosphoinositide 3-kinase (PI3K)-mammalian target of rapamycin complex 1 (mTORC1) pathway. mTORC1 then phosphorylates its substrate ribosomal protein S6 kinase beta-1 (S6K1), which directly phosphorylates PDCD4 as trigger for its ubiquitination and degradation [11]. Inhibition of mTORC1 with rapamycin prevents degradation of PDCD4 [5].
–
Whether other signal transduction pathways in addition to PI3K regulate PDCD4 expression via mTORC1 is largely unknown. We and others have previously found that the mitogen-activated kinase (MAPK) p38α contributes to the activation of mTORC1 [12][13]. Specifically, the p38 substrate MAP kinase-activated protein kinase 2 (MK2) phosphorylates Ser1210 on the tuberous sclerosis complex 2 (TSC2, Tuberin), a negative regulator of mTORC1 signaling, and contributes to inflammatory cytokine expression in macrophages [12]. In the current study, we wanted to investigate whether p38 controls PDCD4 expression.
RESULTS
p38 negatively regulates PDCD4
To study a potential role of p38 on PDCD4, we used the two well-known p38 activators anisomycin and LPS. Anisomycin has an inhibitory effect on protein translation [14], whereas LPS stimulates inflammatory protein synthesis. In bone marrow-derived macrophages (BMDMs) we found that LPS and anisomycin induced the reduction of PDCD4 (Fig. 1A). Interestingly, chemical inhibition of p38 with BIRB796 [15] prevented the LPS- or anisomycin-induced decrease of PDCD4 (Fig. 1A). To genetically corroborate these findings, we analyzed p38α-deficient BMDMs. We detected higher levels of PDCD4 in unstimulated p38α-deficient BMDMs compared to their control cells (Fig. 1B). Of note, PDCD4 was still partially lost in LPS- or anisomycin-stimulated p38α-deficient cells. Moreover, levels of PDCD4 were increased in a macrophage cell line that expressed a catalytic dead mutant of MK2 (K79R) to prevent p38-mediated phosphorylation and activation (Fig. 1C). These data suggest that p38 and its substrate MK2 negatively regulate the expression of PDCD4 in macrophages.
–
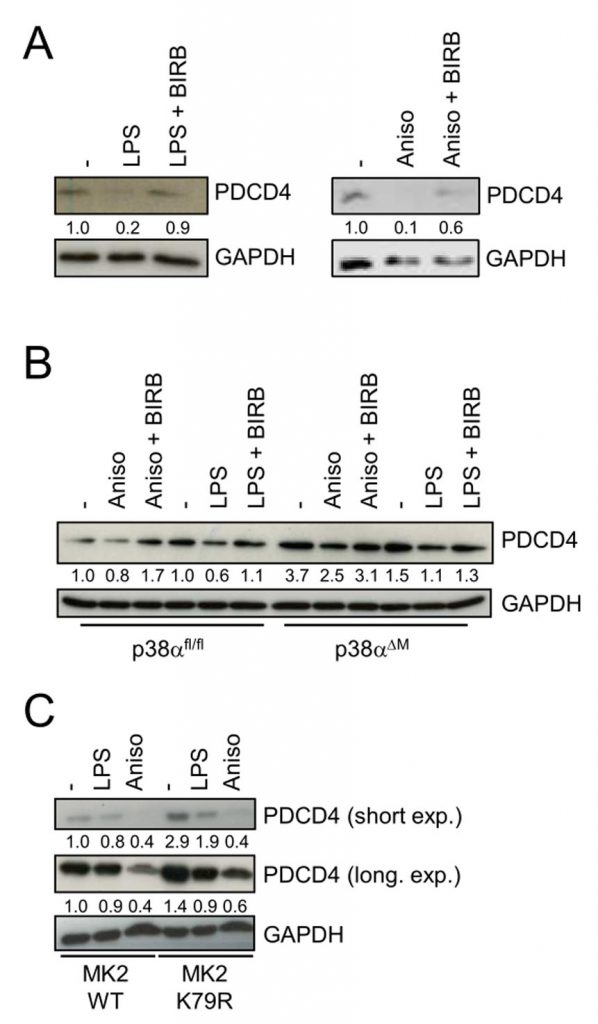 | FIGURE 1: p38 promotes PDCD4 degradation. (A) Bone marrow-derived macrophages (BMDMs) were treated with BIRB796 (BIRB) as depicted and then stimulated with LPS or anisomycin (Aniso) only for 4 hours. (B) p38fl/fl and p38αΔM BMDMs were stimulated with Aniso or LPS for 4 hours. (C) Mk2-/- macrophages reconsti-tuted with either MK2 K79M mutant or WT MK2 were stimulated with Aniso or LPS for 4 hours. Cell lysates were analyzed by im-munoblotting. |
–
p38 controls PDCD4 via TSC1/TSC2
The complex of TSC1 (Hamartin) and TSC2 is a major negative regulator of mTORC1, and its involvement in mTORC1-mediated degradation of PDCD4 has been recently suggested [16]. Indeed, deletion of TSC2 in BMDMs strongly abrogated expression of PDCD4 (Fig. 2A). This effect was reversible by rapamycin and thus dependent on mTORC1 (Fig. 2A). In addition, serum starvation induced the expression of PDCD4 in Tsc1+/+ and Tsc2+/+ fibroblasts (Fig. 2B and D). In contrast, PDCD4 levels were strongly reduced in either non-starved as well as starved Tsc1−/− and Tsc2−/− fibroblasts similar to macrophages (Fig. 2B and D). Inhibition of p38 or mTORC1 prevented anisomycin-induced degradation of PDCD4 in Tsc1+/+ and Tsc2+/+ fibroblasts (Fig. 2C and D) and in Tsc2fl/fl BMDMs stimulated with anisomycin or LPS (Fig. 2E and F). However, BIRB796 failed to rescue PDCD4 degradation in anisomycin-stimulated Tsc1−/− and Tsc2−/− fibroblasts (Fig. 2C and D) and in Tsc2Lyz2 BMDMs (Fig. 2E). These results show that p38 controls PDCD4 expression via TSC1/TSC2. In contrast, rapamycin and the catalytic mTOR inhibitor Torin1 partially restored PDCD4 levels in anisomycin-stimulated Tsc1−/− and Tsc2−/− fibroblasts (Fig. 2C and D). As an ATP-competitive inhibitor, Torin1 effectively prevents both mTORC1 and mTORC2 phosphorylation [17]. Interestingly, neither rapamycin nor Torin1 restored PDCD4 in Tsc1−/− and Tsc2−/− cells to a level that is seen in starved Tsc1+/+ and Tsc2+/+ fibroblasts, suggesting that TSC1/TSC2 promotes basal expression of PDCD4 that is independent of mTORC1 (Fig. 2C and D). Similar results were obtained with anisomycin in BMDMs (Fig. 2E and F). However, we noticed that the inhibitors restored PDCD4 levels in LPS-supplied Tsc2Lyz2 BMDMs to a comparable level as seen in wild type-representing BMDMs. These results support the concept that anisomycin does not just simply block PDCD4 translation but actively promotes degradation of PDCD4. Previous studies have found that activation of Erk contributes to PDCD4 degradation by enhancing proteasome activity [18]. Our experiments revealed an Erk-independent manner of PDCD4 degradation in Tsc2−/− fibroblasts since Erk expression was even reduced in the TSC2-deficient cells (Fig. 2D). The p90 ribosomal S6 kinases (RSKs) act downstream of Erk [19] and were shown to be promote proteasomal degradation of PDCD4 [20]. However, there was not clear association of p90RSK phosphorylation at Ser380 and PDCD4 levels in Tsc2fl/fl and Tsc2Lyz2 BMDMs (Suppl. Fig. 1A). Although PDCD4 can be transcriptionally regulated [21][22], qRT-PCR analysis of PDCD4 mRNA did not reveal significant differences between Tsc2Lyz2 and Tsc2fl/fl BMDMs (Suppl. Fig. 1C).
–
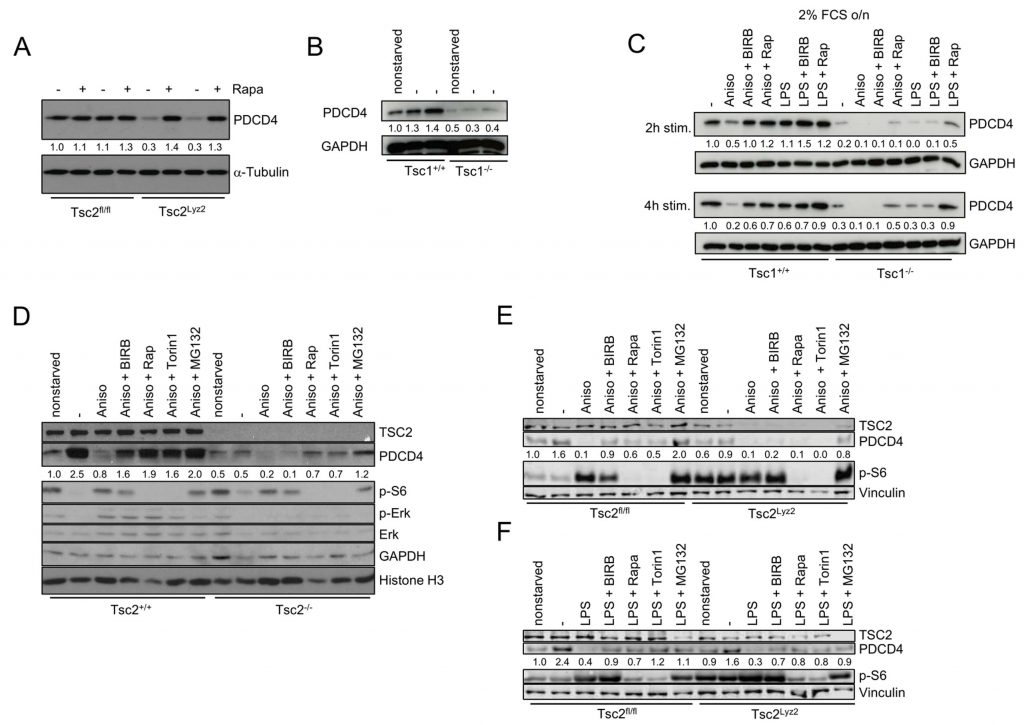 |
FIGURE 2: The TSC complex links p38 to PDCD4. (A) BMDMs from Tsc2fl/fl and Tsc2Lyz2 were cultivated with or without rapamycin for 12 hours. (B) Tsc1+/+ and Tsc1-/- fibroblasts were non-starved or serum-starved as indicated and subsequently, cell lysates were prepared. (C) Tsc1+/+ and Tsc1-/- fibroblasts were serum-starved overnight. Afterwards, cells were treated with the indicated inhibitors for 90 minutes fol-lowed by Aniso stimulation for 2 and 4 hours, respectively. (D) Tsc2+/+ and Tsc2-/- fibroblasts were non-starved where indicated or were se-rum-starved overnight. Inhibitor treatment of cells for 90 minutes preceded 4 hours of Aniso treatment. Cell lysates were analyzed by im-munoblotting. (E, F) BMDMs from Tsc2fl/fl and Tsc2Lyz2 mice were pretreated with the inhibitors BIRB796, rapamycin, Torin 1 or MG-132 for 90 minutes and subsequently elicited with LPS or Aniso for 4 hours. |
–
p38 activation subjects PDCD4 to proteasomal degradation
Finally, we tested whether p38 promotes degradation of PDCD4 via the proteasome. We noticed that the proteasome inhibitor MG-132 restored PDCD4 levels in anisomycin-treated Tsc1+/+ and Tsc2+/+ fibroblasts as well as anisomycin- and LPS-stimulated BMDMs (Fig. 3A, 2D–F). However, in Tsc1−/− and Tsc2−/− fibroblasts as well as Tsc2Lyz2 BMDMs, MG-132 could not fully restore PDCD4 levels arguing again of an TSC1/TSC2-dependent effect that is independent of mTORC1 and proteasomal degradation (Fig. 3A, 2D–F). Treating BMDMs with the p38-activating translation elongation inhibitor cycloheximide (Chx) [23][24] confirmed that PDCD4 translation is under strong control of this MAPK (Fig. 3B).
–
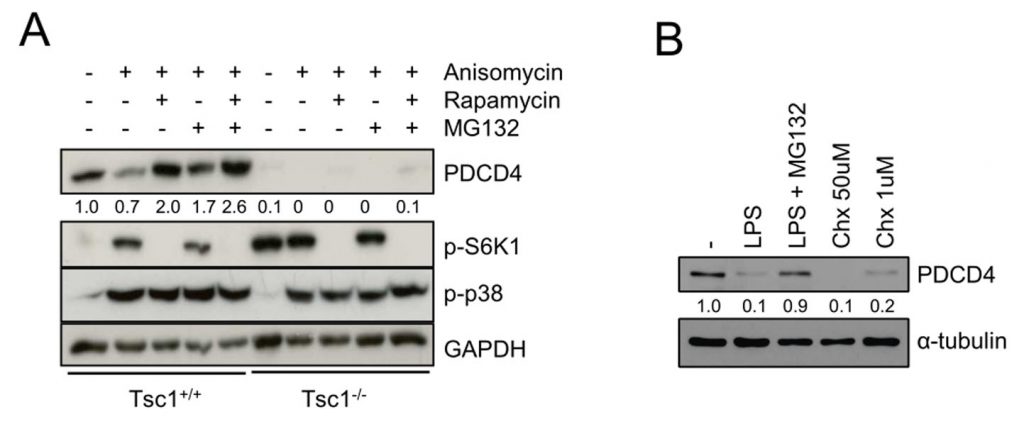 |
FIGURE 3: Reduction of PDCD4 is caused by p38-stimulated pro-teasome degradation. (A) Tsc1+/+ and Tsc1-/- fibroblasts were serum-starved overnight prior to treat-ment with the indicated inhibitors for 90 minutes and subsequent Aniso stimulation for 4 hours. (B) BMDMs were treated with MG-132, LPS or cycloheximide (Chx) as indi-cated for 4 hours. Immunoblots were analyzed as depicted. |
DISCUSSION
The MAPK p38α is ubiquitously expressed in most cell types and regulates diverse functions such as cell proliferation, differentiation, apoptosis, tissue repair, tumorigenesis, or inflammation [25]. Physicochemical stress signals such as heat, osmotic shock, arsenite or anisomycin result in activation of p38 [25]. p38 has been described as either tumor suppressor or oncoprotein depending on the cell type [26]. It will be interesting to evaluate whether PDCD4 contributes to the cell type-specific anti- or pro-tumorigenic functions of p38.
–
PI3K promotes PDCD4 degradation by mTORC1 activity in response to mitogenic signals [11]. Our data now suggests that also p38 induces degradation of PDCD4 via mTORC1 and TSC1/TSC2. We have previously shown that PI3K and p38 coordinately modulate mTORC1 signaling via TSC1/TSC2 in murine macrophages and human monocytes [13]. In agreement, LPS or anisomycin still induced partial degradation of PDCD4 in p38-deficient macrophages suggesting that PI3K and p38 also coordinately control PDCD4 degradation in macrophages (Fig. 3C). PDCD4 is expressed in unstressed, proliferating cells [27] and even though the heterozygous deletion of TSC2 in Tsc2Lys+/- BMDMs creates a more proliferative macrophage type, degradation of PDCD4 by hyperactive mTORC1 in these cells outweighs healthy upregulation of PDCD4. Interestingly, rapamycin and the catalytic inhibitor Torin1, which fully blocks mTORC1 activity, did not restore PDCD4 expression in Tsc1- or Tsc2-deficient fibroblasts to wild-type levels. This indicates an additional positive regulatory role for the TSC complex on PDCD4 expression in fibroblasts independently of mTORC1. The broad PDCD4 network comprises numerous feedback loops, e. g. on PI3K/Akt [28] and dysfunctional recycling of proteins like PDCD4 by the proteasome can be compensated by autophagy [29]. PDCD4 is associated with cell cycle regulation and programmed cell death and is controlled by apoptosis inducers [9][27][30]. Hence, phosphorylation by protein kinases regulating survival pathways, such as casein kinase 2 (CK2), seems plausible. CK2 was already shown to interact with PDCD4 within the nucleus [31][32] with their expression levels being inversely correlated in the tumor setting [33]. This connection would also fit into the overall picture in which PDCD4 acts pro-apoptotic [34]. Of note, CK2 can directly phosphorylate Akt to promote proliferation via mTORC1 [35]. Since PDCD4 is widely known to be regulated by microRNAs, mainly miR-21, their involvement cannot be ruled out. miR-21 is upregulated in the inflammatory and tumor-associated context [36]. However, we did not find a prominent upregulation of miR-21 in Tsc2-/- fibroblasts (data not shown).
–
The precise elucidation of the upstream regulatory network that controls PDCD4 in cancer and immune cells may be important to define novel anti-cancer and anti-inflammatory strategies. In conclusion, we showed that activation of p38 promotes degradation of PDCD4 via the TSC-mTORC1 pathway (Fig. 4).
–
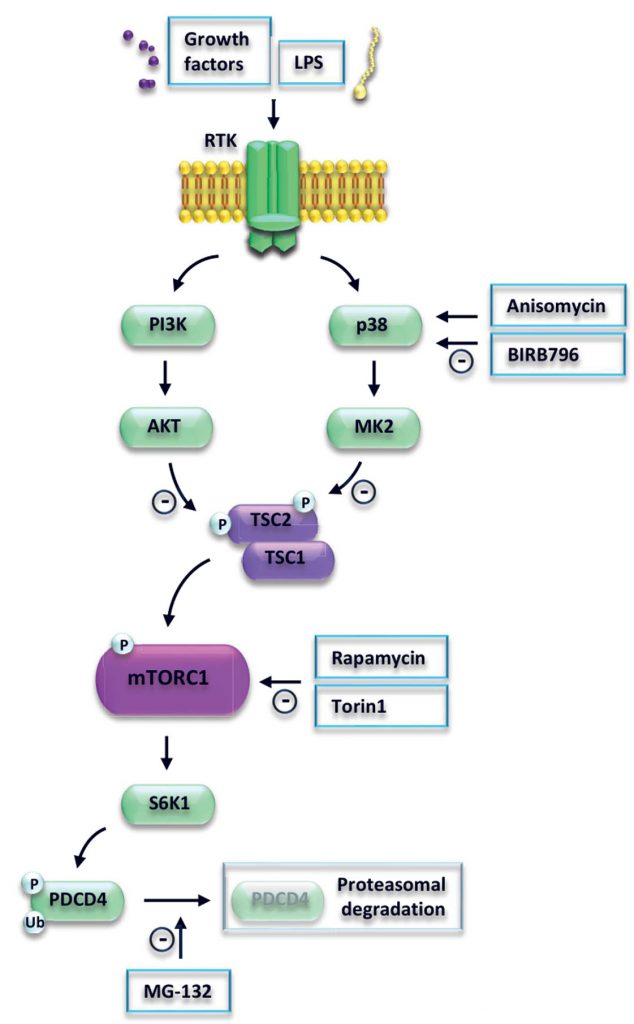 | FIGURE 4: Model of p38 and mTORC1-mediated regulation of PDCD4. LPS activates Toll-like receptor 4 (TLR4) signaling. Subse-quent signal transmission via phosphoinositide 3-kinase (PI3K) and mitogen-activated kinase p38 leads to inhibition of the sup-pressor protein tuberous sclerosis complex 2 (TSC2), followed by activation of mammalian target of rapamycin complex 1 (mTORC1). mTORC1 phosphorylates ribosomal protein S6 kinase beta-1 (S6K1), which in turn phosphorylates programmed cell death protein 4 (PDCD4). PDCD4 is ubiquitinated and degraded by the proteasome. |
MATERIALS AND METHODS
Reagents
LPS (Lipopolysaccharide Escherichia coli serotype O111:B4, #LPS25), anisomycin (#A5862), rapamycin (#553211), MG-132 (#474791) and cycloheximide (#C-0943) were purchased from Sigma, BIRB796 (#5989) and Torin1 (#4247) from Tocris.
–
Cell culture
Mouse embryonic fibroblasts (MEFs) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 4.5 g/L glucose, 2 mM L-glutamine, 100 µg/ml streptomycin, 100 U/ml penicillin and 10% heat-inactivated fetal bovine serum (FBS, Performance Plus, #10082147, Gibco). Tsc2+/+p53−/− and Tsc2−/−p53−/− as well as Tsc1+/+ and Tsc1−/− MEFs were described previously [37]. Mk2−/− immortalized murine macrophages stably reconstituted with MK2 or MK2K79R were kindly provided by Matthias Gaestel, Hannover, Germany. B6;129-Tsc2fl/fl mice were kindly provided by Michael J. Gambello, Atlanta, USA [38] and were crossed to B6.129P2-Lyz2tm1(cre)lfo/J (The Jackson Laboratory) to obtain Tscfl/flLyz2cre/+ (denoted Tsc2Lyz2) or Tscfl/flLyz2+/+ (denoted Tsc2fl/fl) littermates. Animal care was in accord with institutional guidelines. Bone marrow-derived macrophages (BMDMs) were generated as described before [16]. BMDMs from p38fl/fl and p38αΔM mice were isolated and grown as described [39].
–
Analysis of signal transduction events
BMDMs and Mk2−/− macrophages were replated one day prior to stimulation in full medium containing 2% FBS overnight (16 h), whereas 70% confluent MEFs were completely serum-starved overnight if not stated otherwise. The cells were then treated with either 100 nM rapamycin, 200 nM BIRB796, 100 nM Torin1 or 1 µM MG-132 for 90 minutes and subsequently stimulated with 100 ng/ml LPS or 100 ng/ml anisomycin for 2 or 4 hours if not mentioned otherwise. Treatments were performed in full medium with 2% FBS, non-starved samples received 10% FBS during that time. Extract preparation and immunoblotting was done as described [40]. Antibodies were PDCD4 (clone D29C6, #9535, 1:1000 and 1:500), TSC2 (#3612 and clone D93F12, #4308, both 1:1000), p-TSC2 (Ser1254, #3616, 1:1000), p-S6 (Ser240/244, #2215 and clone D68F8, #5364, both 1:1000), p-p38 (Thr180/Tyr182, #9211, 1:1000), Erk1/2 (Thr202/Tyr204, #9102, 1:1000), p-Erk1/2 (Thr202/Tyr204, #9101, 1:1000), p-p90RSK (Ser380, #9341, 1:1000), p-H3 (Ser10, #9701, 1:1000), Vinculin (clone E1E9V, #13901, 1:1000) (all from Cell Signaling Technology) and GAPDH (#2275-PC, 1:1000, Trevigen). Molecular weight of the proteins was determined with PageRuler Prestained Protein Ladder (#26616, Thermo Scientific). With regard to quantification, data was generated either with X-ray or fluorescence detection. For X-ray detection, we applied HRP-conjugated secondary antibodies (1:10000, Bethyl Lab) and the Pierce ECL Western Blotting substrate (Thermo Scientific). Bands were visualized with Amersham Hyperfilm ECL (GE-Healthcare) and the Medical X-ray Processor 2000 system (Kodak). Fluorescence was recorded after secondary antibody incubation (IRDye IgG antibodies, 1:20000, LI-COR Biosciences) with Odyssey CLx Imaging System and analyzed with Image Studio Software (both LI-COR Biosciences). Bands were framed in unchanged manner between the samples. Normalization was performed with respect to the untreated sample of the unmodified genotype.
–
mRNA expression analysis
Total RNA from BMDMs was isolated via the RNeasy Plus Mini Kit (#74134, QIAGEN) according to the manufacturer’s instructions. cDNA synthesis was performed with the RevertAid RT Reverse Transcription Kit (#K1691, ThermoFisher Scientific) prior to proceeding with qRT-PCR using the GoTaq® qPCR Master Mix (#A6001, Promega). Data were acquired with a StepOnePlus Real-Time PCR System (Applied Biosystems). Relative expression was normalized to Peptidyl-prolyl cis-trans isomerase A (PPIA). The following primer pairs were used: Pdcd4, AGTTTTGCCCCTGGATGAGA, GCTAAGGACACTGCCAACAC; PPIA, TCCTGGCATCTTGTCCAT, TGCTGGTGCCATTCCT.
REFERENCES
- Lankat-Buttgereit B and Göke R (2009). The tumour suppressor Pdcd4: recent advances in the elucidation of function and regulation. Biol Cell 101(6): 309–317. 10.1042/BC20080191
- Yang HS, Jansen AP, Nair R, Shibahara K, Verma AK, Cmarik JL, and Colburn NH (2001). A novel transformation suppressor, Pdcd4, inhibits AP-1 transactivation but not NF-kappaB or ODC transactivation. Oncogene 20(6): 669–676. 10.1038/sj.onc.1204137
- Blees JS, Bokesch HR, Rübsamen D, Schulz K, Milke L, Bajer MM, Gustafson KR, Henrich CJ, McMahon JB, Colburn NH, Schmid T, and Brüne B (2012). Erioflorin stabilizes the tumor suppressor Pdcd4 by inhibiting its interaction with the E3-ligase β-TrCP1. PLoS One 7(10): e46567. 10.1371/journal.pone.0046567
- Merline R, Moreth K, Beckmann J, Nastase MV, Zeng-Brouwers J, Tralhão JG, Lemarchand P, Pfeilschifter J, Schaefer RM, Iozzo RV, and Schaefer L (2011). Signaling by the matrix proteoglycan decorin controls inflammation and cancer through PDCD4 and MicroRNA-21. Sci Signal 4(199): ra75. 10.1126/scisignal.2001868
- van den Bosch MWM, Palsson-Mcdermott E, Johnson DS, and O’Neill LAJ (2014). LPS induces the degradation of programmed cell death protein 4 (PDCD4) to release Twist2, activating c-Maf transcription to promote interleukin-10 production. J Biol Chem 289(33): 22980–22990. 10.1074/jbc.M114.573089
- Jiang Y, Gao Q, Wang L, Guo C, Zhu F, Wang B, Wang Q, Gao F, Chen Y, and Zhang L (2016). Deficiency of programmed cell death 4 results in increased IL-10 expression by macrophages and thereby attenuates atherosclerosis in hyperlipidemic mice. Cell Mol Immunol 13(4): 524–534. 10.1038/cmi.2015.47
- Zhong B, Yang X, Sun Q, Liu L, Lan X, Tian J, He Q, Hou W, Liu H, Jiang C, Gao N, and Lu S (2014). Pdcd4 modulates markers of macrophage alternative activation and airway remodeling in antigen-induced pulmonary inflammation. J Leukoc Biol 96(6): 1065–1075. 10.1189/jlb.3A0313-136RRR
- Ozpolat B, Akar U, Steiner M, Zorrilla-Calancha I, Tirado-Gomez M, Colburn N, Danilenko M, Kornblau S, and Berestein GL (2007). Programmed cell death-4 tumor suppressor protein contributes to retinoic acid-induced terminal granulocytic differentiation of human myeloid leukemia cells. Mol Cancer Res 5(1): 95–108. 10.1158/1541-7786.MCR-06-0125
- Shibahara K, Asano M, Ishida Y, Aoki T, Koike T, and Honjo T (1995). Isolation of a novel mouse gene MA-3 that is induced upon programmed cell death. Gene 166(2): 297–301. 10.1016/0378-1119(95)00607-9
- Schmid T, Bajer MM, Blees JS, Eifler LK, Milke L, Rübsamen D, Schulz K, Weigert A, Baker AR, Colburn NH, and Brüne B (2011). Inflammation-induced loss of Pdcd4 is mediated by phosphorylation-dependent degradation. Carcinogenesis 32(10): 1427–1433. 10.1093/carcin/bgr131
- Dorrello NV, Peschiaroli A, Guardavaccaro D, Colburn NH, Sherman NE, and Pagano M (2006). S6K1- and betaTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science 314(5798): 467–471. 10.1126/science.1130276
- Li Y, Inoki K, Vacratsis P, and Guan K-L (2003). The p38 and MK2 kinase cascade phosphorylates tuberin, the tuberous sclerosis 2 gene product, and enhances its interaction with 14-3-3. J Biol Chem 278(16): 13663–13671. 10.1074/jbc.M300862200
- Katholnig K, Kaltenecker CC, Hayakawa H, Rosner M, Lassnig C, Zlabinger GJ, Gaestel M, Müller M, Hengstschläger M, Hörl WH, Park JM, Säemann MD, and Weichhart T (2013). p38α senses environmental stress to control innate immune responses via mechanistic target of rapamycin. J Immunol 190(4): 1519–1527. 10.4049/jimmunol.1202683
- Croons V, Martinet W, Herman AG, Timmermans J-P, and De Meyer GRY (2009). The protein synthesis inhibitor anisomycin induces macrophage apoptosis in rabbit atherosclerotic plaques through p38 mitogen-activated protein kinase. J Pharmacol Exp Ther329(3): 856–864. 10.1124/jpet.108.149948
- Pargellis C, Tong L, Churchill L, Cirillo PF, Gilmore T, Graham AG, Grob PM, Hickey ER, Moss N, Pav S, and Regan J (2002). Inhibition of p38 MAP kinase by utilizing a novel allosteric binding site. Nat Struct Biol 9(4): 268–272. 10.1038/nsb770
- Linke M, Pham HTT, Katholnig K, Schnöller T, Miller A, Demel F, Schütz B, Rosner M, Kovacic B, Sukhbaatar N, Niederreiter B, Blüml S, Kuess P, Sexl V, Müller M, Mikula M, Weckwerth W, Haschemi A, Susani M, Hengstschläger M, Gambello MJ, and Weichhart T (2017). Chronic signaling via the metabolic checkpoint kinase mTORC1 induces macrophage granuloma formation and marks sarcoidosis progression. Nat Immunol 18(3): 293–302. 10.1038/ni.3655
- Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, Reichling LJ, Sim T, Sabatini DM, and Gray NS (2009). An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem 284(12): 8023–8032. 10.1074/jbc.M900301200
- Schmid T, Jansen AP, Baker AR, Hegamyer G, Hagan JP, and Colburn NH (2008). Translation inhibitor Pdcd4 is targeted for degradation during tumor promotion. Cancer Res 68(5): 1254–1260. 10.1158/0008-5472.CAN-07-1719
- Romeo Y, Zhang X, and Roux PP (2012). Regulation and function of the RSK family of protein kinases. Biochem J 441(2): 553–569. 10.1042/BJ20110289
- Galan JA, Geraghty KM, Lavoie G, Kanshin E, Tcherkezian J, Calabrese V, Jeschke GR, Turk BE, Ballif BA, Blenis J, Thibault P, and Roux PP (2014). Phosphoproteomic analysis identifies the tumor suppressor PDCD4 as a RSK substrate negatively regulated by 14-3-3. Proc Natl Acad Sci USA 111(29): E2918–27. 10.1073/pnas.1405601111
- Park SK and Jeong S (2016). SRSF3 represses the expression of PDCD4 protein by coordinated regulation of alternative splicing, export and translation. Biochem Biophys Res Commun 470(2): 431–438. 10.1016/j.bbrc.2016.01.019
- Lu K, Chen Q, Li M, He L, Riaz F, Zhang T, and Li D (2020). Programmed cell death factor 4 (PDCD4), a novel therapy target for metabolic diseases besides cancer. Free Radic Biol Med 159: 150–163. 10.1016/j.freeradbiomed.2020.06.016
- Morinière M, Delhommeau F, Calender A, Ribeiro L, Delaunay J, and Baklouti F (2010). Nonsense-mediated mRNA decay (NMD) blockage promotes nonsense mRNA stabilization in protein 4.1R deficient cells carrying the 4.1R Coimbra variant of hereditary elliptocytosis. Blood Cells Mol Dis 45(4): 284–288. 10.1016/j.bcmd.2010.08.011
- Perkins SD and Angenent LT (2010). Potential pathogenic bacteria in metalworking fluids and aerosols from a machining facility. FEMS Microbiol Ecol 74(3): 643–654. 10.1111/j.1574-6941.2010.00976.x
- Coulthard LR, White DE, Jones DL, McDermott MF, and Burchill SA (2009). p38(MAPK): stress responses from molecular mechanisms to therapeutics. Trends Mol Med 15(8): 369–379. 10.1016/j.molmed.2009.06.005
- Wagner EF and Nebreda AR (2009). Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer 9(8): 537–549. 10.1038/nrc2694
- Haas A, Nilges BS, Leidel SA, and Klempnauer K-H (2020). PDCD4 controls the G1/S-phase transition in a telomerase-immortalized epithelial cell line and affects the expression level and translation of multiple mRNAs. Sci Rep 10(1): 2758. 10.1038/s41598-020-59678-w
- Zhen Y, Fang W, Zhao M, Luo R, Liu Y, Fu Q, Chen Y, Cheng C, Zhang Y, and Liu Z (2017). miR-374a-CCND1-pPI3K/AKT-c-JUN feedback loop modulated by PDCD4 suppresses cell growth, metastasis, and sensitizes nasopharyngeal carcinoma to cisplatin. Oncogene 36(2): 275–285. 10.1038/onc.2016.201
- Manirujjaman M, Ozaki I, Murata Y, Guo J, Xia J, Nishioka K, Perveen R, Takahashi H, Anzai K, and Matsuhashi S (2020). Degradation of the Tumor Suppressor PDCD4 Is Impaired by the Suppression of p62/SQSTM1 and Autophagy. Cells 9(1): 218. 10.3390/cells9010218
- Liwak U, Thakor N, Jordan LE, Roy R, Lewis SM, Pardo OE, Seckl M, and Holcik M (2012). Tumor suppressor PDCD4 represses internal ribosome entry site-mediated translation of antiapoptotic proteins and is regulated by S6 kinase 2. Mol Cell Biol 32(10): 1818–1829. 10.1128/MCB.06317-11
- Homma MK, Shibata T, Suzuki T, Ogura M, Kozuka-Hata H, Oyama M, and Homma Y (2015). Role for protein kinase CK2 on cell proliferation: assessing CK2 complex components in the nucleus during the cell cycle progression. In: Ahmed K, Issinger O-G, Szyszka R, editors. Protein kinase CK2 cellular function in normal and disease states. Springer International Publishing, Cham; pp 197–226.
- Bian Y, Ye M, Wang C, Cheng K, Song C, Dong M, Pan Y, Qin H, and Zou H (2013). Global screening of CK2 kinase substrates by an integrated phosphoproteomics workflow. Sci Rep 3: 3460. 10.1038/srep03460
- Trembley JH, Li B, Kren BT, Gravely AA, Caicedo-Granados E, Klein MA, and Ahmed K (2021). CX-4945 and siRNA-Mediated Knockdown of CK2 Improves Cisplatin Response in HPV(+) and HPV(-) HNSCC Cell Lines. Biomedicines 9(5): 571. 10.3390/biomedicines9050571
- Zhang H, Ozaki I, Mizuta T, Hamajima H, Yasutake T, Eguchi Y, Ideguchi H, Yamamoto K, and Matsuhashi S (2006). Involvement of programmed cell death 4 in transforming growth factor-beta1-induced apoptosis in human hepatocellular carcinoma. Oncogene 25(45): 6101–6112. 10.1038/sj.onc.1209634
- Ponce DP, Maturana JL, Cabello P, Yefi R, Niechi I, Silva E, Armisen R, Galindo M, Antonelli M, and Tapia JC (2011). Phosphorylation of AKT/PKB by CK2 is necessary for the AKT-dependent up-regulation of β-catenin transcriptional activity. J Cell Physiol 226(7): 1953–1959. 10.1002/jcp.22527
- Sheedy FJ, Palsson-McDermott E, Hennessy EJ, Martin C, O’Leary JJ, Ruan Q, Johnson DS, Chen Y, and O’Neill LAJ (2010). Negative regulation of TLR4 via targeting of the proinflammatory tumor suppressor PDCD4 by the microRNA miR-21. Nat Immunol 11(2): 141–147. 10.1038/ni.1828
- Zhang H, Cicchetti G, Onda H, Koon HB, Asrican K, Bajraszewski N, Vazquez F, Carpenter CL, and Kwiatkowski DJ (2003). Loss of Tsc1/Tsc2 activates mTOR and disrupts PI3K-Akt signaling through downregulation of PDGFR. J Clin Invest 112(8): 1223–1233. 10.1172/JCI17222
- Hernandez O, Way S, McKenna J, and Gambello MJ (2007). Generation of a conditional disruption of the Tsc2 gene. Genesis 45(2): 101–106. 10.1002/dvg.20271
- Nishida K, Yamaguchi O, Hirotani S, Hikoso S, Higuchi Y, Watanabe T, Takeda T, Osuka S, Morita T, Kondoh G, Uno Y, Kashiwase K, Taniike M, Nakai A, Matsumura Y, Miyazaki J, Sudo T, Hongo K, Kusakari Y, Kurihara S, Chien KR, Takeda J, Hori M, and Otsu K (2004). p38alpha mitogen-activated protein kinase plays a critical role in cardiomyocyte survival but not in cardiac hypertrophic growth in response to pressure overload. Mol Cell Biol 24(24): 10611–10620. 10.1128/MCB.24.24.10611-10620.2004
- Säemann MD, Weichhart T, Zeyda M, Staffler G, Schunn M, Stuhlmeier KM, Sobanov Y, Stulnig TM, Akira S, von Gabain A, von Ahsen U, Hörl WH, and Zlabinger GJ (2005). Tamm-Horsfall glycoprotein links innate immune cell activation with adaptive immunity via a Toll-like receptor-4-dependent mechanism. J Clin Invest 115(2): 468–475. 10.1172/JCI22720
–
SUPPLEMENTAL INFORMATION
 Download Supplemental Information
Download Supplemental Information
AUTHOR CONTRIBUTIONS
CB, KK, CK, ML, NS designed and per-formed experiments. MH provided materials. TW de-signed experiments. CB, KK and TW wrote the manu-script. All authors edited and approved the final draft.
ACKNOWLEDGMENTS
T.W. is supported by grants from the Austrian Science Fund (FWF) P30857-B28 and P34023-B, by the FWF Sonderforschungsbereich F83, and the Vi-enna Science and Technology Fund (WWTF) LS18-058.
COPYRIGHT
© 2021

p38 regulates the tumor suppressor PDCD4 via the TSC-mTORC1 pathway by Braun et al. is licensed under a Creative Commons Attribution 4.0 International License.


