Reviews:
Cell Stress, Vol. 2, No. 4, pp. 72 - 81; doi: 10.15698/cst2018.04.130
How to use a multipurpose SNARE: The emerging role of Snap29 in cellular health

1 Dipartimento di Bioscienze, Universita’ degli Studi di Milano, Italy.
# Equal contribution.
Keywords: Snap29, membrane trafficking, endocytosis, autophagy, SNARE proteins, SNAP family, cell division.
Abbreviations:
CEDNIK – cerebral dysgenesis, neuropathy, ichtyosis and keratoderma,
HOPS – homotypic fusion and protein sorting,
HPIV – human para influenza virus,
IL – Interleukin,
SNAP – synaptosomal-associated protein,
SNARE - soluble N-ethylmaleimide-sensitive-factor attachment receptor,
Syx – syntaxin.
Received originally: 09/02/2018 Received in revised form: 08/03/2018
Accepted: 08/03/2018
Published: 22/03/2018
Correspondence:
Thomas Vaccari, , Dipartimento di Bioscienze, Universita’ degli Studi di Milano, Via Celoria 26, 20133 Milano, Italy; Tel: +39 02503-14886 ; Fax: +39 02503-14880 thomas.vaccari@unimi.it
Conflict of interest statement: The authors declare no conflict of interest.
Please cite this article as: Valeria Mastrodonato, Elena Morelli and Thomas Vaccari (2018). How to use a multipurpose SNARE: The emerging role of Snap29 in cellular health. Cell Stress 2(4): 72-81. doi: 10.15698/cst2018.04.130
Abstract
Despite extensive study, regulation of membrane trafficking is incompletely understood. In particular, the specific role of SNARE (Soluble NSF Attachment REceptor) proteins for distinct trafficking steps and their mechanism of action, beyond the core function in membrane fusion, are still elusive. Snap29 is a SNARE protein related to Snap25 that gathered a lot of attention in recent years. Here, we review the study of Snap29 and its emerging involvement in autophagy, a self eating process that is key to cell adaptation to changing environments, and in other trafficking pathways. We also discuss Snap29 role in synaptic transmission and in cell division, which might extend the repertoire of SNARE-mediated functions. Finally, we present evidence connecting Snap29 to human disease, highlighting the importance of Snap29 function in tissue development and homeostasis.
 |
INTRODUCTION
The specialized trafficking routes that evolved between compartments of the endomembrane system rely on a wide set of proteins, including primarily SNAREs, Rab GTPases and a large set of effectors and tethering factors, that ensure specificity of cargo delivery to a wide range of different target compartments. Precision and fidelity of targeting is paramount to support cell viability and to prevent disease. However, how such specificity is achieved at a molecular level is incompletely understood.
–
SNAREs are part of the conserved coiled-coil machinery that brings membranes in close proximity, a prerequisite for most membrane fusion events occurring during trafficking [1][2][3]. A stereotypic set of SNARE proteins forming a 4-helix bundle (often referred to as trans-SNARE complex) composed of distinct SNARE domains named Qa-, Qb-, Qc- or R-SNARE are invariantly required for fusion. Usually, a Qa-SNARE-containing protein [often called syntaxin, or target (t)-SNARE] and a R-SNARE -containing protein [often called vesicle associated membrane protein (VAMP) protein, or v-SNARE] are carried by opposing membranes, each providing a SNARE domain to the fusion complex. These proteins are glued together by Qb- and Qc-SNARE domain containing proteins, providing the remaining 2 domains (Fig. 1A). After fusion, all proteins of the 4-helix bundle are associated to the target membrane, in an arrangement often called cis-SNARE complex, that is disassembled by the activity of a number of proteins including the ATPase NSF (N-ethylmaleimide-sensitive factor). The Qb- and Qc-SNAREs domains can also be contributed in a single protein, as is the case of members of the Synaptosomal-Associated Protein (SNAP) protein family. Snap25 and Snap23 are the most extensively studied SNAP family members (For review [4]). Metazoan genomes further include Snap29, while the SNAP family in mammals comprises also Snap47 [5][6]. Here, we will focus on Snap29, whose functions have increasingly come into view in the last two decades.
–
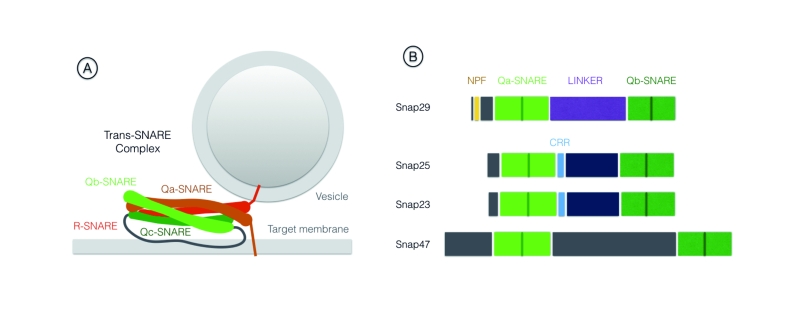 | FIGURE 1: Illustration of the trans-SNARE complex involved in membrane fusion. All SNARE domains are oriented with the N-termini to the left (A). Schematic representation of SNAP family members in mammals (B). The homologous SNARE domains among the 4 paralogs are shown. The position of the central glutamine residue (Q) is indicated by a line. The linker region of Snap29 is (purple) differs from that of other family members and does not contain a cysteine rich region (CRR; light blue) for membrane targeting.The N-Terminal acidic NPF motif, exclusive to Snap29 among family members, is shown in yellow. |
–
Differently from other family members Snap29 possess an N-terminal acidic asparagine-proline-phenylalanine (NPF) motif. Similarly to Snap47, Snap29 also lacks cysteine residues in the spacer region between the two SNARE domains, that in Snap25 and Snap23 are modified to allow membrane anchoring [5] (Fig. 1B). Consistent with the lack of a membrane anchor, early biochemical studies revealed that Snap29 is only partially associated to membranes [5]. They also suggested that Snap29 interacts with a wide range of syntaxins including Syntaxin6 (Syx6) a SNARE protein acting in the Golgi apparatus [5][7][8]. Despite these initial studies, when compared to the advanced understanding of Snap25 and Snap23 function, the specificity and molecular regulation of Snap29 function remained to be explored. Below, we review the growing body of evidence indicating that Snap29 regulates membrane fusion at multiple cellular locales during intracellular trafficking. In addition, we report findings that indicate that Snap29 might not only be involved in membrane fusion, but that it could rather serve a regulatory or structural role in certain contexts. Finally, we discuss the involvement of Snap29 in human disease.
–
ENDOCYTOSIS, RECYCLING AND CILIUM FORMATION REQUIRE SNAP29 AND ITS INTERACTOR EHD1
Together with the NPF interactor EHD1 and with the endocytic adapters AP2, Snap29 was reported to promote endosomal trafficking of the Insulin Growth Factor 1 receptor (IGF-1R) in CHO cells, a process crucial to down-regulate active receptors [9] (Fig. 2A).
–
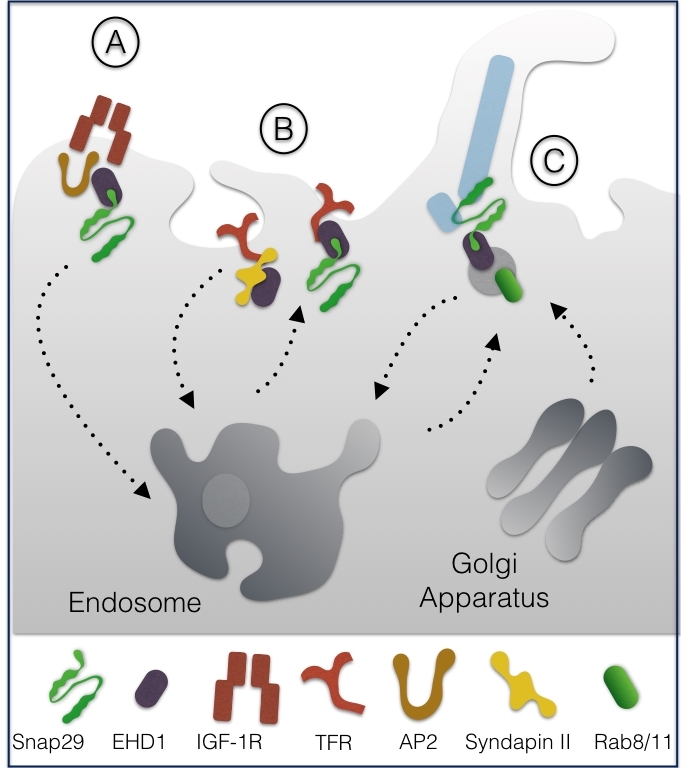 | FIGURE 2: Roles of Snap29 in endocytosis and recycling. Snap29 is required for internalization of the IGF-1R receptor (A), for internalization and/or recycling of TFR (B) and for formation of the basal membrane surrounding the cilium microtubules (light blue; C). All these functions are regulated in association with the endocytic factor EHD1, which binds the NPF motif of Snap29 (see text for details). |
–
Biochemical analyses showed that binding of EHD1 to the NPF motif of Snap29 can occur alternatively to that of the F-BAR protein syndapin II. Upon overexpression in HeLa cells, alternative EHD1 and syndapin II interaction to Snap29 affected internalization of transferrin receptors (TFR) but not of EGFR, suggesting that Snap29 might control trafficking of selected receptors [10] (Fig. 2B).
–
More recently, Snap29 has been shown to participate, both in RPE cells and in Danio rerio, in trafficking of ciliary vesicles, a process that depends on EHD1, on the recycling GTPase Rab11, and on the Golgi apparatus GTPase Rab8 [11]. The authors suggest that EHD1 is required for vesicle formation from preexisting membranes that fuse together and assemble in a membrane structure enveloping the cilium microtubules (MTs), by the action of Snap29-containing SNARE complexes (Fig. 2C). Interaction of Snap29 with EHD1 was also reported in Drosophila melanogaster [12].
SNAP29 IS A KEY REGULATOR OF AUTOPHAGY
Macroautophagy (autophagy here after) is an adaptive cellular pathway operating in house-keeping as well as in stress conditions. It required for the degradation of damaged organelles, aggregated proteins and other potentially toxic waste material. Upon starvation, autophagy is also crucial for maintaining sufficient levels of cellular nutrients by self-digesting cytoplasmic molecules. Autophagic clearance requires the formation of an intermediate double-membrane organelle called autophagosome that fully engulfs the cargo before fusion with lysosome (for review [13]).
–
In 2012, Itakura et al. reported that fusion of starvation-induced autophagosomes with lysosomes in HeLa cells requires Syntaxin17 (Syx17), Snap29 and the lysosomal R-SNARE Vamp8 [14]. In such work, the authors showed that Syx17 is recruited to autophagosomes from the endoplasmic reticulum (ER) and that Snap29 primarily associate to it, possibly from the cytoplasm. Subsequent characterization of Syx17 and Snap29 in D. melanogaster revealed a similar role in autophagy in vivo [15][16].
–
The mechanism of Snap29-dependent regulation of autophagy was further dissected in human cells by showing that the binary Syx17-Snap29 complex on autophagosomes is stabilized by binding of ATG14 oligomers to Syx17. Such tethering function is proposed to be crucial for priming the complex for fusion with Vamp8 on the lysosomes [17].
–
Interestingly, analysis of Caenorhabditis elegans mutants indicated that O-GlcNac modification of Snap29 is crucial to regulate autophagic clearance [18]. In fact, in both worms and in human cells, it was found that in presence of high nutrients Snap29 is modified on 4 Serine residues, thus preventing inclusion in a functional SNARE complex. In contrast, in low nutrient conditions such as starvation, the unmodified Snap29 is readily included in the 4-helix bundle promoting autophagy and nutrient recovery (Fig. 3A).
–
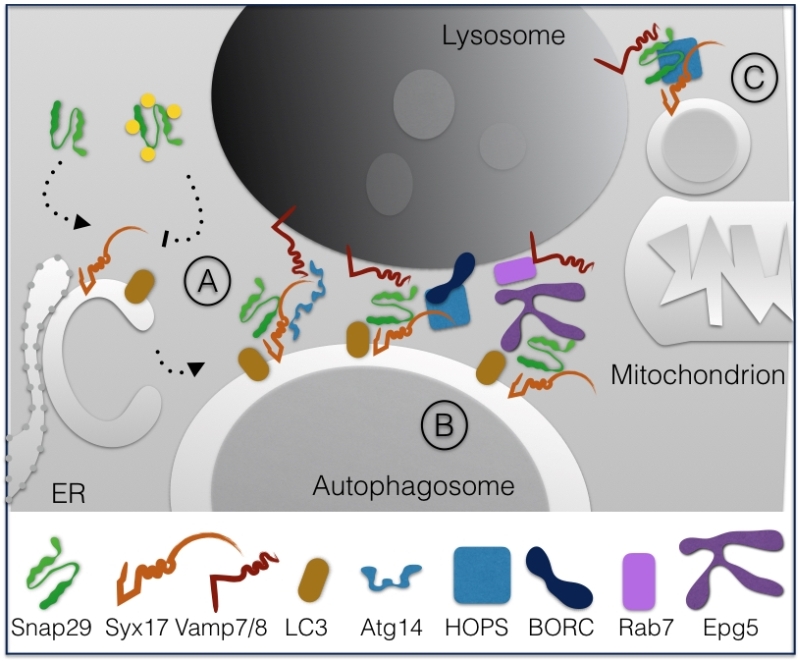 | FIGURE 3: The function of Snap29 during autophagy. Snap29 associates to Syx17, which is recruited from the ER to nascent autophagosomes to regulate their fusion with lysosomes, with the help of Atg14 polymers. O-GlcNac modification of Snap29 in high nutrient conditions prevent inclusion in an fusion complex (A). Fusion involves the HOPS and BORC tethering complexes, Epg5 and Rab7 (B). Snap29, Syx17, Vamp8 and the HOPS complex are also used in fusion of mitochondrial-derived vesicles with lysosomes (C) (see text for details). |
–
Fusion with lysosomes has been further shown to require the homotypic fusion and protein sorting (HOPS) tethering complex that associates with the late endosomal GTPase Rab7, both in D. melanogaster and human cells [19].
–
Fusion and HOPS recruitment is also assisted by the lysosome-associated multiprotein complex named BLOC-1 related complex (BORC) that regulates lysosome positioning [20]. An additional factor controlling fusion of autophagosomes with lysosomes is Vici Syndrome Protein EPG5, a Rab7 effector that is found to interact with LC3/Lgg-1 in C. elegans and human cells to favor formation of the trans-SNARE complex [21]. In absence of EPG5, inappropriate fusion of autophagosomes with early endocytic vesicles is shown to occur (Fig. 3B).
–
Finally, recent data indicate that Snap29 acts in membrane fusion of Syx17-loaded mitochondrial derived vesicles to lysosomes with the help of the HOPS complex [22] (Fig. 3C).
SNAP29 ASSISTS SPECIALIZED SECRETION
In vivo analyses of C. elegans and D. melanogaster lacking Snap29 revealed a wide range of trafficking defects, in addition to alteration of recycling, lysosomal degradation and blocked autophagy. These included a dispersed Golgi morphology, an impairment in Golgi trafficking and secretion, and, finally, inappropriate secretion of autophagosomes, suggesting that Snap29 acts directly during secretion [16][23][24]. Consistent with this in a yeast two hybrid screen and in HeLa cells, it was found that SNAP29 interacts with the Golgi apparatus tethering factor COG6 and with the Golgi SNAREs Syntaxin5, SYX6 and GS27 [25][26].
–
In HeLa cells, Snap29 has also been directly involved in unconventional secretion of the leaderless pro-inflammatory factor interleukin-1β (IL-1β), which occurs upon lysosomal damage [27]. The mode of IL-1β secretion is debated, but it has been proposed to involve formation of autophagosomes [28][29]. Lysosomal damage can cause proteolytic cleavage activation of IL-1β, leading to binding to the receptor TRIM16 residing on the lysosomal membrane. In this way, IL-1β is sequestered within LC3 positive sequestration membranes, eventually forming an autophagosome [27]. Depletion of Snap29, Snap23, Syntaxin 3 or 4 and of the ER-derived R-SNARE Sec22b affect IL-1β secretion, suggesting that autophagosomes containing IL-1β might fuse with the plasma membrane through the formation of a variety of SNARE complexes involving such SNAREs (Fig. 4A).
–
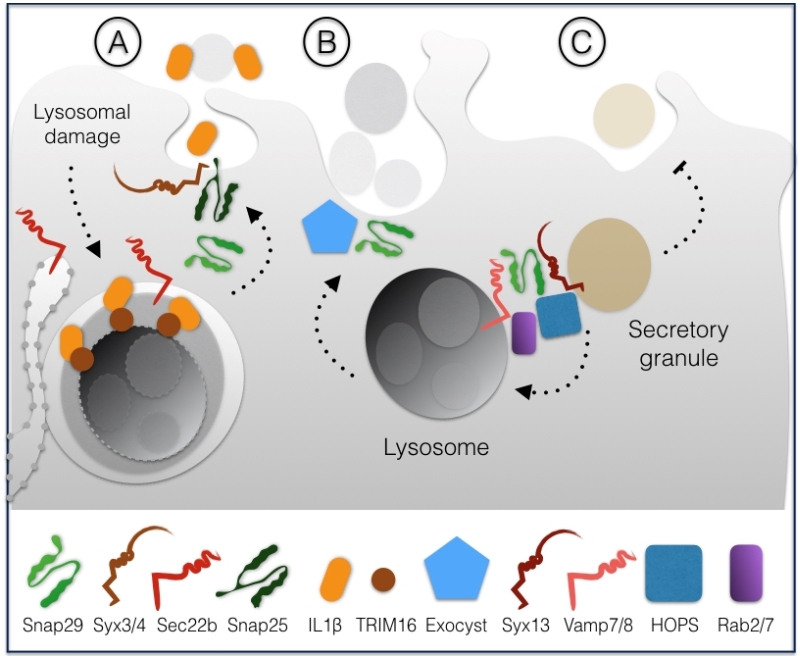 | FIGURE 4: Snap29 activity in secretion. Release of the pro-inflammatory factor IL-1β requires fusion to the plasma membrane mediated by Snap29 (A). Snap29 also regulates secretion of lysosomes during a developmental cell invasion process in C.elegans (B). Regulation of secretion of glue granules in D. melanogaster salivary glands involves Snap29, which acts in fusion of excess granules to lysosomes (C) (see text for details). |
–
In C. elegans anchor cells, a developmental model of cell invasion that relies on secretion of lysosomes to the protruding front, depletion of Snap29 or of components of the exocyst, a complex involved in targeted exocytosis, has been found to inhibit protrusion formation [30]. These data suggest that Snap29-mediated fusion might contribute to promote fusion of lysosomes to the plasma membrane in a highly specialized form of secretion (Fig. 4B).
–
A third specialized secretion pathway involving Snap29 has been recently identified in D. melanogaster [31]. In pupal salivary glands, which secrete large amounts of granule containing glue, excess granules are cleared by lysosomes in a specialized form of autophagy termed crynophagy which was found to be lost upon depletion of Syx13, Snap29, Vamp7, the GTPase Rab2 and Rab7 and components of the HOPS tethering complex (Fig. 4C).
–
Finally, Snap29 is particularly abundant in non excitatory cells of the nervous system, such as rat oligodendrocytes in culture, especially during myelination [32]. In this study, Snap29 was shown to colocalize and interact with the GTPase Rab3a via the N-terminal region of Snap29 and overexpression of Snap29 and Rab3a is reported to enhance cell surface trafficking of tagged myelin components, suggesting that Snap29 might regulate myelin secretion in glial cells [32]. Considering that astrocytes and, in particular, microglia are involved in neuro-inflammation, it might be interesting to determine whether specific functions of Snap29 in these other types of glia cells exists.
A NON TRAFFICKING ROLE OF SNAP29 POINTS TO A NEW MODEL OF OUTER KINETOCHORE FORMATION
Surprisingly, we have recently found that during cell division Snap29 acts as a component of the kinetochore, the mitotic structure that connect condensed chromosomes to spindle microtubules [33]. In D. melanogaster cells, we have observed relocalization of Snap29 to forming kinetochores at the onset of mitosis. Importantly, in both D. melanogaster and HeLa cells Snap29 depletion affects kinetochore formation and chromosome segregation. Electron microscopic analysis revealed that D. melanogaster Snap29 is present at the kinetochore in absence of membrane, suggesting that its association to the kinetochore might be independent of trafficking. Interestingly, RZZ, a well characterized kinetochore complex, shares components with the NRZ tethering complex that assists fusion into the ER of vesicles involved in retrograde trafficking of cargoes from the Golgi apparatus [34][35]. Very recently, the structure of the RZZ complex has been resolved by cryo-electron microscopy and proposed to be similar of that of cytosolic coat scaffolds that mediate membrane trafficking in association with molecular motors and adapters [36]. Thus, Snap29 might be part of a set of peripheral membrane proteins that are repurposed during cell division to mediate interaction of chromosomes with the microtubule cytoskeleton (Fig. 5A). While Snap29 has been found in studies of kinetochore proteins previously [37][38], further analysis is required to understand mechanistically the role of Snap29 at the kinetochore.
–
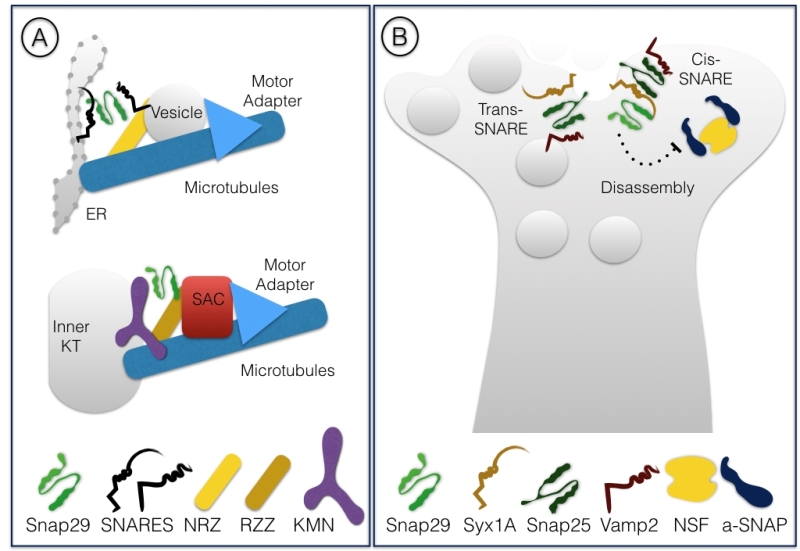 | FIGURE 5: Non canonical roles of Snap29. Schematic representation of the function of Snap29 at synaptic vesicles (A) and at the kinetochore (B). The comparison in A illustrates similarities between membrane trafficking and the membrane-independent process of kinetochore formation. The regulatory activity of SNAP29 during synaptic transmission illustrated in B is likely to involve binding to SNARE bundles containing SNAP25 (see text for details). |
SNAP29 FUNCTION IN THE NERVOUS SYSTEM: A NON CANONICAL MODE OF ACTION?
Snap29 was initially identified in immunoprecipitation experiments using rat brain extracts and in a yeast two-hybrid screen as an interactor of Syntaxin1A (Syx1A), which, together with Snap25, regulates synaptic transmission in neurons [39]. Interestingly, Snap29 was found to be able to associate to a reconstituted SNARE complex consisting of Syx1A alone, Syx1A and Snap25, as well as the synaptic trimeric complex formed by Syx1A, Snap25 and Vamp2. In these experiments, binding of αSNAP, a factor necessary for disassembly of the SNARE complex after fusion to Syx1A complexes was weakened by the presence of Snap29, suggesting that Snap29 competes with αSNAP for binding to Syx1A complexes. However, Snap29 was not found to bind directly αSNAP, indicating that in presence of Snap29 disassembly of the SNARE complex is prevented and that Snap29 is likely to be included in the trimeric Syx1A, Snap25, Vamp2 complex. Injection of Snap29 in neurons in culture decreased the amplitude of synaptic firing, an effect that was reverted by coinjection of increasing amounts of αSNAP [39]. In a subsequent study, the same group reported that hippocampal neurons depleted of Snap29 show increased efficiency of synaptic transmission [40]. These studies suggest that Snap29 acts as a modulator of synaptic vesicle fusion, perhaps by inhibiting complex disassembly or substituting for Snap25 (Fig 5B).
–
D. melanogaster Snap29 also associates to the fly Syx1A homolog Syx1. The authors showed that Snap29 is not included in Snap25-containing cis-SNARE complexes recovered from fly mutants for the disassembly factor NSF, suggesting that Snap29 might be part of unstable SNARE complexes [12]. In addition, Snap29 appears unable to rescue synaptic vesicle fusion in cultured Snap25-deficient neurons, suggesting that it is not functionally redundant with Snap25. In contrast, Snap29 is able to rescue, albeit very poorly, secretion of dense core vesicles containing neurotransmitters, which require prolonged neuron stimulation [41].
SNAP29 AND DISEASE
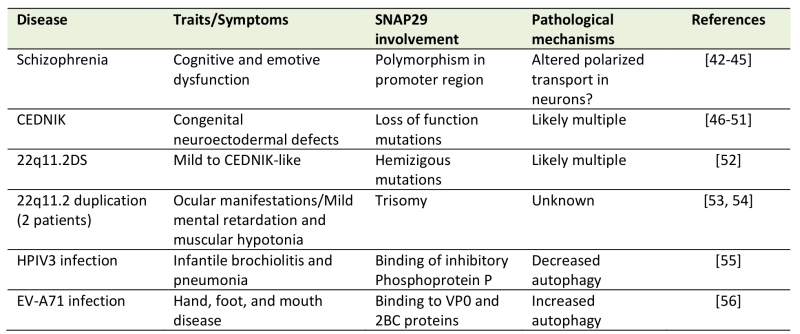
Considering the wide involvement of Snap29 in multiple trafficking and non trafficking processes in diverse cell types, it is not surprising that alteration of Snap29 is associated to human disease (Table 1). Here, we review the consequences of alteration of human SNAP29 activity and we discuss potential mechanisms of pathogenesis.
–
Table 1. List of diseases with proven alterations in SNAP29 (see text for details). |
 |
| [42][43][44][45][46][47][48][49][50][51][52][53][54][55][56] |
–
Could schizophrenia involve an alteration of SNAP29 gene activity?
An association of Snap29 with schizophrenia emerged in two early reports that revealed the presence of a polymorphism in the promoter region of SNAP29 with schizophrenia patients [42][43]. In addition, a study found the SNAP29 promoter among the many bound by β-catenin, a transcription factor regulated by lithium, an antipsychotic drug [44], and a bioinformatic analysis associated SNAP29 with a schizophrenia gene network [45]. While alterations in SNAP29 regulation in schizophrenia remain to be demonstrated, it is interesting to note that dysfunction of synaptic transmission has been proposed to be at the core of schizophrenia pathophysiology [46]. Consistent with the possibility that regulation of SNAP29 gene expression might be altered in schizophrenia, SNAP29 has been found among the interactors of the schizophrenia susceptibility factor dysbindin, a component of the BLOC-1 complex, together with the Golgi adapter COG6 and the polarized transport molecules AP3, SEC6 and SEC8 in human neuroblast cells [47]. Such evidence and that linking Snap29 to secretion and synaptic transmission (see previous chapter) suggest that Snap29 might act in trafficking processes subverted in schizophrenia.
–
CEDNIK and other rare syndromes are caused by mutations in SNAP29
A more direct link of Snap29 with disease emerged in 2005. Indeed, it was reported that loss of SNAP29 cause CEDNIK (cerebral dysgenesis, neuropathy, ichthyosis, and keratoderma), a rare recessive congenital syndrome. In CEDNIK patients, homozygous SNAP29 loss of function mutations cause a typical set of neurocutaneous traits that results in very poor life expectancy [48][49][50][51][52]. The alterations due to lack of SNAP29 have been analyzed in the stratum corneum of skin of patients, which presents an accumulation of glucosylceramides normally secreted towards the upper layers, suggesting the existence of a defect in secretion of lamellar granules containing lipids and proteolytic enzymes important for normal skin development. Consistent with the role that SNAP29 plays in membrane trafficking, fibroblasts of CEDNIK patient possess aberrant morphology of the Golgi apparatus and of recycling endosomes, as well as defects in Golgi trafficking, endocytic recycling and cell motility, overall suggesting that alteration of these process might contribute to the pathogenesis of CEDNIK [53].
–
Patients affected by a 22q11.2 deletion syndrome (22q11.2DS), a relatively common micro-deletion of the region that includes the SNAP29 gene, present a wide range of phenotypic abnormalities including immunodeficiency, palatal anomalies, congenital cardiovascular defects and additional symptoms varying from patient to patient. An exome study of 22q11.2 deletion syndrome patients revealed novel mutations in the remaining copy of SNAP29, suggesting that hemizygous mutations might unmask a recessive CEDNIK-like condition contributing to the phenotypic variety of patients [54]. A similar scenario could apply to a patient affected by a uncommon form of 22q11.2DS [55], and to Di George syndrome, a rare multi-systemic condition also caused by heterozygous de novo deletions of the 22q11.2 region, which contains SNAP29 and other 30-40 genes [56].
–
Finally, duplications in the region containing SNAP29 have been found in 2 patients with congenital ocular, vascular and cranial nerve defects [57] and in 1 patient with mild facial dysmorphism and motor and intellectual delay [58]. Overall, multiple congenital defects are associated to SNAP29 alterations. However, given the pleiotropy of SNAP29 activity, the contribution of altered SNAP29 activity to the traits of these diseases appear complex and requires further examination.
–
Infection is regulated by SNAP29 activity
The utmost importance of SNAP29 for regulation of fusion between autophagosomes and lysosomes is underscored by the finding that the capsid phosphoprotein P of human para influenza virus type 3 (HPIV3) is able to bind both SNAP29 SNARE domains. This interaction prevents binding of SNAP29 with SYX17, possibly preventing the formation of the ternary SNARE complex with VAMP8, required for autophagosome degradation. Since the accumulation of autophagosomes within the host cell is a prerequisite for virus particle release in the extracellular space, the study suggests that SNAP29 activity is a key factor to prevent HPIV3 infection [59].
–
The association of Snap29 with viral infection is not limited to HPIV3. In fact, a recent report showed that SNAP29 binds a structural protein of the enterovirus-A71 (EV-A71), which appear to up regulate autophagy for its replication [60]. While the mechanism of SNAP29 regulation of EV-A71 is not yet clear, these two papers and the emerging notion that subversion of autophagy is a key step in the life cycle of viruses [61][62][63], indicates that SNAP29 might be a crucial factor to prevent virus infection.
–
The activity of SNAP29 to prevent infection is also potentially relevant to bacterial pathogens. Indeed, in mouse mast cells, an immune cell type specialized in clearing bacterial infections, Snap29 associates to E. coli containing phagosomes and its overexpression increases lysosomal clearance, suggesting that Snap29 assists antibacterial phagocytosis [64].
–
The pleiotropy of Snap29 activity and the massive increase in –omics approaches suggests that further association of Snap29 with common and rare disease might emerge. Considering the activities of Snap29 in processes associated to signal transduction, cell motility, cell division, autophagy and synaptic transmission, we predict an involvement in tumorigenesis and neurodegeneration.
CONCLUSION
In the 20 years after Snap29 discovery, multiple studies have revealed a wide range of cellular processes controlled by the SNARE. Despite this, a number of outstanding question regarding the detailed mechanism of Snap29 remain unanswered. Here, we list some of the most outstanding.
–
1. As is the case of its paralog Snap23, Snap29 activity is not restricted a limited set of cellular locales. However, the mode of Snap29 targeting to membrane compartments appears radically different from that of Snap23 (and of Snap25), given the absence of the cysteine rich region and the charged amino acids that regulate membrane association [65]. One factor that could regulate targeting of Snap29, at least during endocytic recycling is its interactor EHD1. However, how Snap29 is recruited to membranes during other trafficking processes remains to be understood.
–
2. So far the only reported post-translational modification of Snap29 is a O-GlcNac modification thought to regulate its activity during autophagy [18]. Whether such modification affects other Snap29 functions is not understood and it remains to be determined whether other modifications might control Snap29 function.
–
3. How Snap29 acts in the SNARE complex is not entirely clear. While Snap29 appear to act positively in membrane fusion in multiple context, the inclusion of Snap29 in a 4-helix bundle has been demonstrated in vitro by X-ray crystallography only using its SNARE domains in separated form [17]. The parallel orientation (N- to C- terminal) of each SNARE domain in the bundle posits that the linker region between the SNARE domains of Snap29 has to be disordered as that of Snap25 or Snap23. This possibility has not been yet tested.
–
4. Whether the 1st SNARE domain of Snap29 corresponds to a Qb and the 2nd to a Qc is also not clear and has been inferred mostly by analogy with Snap25 and Snap23. A large number of bundles employ 4 SNARE proteins, rather than 3, each carrying a SNARE domain. Thus, if the linker region of Snap29 was rigid enough to prevent inclusion of Snap29 in a single bundle, the paradigm of activity based on that of Snap25 would have to be revised. One interesting, albeit currently only theoretical, possibility is that Snap29 could tether multiple SNARE complexes by lending the 1st SNARE domain to a complex and the 2nd to the next. In this scenario, multiple identical bundles could be tethered without violating the Qa-, Qb-, Qc- R-SNARE rule of bundle formation.
–
5. Clarity on the molecular nature of the complexed formed by Snap29 will also aid to understand its role in synaptic transmission, which is likely relevant to the pathogenesis of CEDNIK and perhaps Schizophrenia. As explained above, Snap29 seems to play a regulatory role in neurons. Whether this is due to it being a competitor of Snap25 that is less efficient in membrane fusion, or whether Snap29 plays an inhibitory role in synaptic transmission remains to be determined. Interestingly, inhibitory SNAREs have been described [66] and the cytoplasmic localization of Snap29 could allow recruitment to SNAREs to regulate their availability to form a canonical bundle engaged in fusion. Both scenarios are consistent with the reported propensity of Snap29 to loosely associate to other SNAREs [12][67][68].
–
Remarkably, the yeast S. cerevisiae possesses two SNAP family members Sec9 and Spo20, the latter specifically required during meiosis to connect the prospore membrane to forming gametes during ascospore morphogenesis [69]. Such event involves the enveloping of haploid nuclei by a double membrane, to form spores morphologically similar to autophagosomes. Similar to the case of Snap25 and Snap29, molecular dissection in vitro of Sec9 and Spo20 revealed that the former is a more active and more tightly bound to partner SNAREs [70]. While the evolutionary relationships between the yeast and metazoan SNAP family members are not clear, it is tantalizing to speculate that Snap29 might be a metazoan evolutionary solution to a common eukaryotic necessity to diversify SNAP protein activity, to accommodate cell trafficking and, perhaps, non trafficking needs.
References
- V. Protopopov, B. Govindan, P. Novick, and J.E. Gerst, "Homologs of the synaptobrevin/VAMP family of synaptic vesicle proteins function on the late secretory pathway in S. cerevisiae", Cell, vol. 74, pp. 855-861, 1993. http://dx.doi.org/10.1016/0092-8674(93)90465-3
- T. Söllner, S.W. Whiteheart, M. Brunner, H. Erdjument-Bromage, S. Geromanos, P. Tempst, and J.E. Rothman, "SNAP receptors implicated in vesicle targeting and fusion", Nature, vol. 362, pp. 318-324, 1993. http://dx.doi.org/10.1038/362318a0
- J.E. Rothman, and G. Warren, "Implications of the SNARE hypothesis for intracellular membrane topology and dynamics", Current Biology, vol. 4, pp. 220-233, 1994. http://dx.doi.org/10.1016/s0960-9822(00)00051-8
- W. Hong, "SNAREs and traffic", Biochimica et Biophysica Acta (BBA) - Molecular Cell Research, vol. 1744, pp. 120-144, 2005. http://dx.doi.org/10.1016/j.bbamcr.2005.03.014
- M. Steegmaier, B. Yang, J. Yoo, B. Huang, M. Shen, S. Yu, Y. Luo, and R.H. Scheller, "Three Novel Proteins of the Syntaxin/SNAP-25 Family", Journal of Biological Chemistry, vol. 273, pp. 34171-34179, 1998. http://dx.doi.org/10.1074/jbc.273.51.34171
- M. Holt, F. Varoqueaux, K. Wiederhold, S. Takamori, H. Urlaub, D. Fasshauer, and R. Jahn, "Identification of SNAP-47, a Novel Qbc-SNARE with Ubiquitous Expression", Journal of Biological Chemistry, vol. 281, pp. 17076-17083, 2006. http://dx.doi.org/10.1074/jbc.M513838200
- S.H. Wong, Y. Xu, T. Zhang, G. Griffiths, S.L. Lowe, V.N. Subramaniam, K.T. Seow, and W. Hong, "GS32, a Novel Golgi SNARE of 32 kDa, Interacts Preferentially with Syntaxin 6", Molecular Biology of the Cell, vol. 10, pp. 119-134, 1999. http://dx.doi.org/10.1091/mbc.10.1.119
- A.C. Hohenstein, and P.A. Roche, "SNAP-29 Is a Promiscuous Syntaxin-Binding SNARE", Biochemical and Biophysical Research Communications, vol. 285, pp. 167-171, 2001. http://dx.doi.org/10.1006/bbrc.2001.5141
- R. Rotem-Yehudar, E. Galperin, and M. Horowitz, "Association of Insulin-like Growth Factor 1 Receptor with EHD1 and SNAP29", Journal of Biological Chemistry, vol. 276, pp. 33054-33060, 2001. http://dx.doi.org/10.1074/jbc.M009913200
- Y. Xu, H. Shi, S. Wei, S.H. Wong, and W. Hong, "Mutually exclusive interactions of EHD1 with GS32 and Syndapin II", Molecular Membrane Biology, vol. 21, pp. 269-277, 2004. http://dx.doi.org/10.1080/09687680410001716871
- Q. Lu, C. Insinna, C. Ott, J. Stauffer, P.A. Pintado, J. Rahajeng, U. Baxa, V. Walia, A. Cuenca, Y. Hwang, I.O. Daar, S. Lopes, J. Lippincott-Schwartz, P.K. Jackson, S. Caplan, and C.J. Westlake, "Early steps in primary cilium assembly require EHD1/EHD3-dependent ciliary vesicle formation", Nature Cell Biology, vol. 17, pp. 228-240, 2015. http://dx.doi.org/10.1038/ncb3109
- "Array", . http://dx.doi.org/10.1371/journal.pone.0091471.t001
- N. Mizushima, and M. Komatsu, "Autophagy: Renovation of Cells and Tissues", Cell, vol. 147, pp. 728-741, 2011. http://dx.doi.org/10.1016/j.cell.2011.10.026
- E. Itakura, C. Kishi-Itakura, and N. Mizushima, "The Hairpin-type Tail-Anchored SNARE Syntaxin 17 Targets to Autophagosomes for Fusion with Endosomes/Lysosomes", Cell, vol. 151, pp. 1256-1269, 2012. http://dx.doi.org/10.1016/j.cell.2012.11.001
- S. Takáts, P. Nagy, �. Varga, K. Pircs, M. Kárpáti, K. Varga, A.L. Kovács, K. Hegedűs, and G. Juhász, "Autophagosomal Syntaxin17-dependent lysosomal degradation maintains neuronal function in Drosophila", Journal of Cell Biology, vol. 201, pp. 531-539, 2013. http://dx.doi.org/10.1083/jcb.201211160
- E. Morelli, P. Ginefra, V. Mastrodonato, G.V. Beznoussenko, T.E. Rusten, D. Bilder, H. Stenmark, A.A. Mironov, and T. Vaccari, "Multiple functions of the SNARE protein Snap29 in autophagy, endocytic, and exocytic trafficking during epithelial formation inDrosophila", Autophagy, vol. 10, pp. 2251-2268, 2014. http://dx.doi.org/10.4161/15548627.2014.981913
- J. Diao, R. Liu, Y. Rong, M. Zhao, J. Zhang, Y. Lai, Q. Zhou, L.M. Wilz, J. Li, S. Vivona, R.A. Pfuetzner, A.T. Brunger, and Q. Zhong, "ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes", Nature, vol. 520, pp. 563-566, 2015. http://dx.doi.org/10.1038/nature14147
- B. Guo, Q. Liang, L. Li, Z. Hu, F. Wu, P. Zhang, Y. Ma, B. Zhao, A.L. Kovács, Z. Zhang, D. Feng, S. Chen, and H. Zhang, "O-GlcNAc-modification of SNAP-29 regulates autophagosome maturation", Nature Cell Biology, vol. 16, pp. 1215-1226, 2014. http://dx.doi.org/10.1038/ncb3066
- S. Takáts, K. Pircs, P. Nagy, �. Varga, M. Kárpáti, K. Hegedűs, H. Kramer, A.L. Kovács, M. Sass, and G. Juhász, "Interaction of the HOPS complex with Syntaxin 17 mediates autophagosome clearance inDrosophila", Molecular Biology of the Cell, vol. 25, pp. 1338-1354, 2014. http://dx.doi.org/10.1091/mbc.E13-08-0449
- R. Jia, C.M. Guardia, J. Pu, Y. Chen, and J.S. Bonifacino, "BORC coordinates encounter and fusion of lysosomes with autophagosomes", Autophagy, vol. 13, pp. 1648-1663, 2017. http://dx.doi.org/10.1080/15548627.2017.1343768
- Z. Wang, G. Miao, X. Xue, X. Guo, C. Yuan, Z. Wang, G. Zhang, Y. Chen, D. Feng, J. Hu, and H. Zhang, "The Vici Syndrome Protein EPG5 Is a Rab7 Effector that Determines the Fusion Specificity of Autophagosomes with Late Endosomes/Lysosomes", Molecular Cell, vol. 63, pp. 781-795, 2016. http://dx.doi.org/10.1016/j.molcel.2016.08.021
- G. McLelland, S.A. Lee, H.M. McBride, and E.A. Fon, "Syntaxin-17 delivers PINK1/parkin-dependent mitochondrial vesicles to the endolysosomal system", Journal of Cell Biology, vol. 214, pp. 275-291, 2016. http://dx.doi.org/10.1083/jcb.201603105
- J. Kang, Z. Bai, M.H. Zegarek, B.D. Grant, and J. Lee, "Essential roles of snap-29 in C. elegans", Developmental Biology, vol. 355, pp. 77-88, 2011. http://dx.doi.org/10.1016/j.ydbio.2011.04.013
- M. Sato, K. Saegusa, K. Sato, T. Hara, A. Harada, and K. Sato, "Caenorhabditis elegansSNAP-29 is required for organellar integrity of the endomembrane system and general exocytosis in intestinal epithelial cells", Molecular Biology of the Cell, vol. 22, pp. 2579-2587, 2011. http://dx.doi.org/10.1091/mbc.E11-04-0279
-
T. Kudlyk, R. Willett, I.D. Pokrovskaya, and V. Lupashin, "
COG6 Interacts with a Subset of the GolgiSNAREs and Is Important for the Golgi Complex Integrity", Traffic, vol. 14, pp. 194-204, 2012. http://dx.doi.org/10.1111/tra.12020 - R. Willett, T. Kudlyk, I. Pokrovskaya, R. Schönherr, D. Ungar, R. Duden, and V. Lupashin, "COG complexes form spatial landmarks for distinct SNARE complexes", Nature Communications, vol. 4, 2013. http://dx.doi.org/10.1038/ncomms2535
-
T. Kimura, J. Jia, S. Kumar, S.W. Choi, Y. Gu, M. Mudd, N. Dupont, S. Jiang, R. Peters, F. Farzam, A. Jain, K.A. Lidke, C.M. Adams, T. Johansen, and V. Deretic, "Dedicated
SNARE s and specializedTRIM cargo receptors mediate secretory autophagy", The EMBO Journal, vol. 36, pp. 42-60, 2016. http://dx.doi.org/10.15252/embj.201695081 - N. Dupont, S. Jiang, M. Pilli, W. Ornatowski, D. Bhattacharya, and V. Deretic, "Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β", The EMBO Journal, vol. 30, pp. 4701-4711, 2011. http://dx.doi.org/10.1038/emboj.2011.398
- M. Zhang, S.J. Kenny, L. Ge, K. Xu, and R. Schekman, "Translocation of interleukin-1β into a vesicle intermediate in autophagy-mediated secretion", eLife, vol. 4, 2015. http://dx.doi.org/10.7554/eLife.11205
- K.M. Naegeli, E. Hastie, A. Garde, Z. Wang, D.P. Keeley, K.L. Gordon, A.M. Pani, L.C. Kelley, M.A. Morrissey, Q. Chi, B. Goldstein, and D.R. Sherwood, "Cell Invasion In Vivo via Rapid Exocytosis of a Transient Lysosome-Derived Membrane Domain", Developmental Cell, vol. 43, pp. 403-417.e10, 2017. http://dx.doi.org/10.1016/j.devcel.2017.10.024
- T. Csizmadia, P. Lőrincz, K. Hegedűs, S. Széplaki, P. Lőw, and G. Juhász, "Molecular mechanisms of developmentally programmed crinophagy in Drosophila", Journal of Cell Biology, vol. 217, pp. 361-374, 2017. http://dx.doi.org/10.1083/jcb.201702145
- A. Schardt, B.G. Brinkmann, M. Mitkovski, M.W. Sereda, H.B. Werner, and K. Nave, "The SNARE protein SNAP‐29 interacts with the GTPase Rab3A: Implications for membrane trafficking in myelinating glia", Journal of Neuroscience Research, vol. 87, pp. 3465-3479, 2009. http://dx.doi.org/10.1002/jnr.22005
-
E. Morelli, V. Mastrodonato, G.V. Beznoussenko, A.A. Mironov, E. Tognon, and T. Vaccari, "An essential step of kinetochore formation controlled by the
SNARE protein Snap29", The EMBO Journal, vol. 35, pp. 2223-2237, 2016. http://dx.doi.org/10.15252/embj.201693991 - F. Çivril, A. Wehenkel, F.M. Giorgi, S. Santaguida, A. Di Fonzo, G. Grigorean, F.D. Ciccarelli, and A. Musacchio, "Structural Analysis of the RZZ Complex Reveals Common Ancestry with Multisubunit Vesicle Tethering Machinery", Structure, vol. 18, pp. 616-626, 2010. http://dx.doi.org/10.1016/j.str.2010.02.014
- A. Wainman, M.G. Giansanti, M.L. Goldberg, and M. Gatti, "TheDrosophilaRZZ complex: roles in membrane traffic and cytokinesis", Journal of Cell Science, 2012. http://dx.doi.org/10.1242/jcs.099820
- S. Mosalaganti, J. Keller, A. Altenfeld, M. Winzker, P. Rombaut, M. Saur, A. Petrovic, A. Wehenkel, S. Wohlgemuth, F. Müller, S. Maffini, T. Bange, F. Herzog, H. Waldmann, S. Raunser, and A. Musacchio, "Structure of the RZZ complex and molecular basis of its interaction with Spindly", Journal of Cell Biology, vol. 216, pp. 961-981, 2017. http://dx.doi.org/10.1083/jcb.201611060
- J.P. Welburn, E.L. Grishchuk, C.B. Backer, E.M. Wilson-Kubalek, J.R. Yates, and I.M. Cheeseman, "The Human Kinetochore Ska1 Complex Facilitates Microtubule Depolymerization-Coupled Motility", Developmental Cell, vol. 16, pp. 374-385, 2009. http://dx.doi.org/10.1016/j.devcel.2009.01.011
- J.R.A. Hutchins, Y. Toyoda, B. Hegemann, I. Poser, J. Hériché, M.M. Sykora, M. Augsburg, O. Hudecz, B.A. Buschhorn, J. Bulkescher, C. Conrad, D. Comartin, A. Schleiffer, M. Sarov, A. Pozniakovsky, M.M. Slabicki, S. Schloissnig, I. Steinmacher, M. Leuschner, A. Ssykor, S. Lawo, L. Pelletier, H. Stark, K. Nasmyth, J. Ellenberg, R. Durbin, F. Buchholz, K. Mechtler, A.A. Hyman, and J. Peters, "Systematic Analysis of Human Protein Complexes Identifies Chromosome Segregation Proteins", Science, vol. 328, pp. 593-599, 2010. http://dx.doi.org/10.1126/science.1181348
- Q. Su, S. Mochida, J. Tian, R. Mehta, and Z. Sheng, "SNAP-29: A general SNARE protein that inhibits SNARE disassembly and is implicated in synaptic transmission", Proceedings of the National Academy of Sciences, vol. 98, pp. 14038-14043, 2001. http://dx.doi.org/10.1073/pnas.251532398
- P. Pan, Q. Cai, L. Lin, P. Lu, S. Duan, and Z. Sheng, "SNAP-29-mediated Modulation of Synaptic Transmission in CulturedHippocampalNeurons", Journal of Biological Chemistry, vol. 280, pp. 25769-25779, 2005. http://dx.doi.org/10.1074/jbc.M502356200
- S. Arora, I. Saarloos, R. Kooistra, R. van de Bospoort, M. Verhage, and R.F. Toonen, "SNAP-25 gene family members differentially support secretory vesicle fusion", Journal of Cell Science, vol. 130, pp. 1877-1889, 2017. http://dx.doi.org/10.1242/jcs.201889
- T. Saito, F. Guan, D.F. Papolos, N. Rajouria, C.S.J. Fann, and H.M. Lachman, "Polymorphism in SNAP29 gene promoter region associated with schizophrenia", Molecular Psychiatry, vol. 6, pp. 193-201, 2001. http://dx.doi.org/10.1038/sj.mp.4000825
- I. Wonodi, L.E. Hong, M.T. Avila, R.W. Buchanan, W.T. Carpenter, O.C. Stine, B.D. Mitchell, and G.K. Thaker, "Association between polymorphism of the SNAP29 gene promoter region and schizophrenia", Schizophrenia Research, vol. 78, pp. 339-341, 2005. http://dx.doi.org/10.1016/j.schres.2005.03.023
- E. Pedrosa, A. Shah, C. Tenore, M. Capogna, C. Villa, X. Guo, D. Zheng, and H.M. Lachman, "β-Catenin Promoter ChIP-Chip Reveals Potential Schizophrenia and Bipolar Disorder Gene Network", Journal of Neurogenetics, vol. 24, pp. 182-193, 2010. http://dx.doi.org/10.3109/01677063.2010.495182
- X. Luo, L. Huang, L. Han, Z. Luo, F. Hu, R. Tieu, and L. Gan, "Systematic Prioritization and Integrative Analysis of Copy Number Variations in Schizophrenia Reveal Key Schizophrenia Susceptibility Genes", Schizophrenia Bulletin, vol. 40, pp. 1285-1299, 2014. http://dx.doi.org/10.1093/schbul/sbu045
- W. Frankle, J. Lerma, and M. Laruelle, "The Synaptic Hypothesis of Schizophrenia", Neuron, vol. 39, pp. 205-216, 2003. http://dx.doi.org/10.1016/s0896-6273(03)00423-9
- A. Gokhale, J. Larimore, E. Werner, L. So, A. Moreno-De-Luca, C. Lese-Martin, V.V. Lupashin, Y. Smith, and V. Faundez, "Quantitative Proteomic and Genetic Analyses of the Schizophrenia Susceptibility Factor Dysbindin Identify Novel Roles of the Biogenesis of Lysosome-Related Organelles Complex 1", The Journal of Neuroscience, vol. 32, pp. 3697-3711, 2012. http://dx.doi.org/10.1523/jneurosci.5640-11.2012
- E. Sprecher, A. Ishida-Yamamoto, M. Mizrahi-Koren, D. Rapaport, D. Goldsher, M. Indelman, O. Topaz, I. Chefetz, H. Keren, T.J. O’Brien, D. Bercovich, S. Shalev, D. Geiger, R. Bergman, M. Horowitz, and H. Mandel, "A Mutation in SNAP29, Coding for a SNARE Protein Involved in Intracellular Trafficking, Causes a Novel Neurocutaneous Syndrome Characterized by Cerebral Dysgenesis, Neuropathy, Ichthyosis, and Palmoplantar Keratoderma", The American Journal of Human Genetics, vol. 77, pp. 242-251, 2005. http://dx.doi.org/10.1086/432556
- D. Fuchs-Telem, H. Stewart, D. Rapaport, J. Nousbeck, A. Gat, M. Gini, Y. Lugassy, S. Emmert, K. Eckl, H. Hennies, O. Sarig, D. Goldsher, B. Meilik, A. Ishida-Yamamoto, M. Horowitz, and E. Sprecher, "CEDNIK syndrome results from loss-of-function mutations in SNAP29", British Journal of Dermatology, pp. no-no, 2011. http://dx.doi.org/10.1111/j.1365-2133.2010.10133.x
- S. Ben‐Salem, S. Nara, A.M. Al‐Shamsi, D. Valle, B.R. Ali, and L. Al‐Gazali, "New Arab family with cerebral dysgenesis, neuropathy, ichthyosis and keratoderma syndrome suggests a possible founder effect for the c.223delG mutation", The Journal of Dermatology, vol. 42, pp. 821-822, 2015. http://dx.doi.org/10.1111/1346-8138.12917
- C.P. Diggle, I. Martinez-Garay, Z. Molnar, M.H. Brinkworth, E. White, E. Fowler, R. Hughes, B.E. Hayward, I.M. Carr, C.M. Watson, L. Crinnion, A. Asipu, B. Woodman, P.L. Coletta, A.F. Markham, T.N. Dear, D.T. Bonthron, M. Peckham, E.E. Morrison, and E. Sheridan, "A tubulin alpha 8 mouse knockout model indicates a likely role in spermatogenesis but not in brain development", PLOS ONE, vol. 12, pp. e0174264, 2017. http://dx.doi.org/10.1371/journal.pone.0174264
- T. Hsu, C.C. Coughlin, K.G. Monaghan, E. Fiala, R.C. McKinstry, A.R. Paciorkowski, and M. Shinawi, "CEDNIK", Child Neurology Open, vol. 4, pp. 2329048X1773321, 2017. http://dx.doi.org/10.1177/2329048X17733214
- D. Rapaport, Y. Lugassy, E. Sprecher, and M. Horowitz, "Loss of SNAP29 Impairs Endocytic Recycling and Cell Motility", PLoS ONE, vol. 5, pp. e9759, 2010. http://dx.doi.org/10.1371/journal.pone.0009759
- D.M. McDonald-McGinn, S. Fahiminiya, T. Revil, B.A. Nowakowska, J. Suhl, A. Bailey, E. Mlynarski, D.R. Lynch, A.C. Yan, L.T. Bilaniuk, K.E. Sullivan, S.T. Warren, B.S. Emanuel, J.R. Vermeesch, E.H. Zackai, and L.A. Jerome-Majewska, "Hemizygous mutations in SNAP29 unmask autosomal recessive conditions and contribute to atypical findings in patients with 22q11.2DS", Journal of Medical Genetics, vol. 50, pp. 80-90, 2012. http://dx.doi.org/10.1136/jmedgenet-2012-101320
- D.C. de Queiroz Soares, R.L. Dutra, C.R. D'angioli Costa Quaio, M.I. Melaragno, L.D. Kulikowski, L.C. Torres, and C.A. Kim, "Role of SNAP29, LZTR1 and P2RXL1 genes on immune regulation in a patient with atypical 0.5Mb deletion in 22q11.2 region", Clinical Immunology, vol. 145, pp. 55-58, 2012. http://dx.doi.org/10.1016/j.clim.2012.07.013
- E. Lopez-Rivera, Y.P. Liu, M. Verbitsky, B.R. Anderson, V.P. Capone, E.A. Otto, Z. Yan, A. Mitrotti, J. Martino, N.J. Steers, D.A. Fasel, K. Vukojevic, R. Deng, S.E. Racedo, Q. Liu, M. Werth, R. Westland, A. Vivante, G.S. Makar, M. Bodria, M.G. Sampson, C.E. Gillies, V. Vega-Warner, M. Maiorana, D.S. Petrey, B. Honig, V.J. Lozanovski, R. Salomon, L. Heidet, W. Carpentier, D. Gaillard, A. Carrea, L. Gesualdo, D. Cusi, C. Izzi, F. Scolari, J.A. van Wijk, A. Arapovic, M. Saraga-Babic, M. Saraga, N. Kunac, A. Samii, D.M. McDonald-McGinn, T.B. Crowley, E.H. Zackai, D. Drozdz, M. Miklaszewska, M. Tkaczyk, P. Sikora, M. Szczepanska, M. Mizerska-Wasiak, G. Krzemien, A. Szmigielska, M. Zaniew, J.M. Darlow, P. Puri, D. Barton, E. Casolari, S.L. Furth, B.A. Warady, Z. Gucev, H. Hakonarson, H. Flogelova, V. Tasic, A. Latos-Bielenska, A. Materna-Kiryluk, L. Allegri, C.S. Wong, I.A. Drummond, V. D’Agati, A. Imamoto, J.M. Barasch, F. Hildebrandt, K. Kiryluk, R.P. Lifton, B.E. Morrow, C. Jeanpierre, V.E. Papaioannou, G.M. Ghiggeri, A.G. Gharavi, N. Katsanis, and S. Sanna-Cherchi, "Genetic Drivers of Kidney Defects in the DiGeorge Syndrome", New England Journal of Medicine, vol. 376, pp. 742-754, 2017. http://dx.doi.org/10.1056/NEJMoa1609009
- J.A. Cordovez, J. Capasso, M.D. Lingao, K.A. Sadagopan, G.L. Spaeth, B.N. Wasserman, and A.V. Levin, "Ocular Manifestations of 22q11.2 Microduplication", Ophthalmology, vol. 121, pp. 392-398, 2014. http://dx.doi.org/10.1016/j.ophtha.2013.06.040
- C. Pebrel-Richard, S. Kemeny, L. Gouas, E. Eymard-Pierre, N. Blanc, C. Francannet, A. Tchirkov, C. Goumy, and P. Vago, "An atypical 0.8 Mb inherited duplication of 22q11.2 associated with psychomotor impairment", European Journal of Medical Genetics, vol. 55, pp. 650-655, 2012. http://dx.doi.org/10.1016/j.ejmg.2012.06.014
- B. Ding, G. Zhang, X. Yang, S. Zhang, L. Chen, Q. Yan, M. Xu, A. Banerjee, and M. Chen, "Phosphoprotein of Human Parainfluenza Virus Type 3 Blocks Autophagosome-Lysosome Fusion to Increase Virus Production", Cell Host & Microbe, vol. 15, pp. 564-577, 2014. http://dx.doi.org/10.1016/j.chom.2014.04.004
- J. Lai, I. Sam, P. Verlhac, J. Baguet, E. Eskelinen, M. Faure, and Y. Chan, "2BC Non-Structural Protein of Enterovirus A71 Interacts with SNARE Proteins to Trigger Autolysosome Formation", Viruses, vol. 9, pp. 169, 2017. http://dx.doi.org/10.3390/v9070169
- J. Wong, J. Zhang, X. Si, G. Gao, I. Mao, B.M. McManus, and H. Luo, "Autophagosome Supports Coxsackievirus B3 Replication in Host Cells", Journal of Virology, vol. 82, pp. 9143-9153, 2008. http://dx.doi.org/10.1128/jvi.00641-08
- M. Dreux, P. Gastaminza, S.F. Wieland, and F.V. Chisari, "The autophagy machinery is required to initiate hepatitis C virus replication", Proceedings of the National Academy of Sciences, vol. 106, pp. 14046-14051, 2009. http://dx.doi.org/10.1073/pnas.0907344106
- Y. Shi, X. He, G. Zhu, H. Tu, Z. Liu, W. Li, S. Han, J. Yin, B. Peng, and W. Liu, "Coxsackievirus A16 Elicits Incomplete Autophagy Involving the mTOR and ERK Pathways", PLOS ONE, vol. 10, pp. e0122109, 2015. http://dx.doi.org/10.1371/journal.pone.0122109
- J. Wesolowski, V. Caldwell, and F. Paumet, "A Novel Function for SNAP29 (Synaptosomal-Associated Protein of 29 kDa) in Mast Cell Phagocytosis", PLoS ONE, vol. 7, pp. e49886, 2012. http://dx.doi.org/10.1371/journal.pone.0049886
- P. Weber, H. Batoulis, K.M. Rink, S. Dahlhoff, K. Pinkwart, T.H. Söllner, and T. Lang, "Electrostatic anchoring precedes stable membrane attachment of SNAP25/SNAP23 to the plasma membrane", eLife, vol. 6, 2017. http://dx.doi.org/10.7554/eLife.19394
- "Array", . http://dx.doi.org/10.1371/journal.pone.0007375.t002
- B. Yang, L. Gonzalez, R. Prekeris, M. Steegmaier, R.J. Advani, and R.H. Scheller, "SNARE Interactions Are Not Selective", Journal of Biological Chemistry, vol. 274, pp. 5649-5653, 1999. http://dx.doi.org/10.1074/jbc.274.9.5649
- S.J. Scales, Y.A. Chen, B.Y. Yoo, S.M. Patel, Y. Doung, and R.H. Scheller, "SNAREs Contribute to the Specificity of Membrane Fusion", Neuron, vol. 26, pp. 457-464, 2000. http://dx.doi.org/10.1016/s0896-6273(00)81177-0
- A.M. Neiman, "Prospore Membrane Formation Defines a Developmentally Regulated Branch of the Secretory Pathway in Yeast", The Journal of Cell Biology, vol. 140, pp. 29-37, 1998. http://dx.doi.org/10.1083/jcb.140.1.29
- H. Yang, H. Nakanishi, S. Liu, J.A. McNew, and A.M. Neiman, "Binding interactions control SNARE specificity in vivo", The Journal of Cell Biology, vol. 183, pp. 1089-1100, 2008. http://dx.doi.org/10.1083/jcb.200809178
ACKNOWLEDGMENTS
T.V. is supported by investigator grant 20661 from Associazione Italiana Ricerca su Cancro (AIRC) and investigator grant 18-0399 from Worldwide Cancer Research (WCR). E. M. receives support from a Fondazione Italiana Ricerca sul Cancro (FIRC) and V.M. is supported by a fellowship from Fondazione Veronesi.
COPYRIGHT
© 2018

How to use a multipurpose SNARE: The emerging role of Snap29 in cellular health by Mastrodonato et al. is licensed under a Creative Commons Attribution 4.0 International License.


